
Ghrelin Alleviates Experimental Ulcerative Colitis in Old Mice and Modulates Colonocyte Metabolism via PPARγ Pathway


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Ghrelin Treatment Attenuated Some Clinical Symptoms of DSS-Induced Colitis in Old Mice
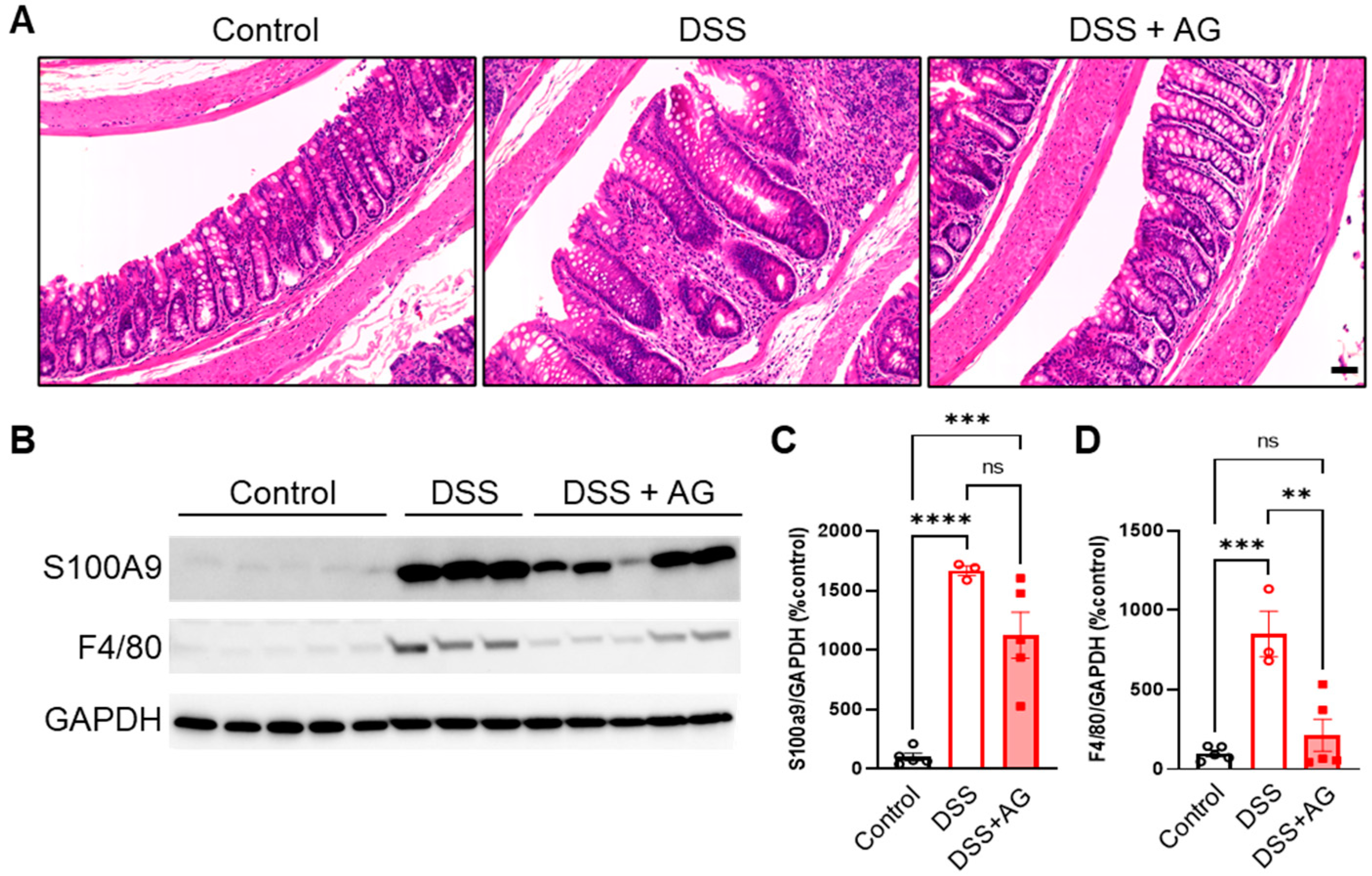
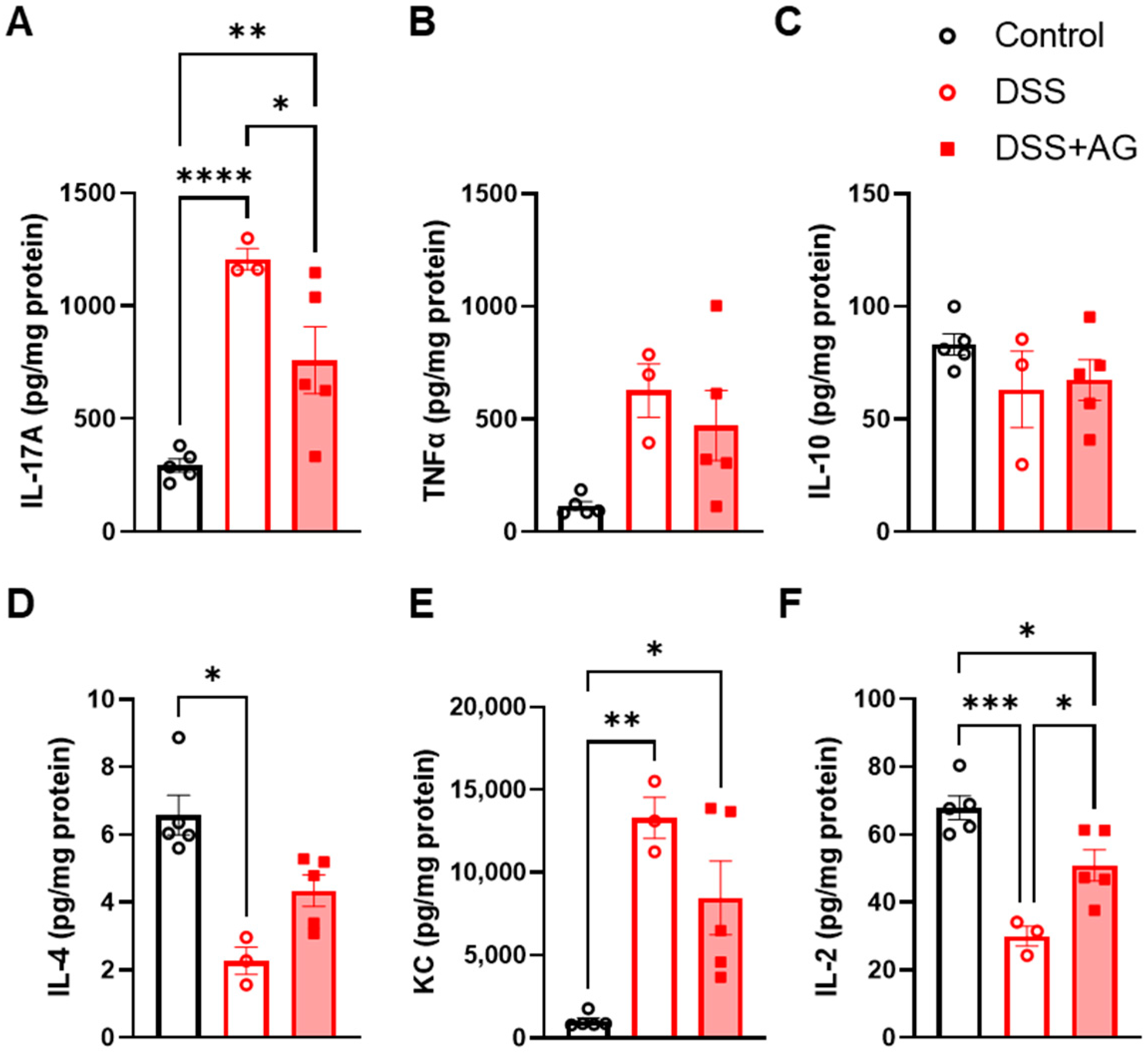
2.2. Ghrelin Treatment Reduced Colon Inflammation
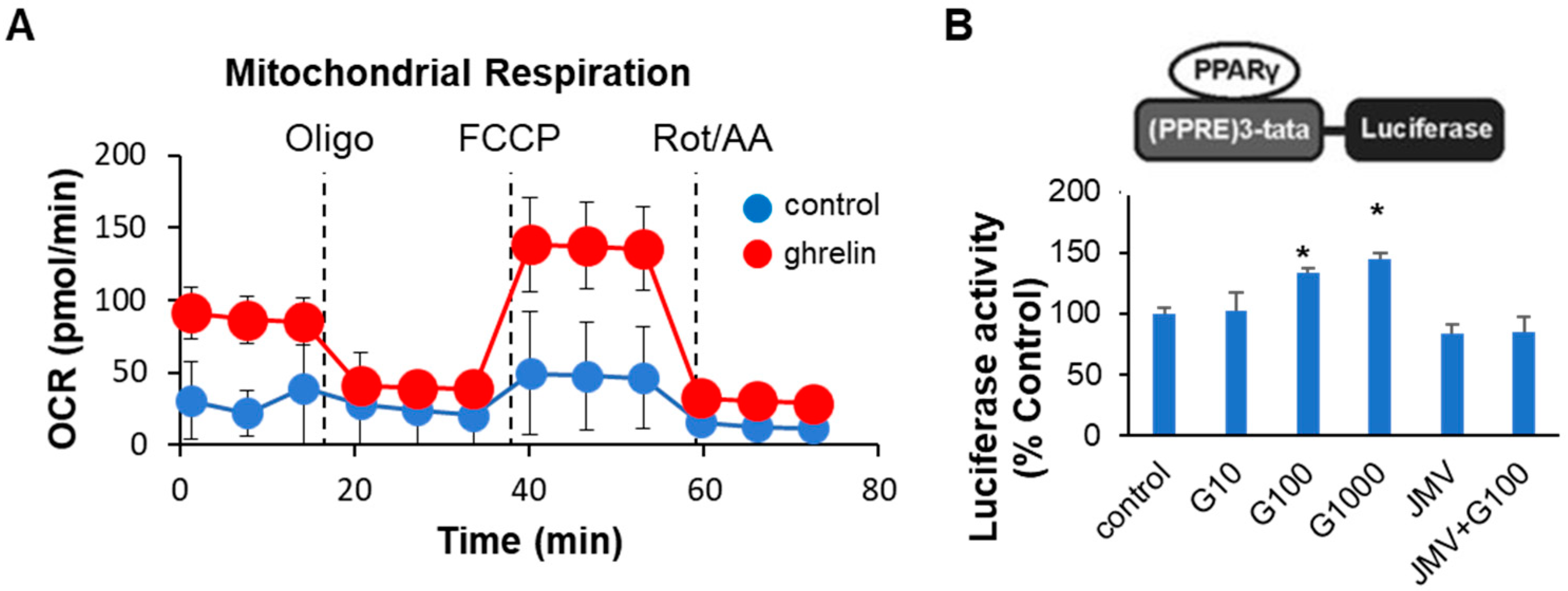
2.3. Ghrelin Increased Colonocyte Mitochondrial Function and PPARγ Transcriptional Activity
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Experimental Design
4.3. Cytokine Analyses
4.4. Western Blot Analysis
4.5. Cell Culture and Mitochondrial Function Analysis by Seahorse Flux Assay
4.6. PPARγ Reporter Assay
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ng, S.C.; Shi, H.Y.; Hamidi, N.; Underwood, F.E.; Tang, W.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Wu, J.C.Y.; Chan, F.K.L.; et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: A systematic review of population-based studies. Lancet 2017, 390, 2769–2778. [Google Scholar] [CrossRef] [PubMed]
- Nimmons, D.; Limdi, J.K. Elderly patients and inflammatory bowel disease. World J. Gastrointest. Pharm. 2016, 7, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Taleban, S.; Colombel, J.F.; Mohler, M.J.; Fain, M.J. Inflammatory bowel disease and the elderly: A review. J. Crohn’s Colitis 2015, 9, 507–515. [Google Scholar] [CrossRef] [PubMed]
- del Val, J.H. Old-age inflammatory bowel disease onset: A different problem? World J. Gastroenterol. WJG 2011, 17, 2734–2739. [Google Scholar] [CrossRef] [PubMed]
- Halfvarson, J.; Brislawn, C.J.; Lamendella, R.; Vazquez-Baeza, Y.; Walters, W.A.; Bramer, L.M.; D’Amato, M.; Bonfiglio, F.; McDonald, D.; Gonzalez, A.; et al. Dynamics of the human gut microbiome in inflammatory bowel disease. Nat. Microbiol. 2017, 2, 17004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franzosa, E.A.; Sirota-Madi, A.; Avila-Pacheco, J.; Fornelos, N.; Haiser, H.J.; Reinker, S.; Vatanen, T.; Hall, A.B.; Mallick, H.; McIver, L.J.; et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat. Microbiol. 2019, 4, 293–305. [Google Scholar] [CrossRef]
- Duvallet, C.; Gibbons, S.M.; Gurry, T.; Irizarry, R.A.; Alm, E.J. Meta-analysis of gut microbiome studies identifies disease-specific and shared responses. Nat. Commun. 2017, 8, 1784. [Google Scholar] [CrossRef] [Green Version]
- Schirmer, M.; Denson, L.; Vlamakis, H.; Franzosa, E.A.; Thomas, S.; Gotman, N.M.; Rufo, P.; Baker, S.S.; Sauer, C.; Markowitz, J.; et al. Compositional and Temporal Changes in the Gut Microbiome of Pediatric Ulcerative Colitis Patients Are Linked to Disease Course. Cell Host Microbe 2018, 24, 600–610.e4. [Google Scholar] [CrossRef] [Green Version]
- Morgan, X.C.; Tickle, T.L.; Sokol, H.; Gevers, D.; Devaney, K.L.; Ward, D.V.; Reyes, J.A.; Shah, S.A.; LeLeiko, N.; Snapper, S.B.; et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012, 13, R79. [Google Scholar] [CrossRef]
- Shaw, K.A.; Bertha, M.; Hofmekler, T.; Chopra, P.; Vatanen, T.; Srivatsa, A.; Prince, J.; Kumar, A.; Sauer, C.; Zwick, M.E.; et al. Dysbiosis, inflammation, and response to treatment: A longitudinal study of pediatric subjects with newly diagnosed inflammatory bowel disease. Genome Med. 2016, 8, 75. [Google Scholar] [CrossRef]
- Atarashi, K.; Suda, W.; Luo, C.; Kawaguchi, T.; Motoo, I.; Narushima, S.; Kiguchi, Y.; Yasuma, K.; Watanabe, E.; Tanoue, T.; et al. Ectopic colonization of oral bacteria in the intestine drives TH1 cell induction and inflammation. Science 2017, 358, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.B.; Yassour, M.; Sauk, J.; Garner, A.; Jiang, X.; Arthur, T.; Lagoudas, G.K.; Vatanen, T.; Fornelos, N.; Wilson, R.; et al. A novel Ruminococcus gnavus clade enriched in inflammatory bowel disease patients. Genome Med. 2017, 9, 103. [Google Scholar] [CrossRef] [PubMed]
- Cevallos, S.A.; Lee, J.Y.; Tiffany, C.R.; Byndloss, A.J.; Johnston, L.; Byndloss, M.X.; Baumler, A.J. Increased Epithelial Oxygenation Links Colitis to an Expansion of Tumorigenic Bacteria. mBio 2019, 10, e02244-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cevallos, S.A.; Lee, J.Y.; Velazquez, E.M.; Foegeding, N.J.; Shelton, C.D.; Tiffany, C.R.; Parry, B.H.; Stull-Lane, A.R.; Olsan, E.E.; Savage, H.P.; et al. 5-Aminosalicylic Acid Ameliorates Colitis and Checks Dysbiotic Escherichia coli Expansion by Activating PPAR-gamma Signaling in the Intestinal Epithelium. mBio 2021, 12, e03227-20. [Google Scholar] [CrossRef]
- Date, Y.; Kojima, M.; Hosoda, H.; Sawaguchi, A.; Mondal, M.S.; Suganuma, T.; Matsukura, S.; Kangawa, K.; Nakazato, M. Ghrelin, a novel growth hormone-releasing acylated peptide, is synthesized in a distinct endocrine cell type in the gastrointestinal tracts of rats and humans. Endocrinology 2000, 141, 4255–4261. [Google Scholar] [CrossRef]
- Kojima, M.; Hosoda, H.; Date, Y.; Nakazato, M.; Matsuo, H.; Kangawa, K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 1999, 402, 656–660. [Google Scholar] [CrossRef]
- Pradhan, G.; Samson, S.L.; Sun, Y. Ghrelin: Much more than a hunger hormone. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 619–624. [Google Scholar] [CrossRef] [Green Version]
- Collden, G.; Tschop, M.H.; Muller, T.D. Therapeutic Potential of Targeting the Ghrelin Pathway. Int. J. Mol. Sci. 2017, 18, 798. [Google Scholar] [CrossRef] [Green Version]
- Tuchaai, E.; Endres, V.; Jones, B.; Shankar, S.; Klemashevich, C.; Sun, Y.; Wu, C.S. Deletion of ghrelin alters tryptophan metabolism and exacerbates experimental ulcerative colitis in aged mice. Exp. Biol. Med. 2022, 247, 1558–1569. [Google Scholar] [CrossRef]
- Chang, L.; Zhao, J.; Yang, J.; Zhang, Z.; Du, J.; Tang, C. Therapeutic effects of ghrelin on endotoxic shock in rats. Eur. J. Pharmacol. 2003, 473, 171–176. [Google Scholar] [CrossRef]
- Chang, L.; Du, J.B.; Gao, L.R.; Pang, Y.Z.; Tang, C.S. Effect of ghrelin on septic shock in rats. Acta Pharm. Sin. 2003, 24, 45–49. [Google Scholar]
- Gonzalez-Rey, E.; Chorny, A.; Delgado, M. Therapeutic action of ghrelin in a mouse model of colitis. Gastroenterology 2006, 130, 1707–1720. [Google Scholar] [CrossRef] [PubMed]
- Konturek, P.C.; Brzozowski, T.; Engel, M.; Burnat, G.; Gaca, P.; Kwiecien, S.; Pajdo, R.; Konturek, S.J. Ghrelin ameliorates colonic inflammation. Role of nitric oxide and sensory nerves. J. Physiol. Pharm. 2009, 60, 41–47. [Google Scholar]
- Matuszyk, A.; Ceranowicz, P.; Warzecha, Z.; Cieszkowski, J.; Ceranowicz, D.; Galazka, K.; Bonior, J.; Jaworek, J.; Bartus, K.; Gil, K.; et al. Exogenous Ghrelin Accelerates the Healing of Acetic Acid-Induced Colitis in Rats. Int. J. Mol. Sci. 2016, 17, 1455. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Cheng, J.; Shen, J.; Wang, S.; Guo, C.; Fan, X. Ghrelin Inhibits Intestinal Epithelial Cell Apoptosis Through the Unfolded Protein Response Pathway in Ulcerative Colitis. Front. Pharm. 2021, 12, 661853. [Google Scholar] [CrossRef]
- Wu, C.S.; Wei, Q.; Wang, H.; Kim, D.M.; Balderas, M.; Wu, G.; Lawler, J.; Safe, S.; Guo, S.; Devaraj, S.; et al. Protective Effects of Ghrelin on Fasting-Induced Muscle Atrophy in Aging Mice. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, 621–630. [Google Scholar] [CrossRef]
- Kedia, S.; Ahuja, V.; Tandon, R. Management of acute severe ulcerative colitis. World J. Gastrointest. Pathophysiol. 2014, 5, 579–588. [Google Scholar] [CrossRef]
- Czarnewski, P.; Parigi, S.M.; Sorini, C.; Diaz, O.E.; Das, S.; Gagliani, N.; Villablanca, E.J. Conserved transcriptomic profile between mouse and human colitis allows unsupervised patient stratification. Nat. Commun. 2019, 10, 2892. [Google Scholar] [CrossRef] [Green Version]
- Laroui, H.; Ingersoll, S.A.; Liu, H.C.; Baker, M.T.; Ayyadurai, S.; Charania, M.A.; Laroui, F.; Yan, Y.; Sitaraman, S.V.; Merlin, D. Dextran sodium sulfate (DSS) induces colitis in mice by forming nano-lipocomplexes with medium-chain-length fatty acids in the colon. PLoS ONE 2012, 7, e32084. [Google Scholar] [CrossRef]
- Chassaing, B.; Aitken, J.D.; Malleshappa, M.; Vijay-Kumar, M. Dextran sulfate sodium (DSS)-induced colitis in mice. Curr. Protoc. Immunol. 2014, 104, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Leppkes, M.; Lindemann, A.; Gosswein, S.; Paulus, S.; Roth, D.; Hartung, A.; Liebing, E.; Zundler, S.; Gonzalez-Acera, M.; Patankar, J.V.; et al. Neutrophils prevent rectal bleeding in ulcerative colitis by peptidyl-arginine deiminase-4-dependent immunothrombosis. Gut 2022, 71, 2414–2429. [Google Scholar] [PubMed]
- Schupp, M.; Lazar, M.A. Endogenous ligands for nuclear receptors: Digging deeper. J. Biol. Chem. 2010, 285, 40409–40415. [Google Scholar] [CrossRef] [PubMed]
- Bensinger, S.J.; Tontonoz, P. Integration of metabolism and inflammation by lipid-activated nuclear receptors. Nature 2008, 454, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Mansen, A.; Guardiola-Diaz, H.; Rafter, J.; Branting, C.; Gustafsson, J.A. Expression of the peroxisome proliferator-activated receptor (PPAR) in the mouse colonic mucosa. Biochem. Biophys. Res. Commun. 1996, 222, 844–851. [Google Scholar] [CrossRef] [PubMed]
- Escher, P.; Braissant, O.; Basu-Modak, S.; Michalik, L.; Wahli, W.; Desvergne, B. Rat PPARs: Quantitative analysis in adult rat tissues and regulation in fasting and refeeding. Endocrinology 2001, 142, 4195–4202. [Google Scholar] [CrossRef]
- Kliewer, S.A.; Lenhard, J.M.; Willson, T.M.; Patel, I.; Morris, D.C.; Lehmann, J.M. A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor gamma and promotes adipocyte differentiation. Cell 1995, 83, 813–819. [Google Scholar] [CrossRef] [Green Version]
- Nagy, L.; Tontonoz, P.; Alvarez, J.G.; Chen, H.; Evans, R.M. Oxidized LDL regulates macrophage gene expression through ligand activation of PPARgamma. Cell 1998, 93, 229–240. [Google Scholar] [CrossRef] [Green Version]
- Evans, R.M.; Barish, G.D.; Wang, Y.X. PPARs and the complex journey to obesity. Nat. Med. 2004, 10, 355–361. [Google Scholar] [CrossRef]
- Lazar, M.A. PPAR gamma, 10 years later. Biochimie 2005, 87, 9–13. [Google Scholar] [CrossRef]
- Ohno, H.; Shinoda, K.; Spiegelman, B.M.; Kajimura, S. PPARgamma agonists induce a white-to-brown fat conversion through stabilization of PRDM16 protein. Cell Metab. 2012, 15, 395–404. [Google Scholar] [CrossRef] [Green Version]
- Tontonoz, P.; Spiegelman, B.M. Fat and beyond: The diverse biology of PPARgamma. Annu. Rev. Biochem. 2008, 77, 289–312. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Banks, A.S.; Estall, J.L.; Kajimura, S.; Bostrom, P.; Laznik, D.; Ruas, J.L.; Chalmers, M.J.; Kamenecka, T.M.; Bluher, M.; et al. Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by Cdk5. Nature 2010, 466, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Bassaganya-Riera, J.; Reynolds, K.; Martino-Catt, S.; Cui, Y.; Hennighausen, L.; Gonzalez, F.; Rohrer, J.; Benninghoff, A.U.; Hontecillas, R. Activation of PPAR gamma and delta by conjugated linoleic acid mediates protection from experimental inflammatory bowel disease. Gastroenterology 2004, 127, 777–791. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.D.; Lichtenstein, G.R.; Deren, J.J.; Sands, B.E.; Hanauer, S.B.; Katz, J.A.; Lashner, B.; Present, D.H.; Chuai, S.; Ellenberg, J.H.; et al. Rosiglitazone for Ulcerative Colitis Study, G., Rosiglitazone for active ulcerative colitis: A randomized placebo-controlled trial. Gastroenterology 2008, 134, 688–695. [Google Scholar] [CrossRef] [Green Version]
- Adachi, M.; Kurotani, R.; Morimura, K.; Shah, Y.; Sanford, M.; Madison, B.B.; Gumucio, D.L.; Marin, H.E.; Peters, J.M.; Young, H.A.; et al. Peroxisome proliferator activated receptor gamma in colonic epithelial cells protects against experimental inflammatory bowel disease. Gut 2006, 55, 1104–1113. [Google Scholar] [CrossRef] [Green Version]
- Mohapatra, S.K.; Guri, A.J.; Climent, M.; Vives, C.; Carbo, A.; Horne, W.T.; Hontecillas, R.; Bassaganya-Riera, J. Immunoregulatory actions of epithelial cell PPAR gamma at the colonic mucosa of mice with experimental inflammatory bowel disease. PLoS ONE 2010, 5, e10215. [Google Scholar] [CrossRef]
- Byndloss, M.X.; Olsan, E.E.; Rivera-Chavez, F.; Tiffany, C.R.; Cevallos, S.A.; Lokken, K.L.; Torres, T.P.; Byndloss, A.J.; Faber, F.; Gao, Y.; et al. Microbiota-activated PPAR-gamma signaling inhibits dysbiotic Enterobacteriaceae expansion. Science 2017, 357, 570–575. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Tian, H.; Jiang, H.J.; Han, B. Interleukin-17 SNPs and serum levels increase ulcerative colitis risk: A meta-analysis. World J. Gastroenterol. WJG 2014, 20, 15899–15909. [Google Scholar] [CrossRef]
- He, J.; Zhang, X.; Wei, Y.; Sun, X.; Chen, Y.; Deng, J.; Jin, Y.; Gan, Y.; Hu, X.; Jia, R.; et al. Low-dose interleukin-2 treatment selectively modulates CD4(+) T cell subsets in patients with systemic lupus erythematosus. Nat. Med. 2016, 22, 991–993. [Google Scholar] [CrossRef]
- Koreth, J.; Matsuoka, K.; Kim, H.T.; McDonough, S.M.; Bindra, B.; Alyea, E.P., 3rd; Armand, P.; Cutler, C.; Ho, V.T.; Treister, N.S.; et al. Interleukin-2 and regulatory T cells in graft-versus-host disease. N. Engl. J. Med. 2011, 365, 2055–2066. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Son, Y.S.; Lee, M.O.; Ryu, J.W.; Park, K.; Kwon, O.; Jung, K.B.; Kim, K.; Ryu, T.Y.; Baek, A.; et al. Low-dose interleukin-2 alleviates dextran sodium sulfate-induced colitis in mice by recovering intestinal integrity and inhibiting AKT-dependent pathways. Theranostics 2020, 10, 5048–5063. [Google Scholar] [CrossRef] [PubMed]
- Noh, J.Y.; Wu, C.S.; DeLuca, J.A.A.; Devaraj, S.; Jayaraman, A.; Alaniz, R.C.; Tan, X.D.; Allred, C.D.; Sun, Y. Novel Role of Ghrelin Receptor in Gut Dysbiosis and Experimental Colitis in Aging. Int. J. Mol. Sci. 2022, 23, 2219. [Google Scholar] [CrossRef] [PubMed]
- Degrelle, S.A.; Shoaito, H.; Fournier, T. New Transcriptional Reporters to Quantify and Monitor PPARgamma Activity. PPAR Res. 2017, 2017, 6139107. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muthyala, S.; Chapkin, R.S.; Wu, C.; Wu, C.-S. Ghrelin Alleviates Experimental Ulcerative Colitis in Old Mice and Modulates Colonocyte Metabolism via PPARγ Pathway. Int. J. Mol. Sci. 2023, 24, 565. https://doi.org/10.3390/ijms24010565
Muthyala S, Chapkin RS, Wu C, Wu C-S. Ghrelin Alleviates Experimental Ulcerative Colitis in Old Mice and Modulates Colonocyte Metabolism via PPARγ Pathway. International Journal of Molecular Sciences. 2023; 24(1):565. https://doi.org/10.3390/ijms24010565
Chicago/Turabian StyleMuthyala, Srilakshmi, Robert S. Chapkin, Chaodong Wu, and Chia-Shan Wu. 2023. "Ghrelin Alleviates Experimental Ulcerative Colitis in Old Mice and Modulates Colonocyte Metabolism via PPARγ Pathway" International Journal of Molecular Sciences 24, no. 1: 565. https://doi.org/10.3390/ijms24010565
APA StyleMuthyala, S., Chapkin, R. S., Wu, C., & Wu, C. -S. (2023). Ghrelin Alleviates Experimental Ulcerative Colitis in Old Mice and Modulates Colonocyte Metabolism via PPARγ Pathway. International Journal of Molecular Sciences, 24(1), 565. https://doi.org/10.3390/ijms24010565