
Gut Microbial Perturbation and Host Response Induce Redox Pathway Upregulation along the Gut–Liver Axis during Giardiasis in C57BL/6J Mouse Model

 , , , ,
, , , ,  ,
,  and
and 
Abstract
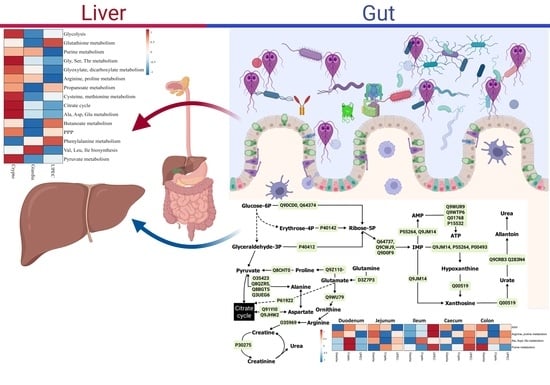
:
1. Introduction
2. Results
2.1. Infection Analysis and Mouse Strain Selection
2.2. Gut Microbial Distribution during Giardiasis
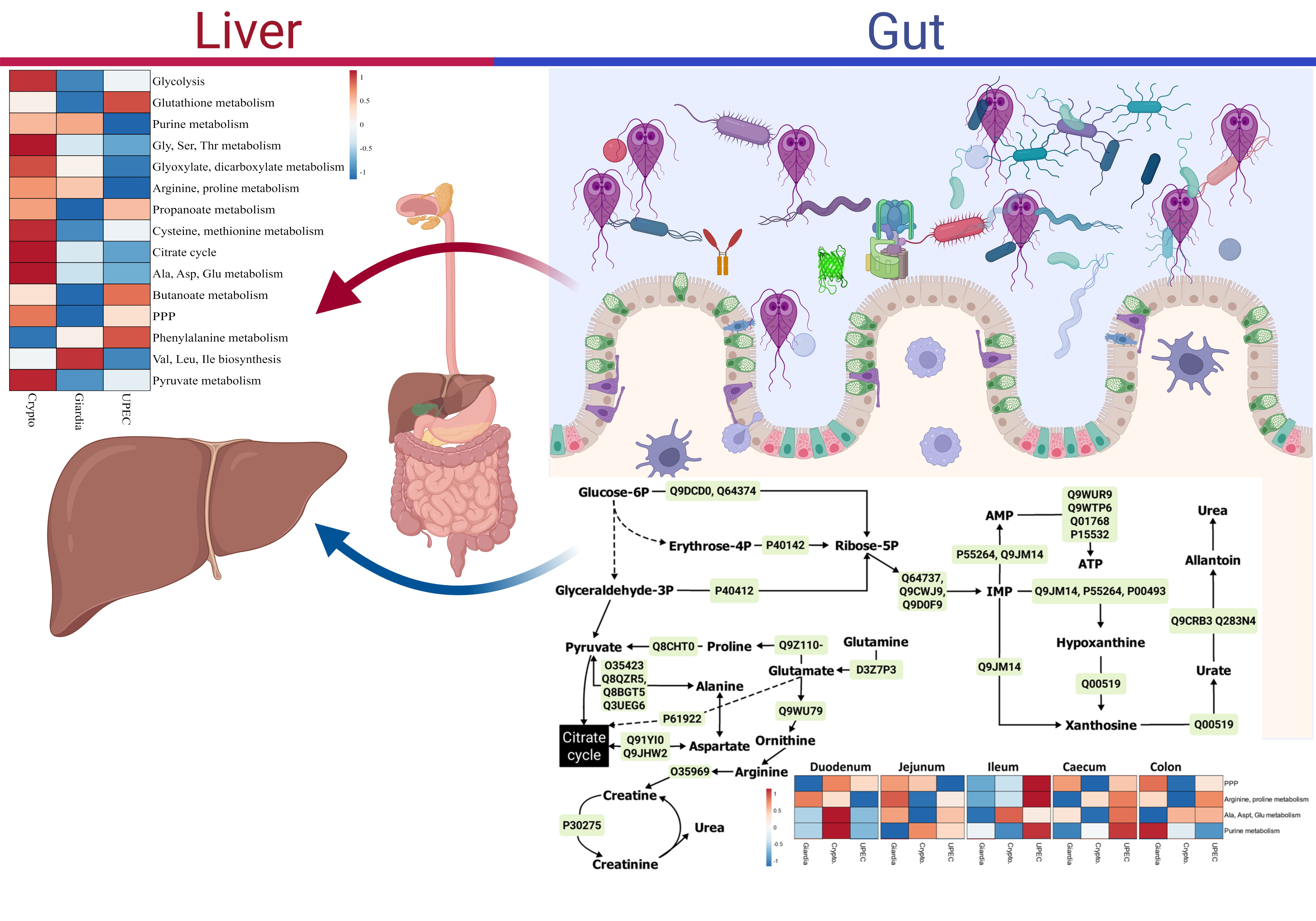
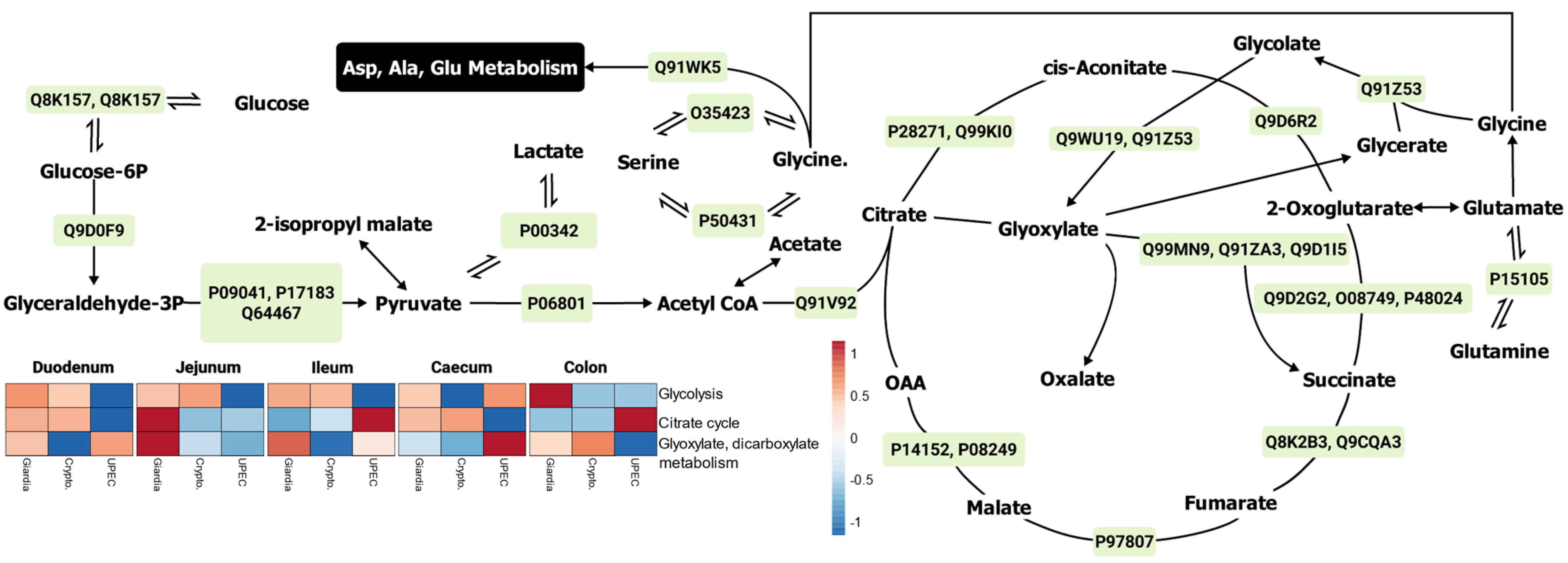
2.3. Gut Metabolism during Giardiasis
2.3.1. Energy Pathways
2.3.2. Pentose Phosphate Pathways (PPP) and Amino Acid–Sugar Interconversions
2.3.3. Redox (Sulphur-Related) Metabolism and Pathways
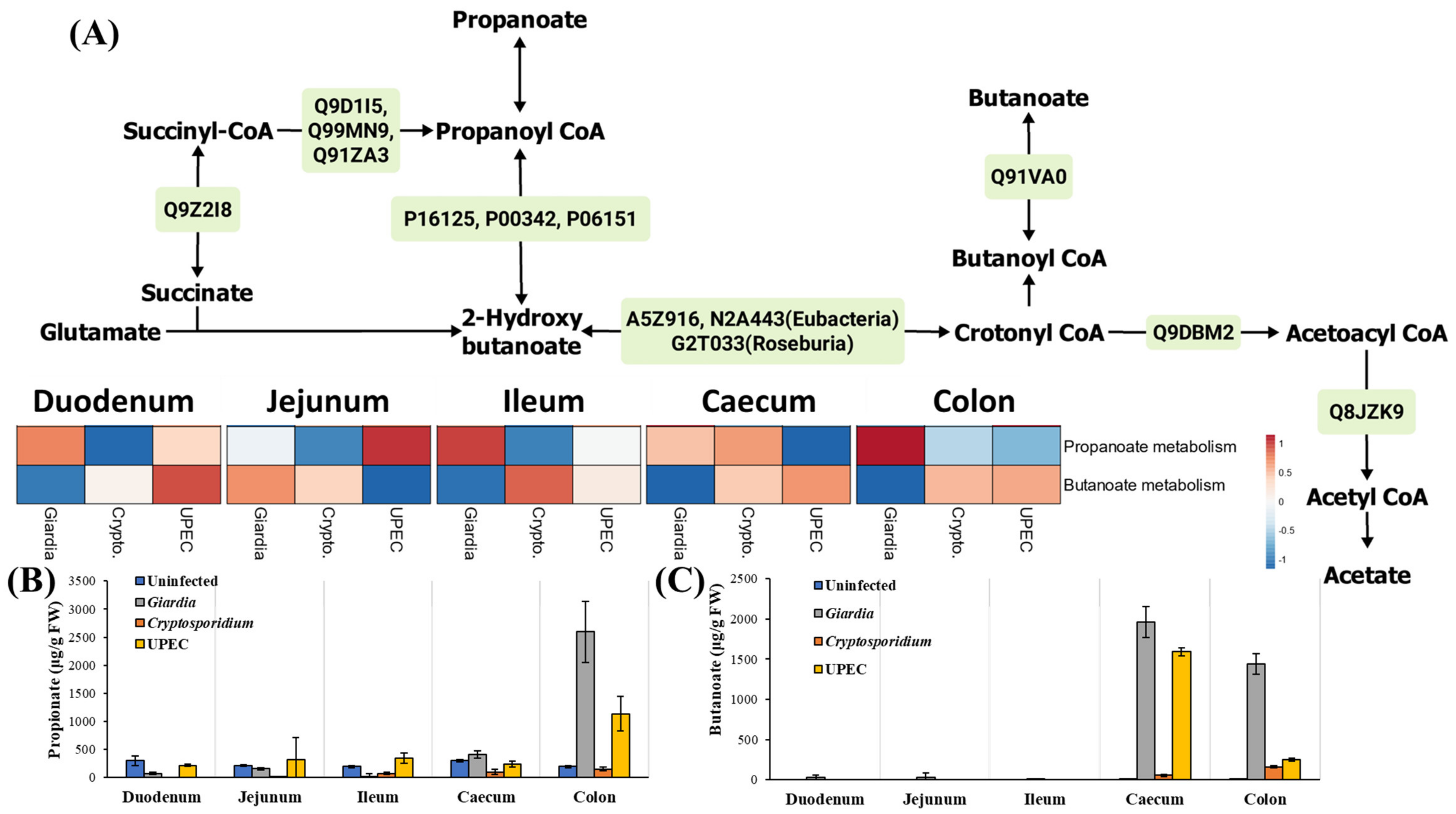
2.3.4. Short-Chained Fatty Acid (SCFA) and D-Amino Acid Metabolism
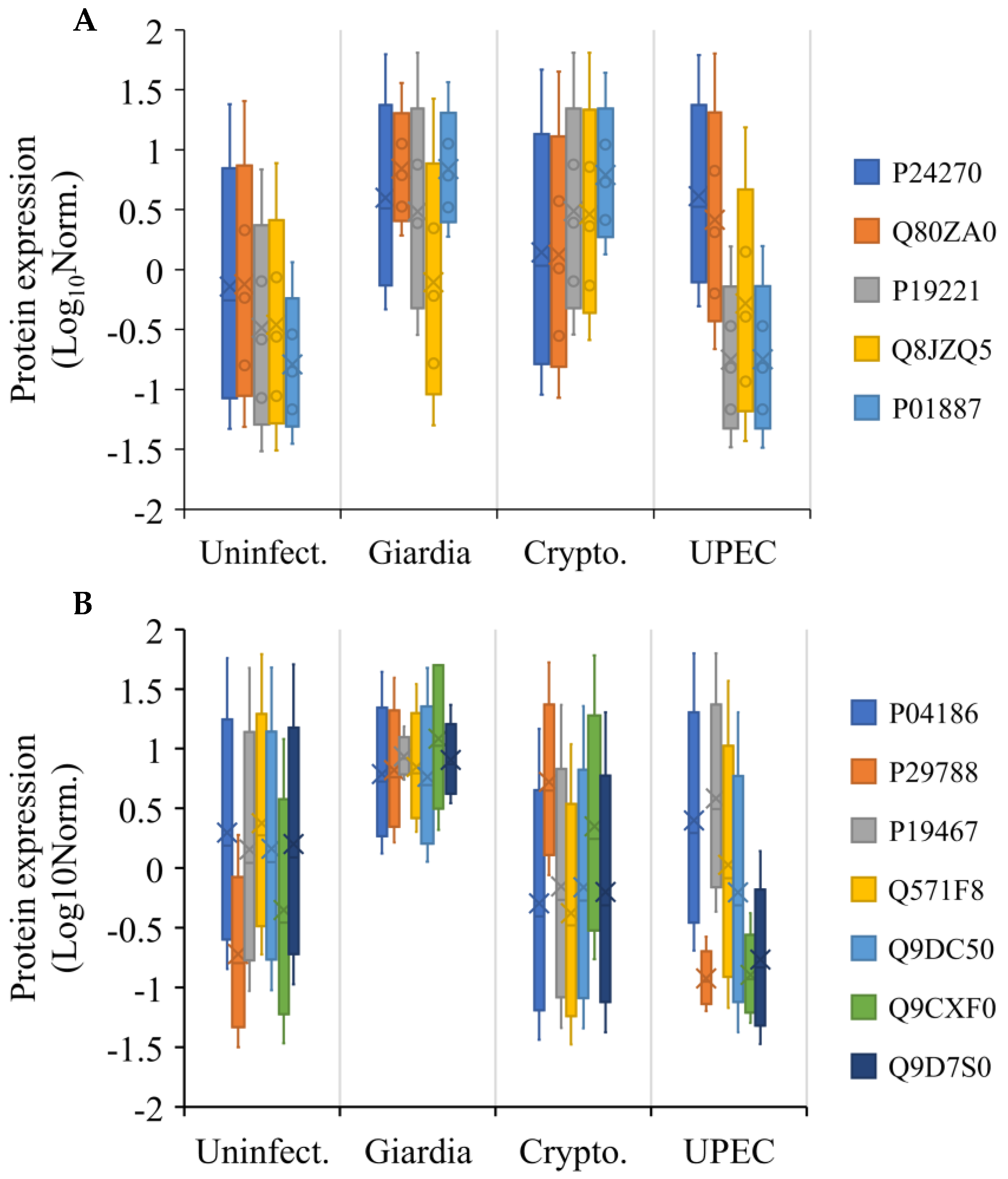
2.4. Extra-Gut Effects of Giardiasis
3. Discussion
4. Materials and Methods
4.1. Animal Ethics Approval
4.2. Mouse Infection and Strain Selection
4.3. Microbiome, Proteomics, and Metabolome: Extraction and Processing
4.4. Statistical Analysis and Quality Control
4.5. Multi-Omics Integration
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Feng, Y.; Xiao, L. Zoonotic potential and molecular epidemiology of Giardia species and giardiasis. Clin. Microbiol. Rev. 2011, 24, 110–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, S. Giardiasis: Global Status; GIDEON Informatics Inc.: Los Angeles, CA, USA, 2017. [Google Scholar]
- Betancourt, W.Q.; Rose, J.B. Drinking water treatment processes for removal of Cryptosporidium and Giardia. Vet. Parasitol. 2004, 126, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.K.; Gupta, P. Opportunistic Parasitic Infections. In Pathology of Opportunistic Infections: An Illustrative Atlas; Gupta, R.K., Gupta, P., Eds.; Springer: Singapore, 2017; pp. 131–145. [Google Scholar]
- Lane, S.; Lloyd, D. Current trends in research into the waterborne parasite Giardia. Crit. Rev. Microbiol. 2002, 28, 123–147. [Google Scholar] [CrossRef] [PubMed]
- Ryan, U.; Hijjawi, N. New developments in Cryptosporidium research. Int. J. Parasitol. 2015, 45, 367–373. [Google Scholar] [PubMed] [Green Version]
- Tzipori, S.; Widmer, G. A hundred-year retrospective on cryptosporidiosis. Trends Parasitol. 2008, 24, 184–189. [Google Scholar] [CrossRef] [Green Version]
- Painter, J.E.; Collier, S.A.; Gargano, J.W. Association between Giardia and arthritis or joint pain in a large health insurance cohort: Could it be reactive arthritis? Epidemiol. Infect. 2016, 145, 471–477. [Google Scholar] [CrossRef] [Green Version]
- Halliez, M.C.M.; Buret, A.G. Extra-intestinal and long term consequences of Giardia duodenalis infections. World J. Gastroenterol. WJG 2013, 19, 8974–8985. [Google Scholar] [CrossRef]
- Berrilli, F.; Di Cave, D.; Cavallero, S.; D’Amelio, S. Interactions between parasites and microbial communities in the human gut. Front. Cell. Infect. Microbiol. 2012, 2, 141. [Google Scholar] [CrossRef] [Green Version]
- Gerbaba, T.K.; Gupta, P.; Rioux, K.; Hansen, D.; Buret, A.G. Giardia duodenalis-induced alterations of commensal bacteria kill Caenorhabditis elegans: A new model to study microbial-microbial interactions in the gut. Am. J. Physiol.-Gastrointest. Liver Physiol. 2015, 308, G550–G561. [Google Scholar] [CrossRef] [Green Version]
- Barash, N.R.; Maloney, J.G.; Singer, S.M.; Dawson, S.C. Giardia alters commensal microbial diversity throughout the murine gut. Infect. Immun. 2017, 85, e00948-16. [Google Scholar] [CrossRef]
- Gyawali, P.; Karpe, A.V.; Hillyer, K.E.; Nguyen, T.V.; Hewitt, J.; Beale, D.J. A multi-platform metabolomics approach to identify possible biomarkers for human faecal contamination in Greenshell™ mussels (Perna canaliculus). Sci. Total Environ. 2021, 771, 145363. [Google Scholar] [CrossRef]
- Beale, D.J.; Oh, D.Y.; Karpe, A.V.; Tai, C.; Dunn, M.S.; Tilmanis, D.; Palombo, E.A.; Hurt, A.C. Untargeted metabolomics analysis of the upper respiratory tract of ferrets following influenza A virus infection and oseltamivir treatment. Metabolomics 2019, 15, 33. [Google Scholar] [CrossRef]
- Karaduta, O.; Dvanajscak, Z.; Zybailov, B. Metaproteomics—An Advantageous Option in Studies of Host-Microbiota Interaction. Microorganisms 2021, 9, 980. [Google Scholar] [CrossRef]
- Shinde, T.; Vemuri, R.; Shastri, S.; Perera, A.P.; Gondalia, S.V.; Beale, D.J.; Karpe, A.V.; Eri, R.; Stanley, R. Modulating the Microbiome and Immune Responses Using Whole Plant Fibre in Synbiotic Combination with Fibre-Digesting Probiotic Attenuates Chronic Colonic Inflammation in Spontaneous Colitic Mice Model of IBD. Nutrients 2020, 12, 2380. [Google Scholar] [CrossRef]
- Shinde, T.; Perera, A.P.; Vemuri, R.; Gondalia, S.V.; Beale, D.J.; Karpe, A.V.; Shastri, S.; Basheer, W.; Southam, B.; Eri, R. Synbiotic supplementation with prebiotic green banana resistant starch and probiotic Bacillus coagulans spores ameliorates gut inflammation in mouse model of inflammatory bowel diseases. Eur. J. Nutr. 2020, 59, 3669–3689. [Google Scholar] [CrossRef] [Green Version]
- Robinson, A.M.; Gondalia, S.V.; Karpe, A.V.; Eri, R.; Beale, D.J.; Morrison, P.D.; Palombo, E.A.; Nurgali, K. Fecal Microbiota and Metabolome in a Mouse Model of Spontaneous Chronic Colitis: Relevance to Human Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2016, 22, 2767–2787. [Google Scholar] [CrossRef]
- Shah, R.M.; Hillyer, K.E.; Stephenson, S.; Crosswell, J.; Karpe, A.V.; Palombo, E.A.; Jones, O.A.H.; Gorman, D.; Bodrossy, L.; van de Kamp, J.; et al. Functional analysis of pristine estuarine marine sediments. Sci. Total Environ. 2021, 781, 146526. [Google Scholar] [CrossRef]
- Beale, D.J.; Crosswell, J.; Karpe, A.V.; Metcalfe, S.S.; Morrison, P.D.; Staley, C.; Ahmed, W.; Sadowsky, M.J.; Palombo, E.A.; Steven, A.D.L. Seasonal metabolic analysis of marine sediments collected from Moreton Bay in South East Queensland, Australia, using a multi-omics-based approach. Sci. Total Environ. 2018, 631–632, 1328–1341. [Google Scholar] [CrossRef]
- Beale, D.J.; Karpe, A.V.; Ahmed, W. Beyond Metabolomics: A Review of Multi-Omics-Based Approaches. In Microbial Metabolomics: Applications in Clinical, Environmental, and Industrial Microbiology; Beale, D., Kouremenos, K., Palombo, E., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 295–319. [Google Scholar]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- McNally, C.P.; Eng, A.; Noecker, C.; Gagne-Maynard, W.C.; Borenstein, E. BURRITO: An interactive multi-omic tool for visualizing taxa–function relationships in microbiome data. Front. Microbiol. 2018, 9, 365. [Google Scholar] [CrossRef]
- Metsalu, T.; Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef] [PubMed]
- Emery-Corbin, S.J.; Hamey, J.J.; Balan, B.; Rojas-López, L.; Svärd, S.G.; Jex, A.R. Eukaryote-conserved histone post-translational modification landscape in Giardia duodenalis revealed by mass spectrometry. Int. J. Parasitol. 2021, 51, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Barash, N.R.; Nosala, C.; Pham, J.K.; McInally, S.G.; Gourguechon, S.; McCarthy-Sinclair, B.; Dawson, S.C. Giardia colonizes and encysts in high-density foci in the murine small intestine. mSphere 2017, 2, e00343-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karpe, A.V.; Hutton, M.L.; Mileto, S.J.; James, M.L.; Evans, C.; Shah, R.M.; Ghodke, A.B.; Hillyer, K.E.; Metcalfe, S.S.; Liu, J.-W.; et al. Cryptosporidiosis Modulates the Gut Microbiome and Metabolism in a Murine Infection Model. Metabolites 2021, 11, 380. [Google Scholar] [CrossRef]
- Ohtani, N.; Kawada, N. Role of the Gut–Liver Axis in Liver Inflammation, Fibrosis, and Cancer: A Special Focus on the Gut Microbiota Relationship. Hepatol. Commun. 2019, 3, 456–470. [Google Scholar] [CrossRef] [Green Version]
- Kang, D.J.; Betrapally, N.S.; Ghosh, S.A.; Sartor, R.B.; Hylemon, P.B.; Gillevet, P.M.; Sanyal, A.J.; Heuman, D.M.; Carl, D.; Zhou, H.; et al. Gut microbiota drive the development of neuroinflammatory response in cirrhosis in mice. Hepatology 2016, 64, 1232–1248. [Google Scholar] [CrossRef] [Green Version]
- Shang, Z.-H.; Huang, M.; Wu, M.-X.; Mi, D.; You, K.; Zhang, Y.-L. Transcriptomic analyses of the acute aerial and ammonia stress response in the gill and liver of large-scale loach (Paramisgurnus dabryanus). Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2021, 250, 109185. [Google Scholar] [CrossRef]
- Milner, J. Arginine: A dietary modifier of ammonia detoxification and pyrimidine biosynthesis. In Absorption and Utilization of Amino Acids; CRC Press: Boca Raton, FL, USA, 2018; pp. 25–40. [Google Scholar]
- Azzouz, L.L.; Sharma, S. Physiology, Large Intestine. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Reid, G.K.; Berardinelli, A.J.; Ray, L.; Jackson, A.R.; Neish, A.S.; Hansen, J.M.; Denning, P.W. Timing of developmental reduction in epithelial glutathione redox potential is associated with increased epithelial proliferation in the immature murine intestine. Pediatr. Res. 2017, 82, 362–369. [Google Scholar] [CrossRef] [Green Version]
- Camerini, S.; Bocedi, A.; Cecchetti, S.; Casella, M.; Carbo, M.; Morea, V.; Pozio, E.; Ricci, G.; Lalle, M. Proteomic and functional analyses reveal pleiotropic action of the anti-tumoral compound NBDHEX in Giardia duodenalis. Int. J. Parasitol. Drugs Drug Resist. 2017, 7, 147–158. [Google Scholar] [CrossRef]
- Million, M.; Armstrong, N.; Khelaifia, S.; Guilhot, E.; Richez, M.; Lagier, J.-C.; Dubourg, G.; Chabriere, E.; Raoult, D. The antioxidants glutathione, ascorbic acid and uric acid maintain butyrate production by human gut clostridia in the presence of oxygen in vitro. Sci. Rep. 2020, 10, 7705. [Google Scholar] [CrossRef]
- Ansell, B.R.E.; McConville, M.J.; Baker, L.; Korhonen, P.K.; Young, N.D.; Hall, R.S.; Rojas, C.A.A.; Svärd, S.G.; Gasser, R.B.; Jex, A.R. Time-Dependent Transcriptional Changes in Axenic Giardia duodenalis Trophozoites. PLOS Negl. Trop. Dis. 2015, 9, e0004261. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Yin, L.; Li, J.-Y.; Li, Q.; Shi, D.; Feng, L.; Liu, Y.; Jiang, W.-D.; Wu, P.; Zhao, Y.; et al. Glutamate attenuates lipopolysaccharide-induced oxidative damage and mRNA expression changes of tight junction and defensin proteins, inflammatory and apoptosis response signaling molecules in the intestine of fish. Fish Shellfish. Immunol. 2017, 70, 473–484. [Google Scholar] [CrossRef]
- Ji, Y.; Dai, Z.; Wu, G.; Wu, Z. 4-Hydroxy-2-nonenal induces apoptosis by activating ERK1/2 signaling and depleting intracellular glutathione in intestinal epithelial cells. Sci. Rep. 2016, 6, 32929. [Google Scholar] [CrossRef] [Green Version]
- Kushkevych, I.; Cejnar, J.; Treml, J.; Dordević, D.; Kollar, P.; Vítězová, M.J.C. Recent advances in metabolic pathways of sulfate reduction in intestinal bacteria. Cells 2020, 9, 698. [Google Scholar] [CrossRef] [Green Version]
- Coutinho, C.M.L.M.; Coutinho-Silva, R.; Zinkevich, V.; Pearce, C.B.; Ojcius, D.M.; Beech, I. Sulphate-reducing bacteria from ulcerative colitis patients induce apoptosis of gastrointestinal epithelial cells. Microb. Pathog. 2017, 112, 126–134. [Google Scholar] [CrossRef] [Green Version]
- Dordević, D.; Jančíková, S.; Vítězová, M.; Kushkevych, I. Hydrogen sulfide toxicity in the gut environment: Meta-analysis of sulfate-reducing and lactic acid bacteria in inflammatory processes. J. Adv. Res. 2020, 27, 55–69. [Google Scholar] [CrossRef]
- Zhai, R.; Xue, X.; Zhang, L.; Yang, X.; Zhao, L.; Zhang, C. Strain-Specific Anti-inflammatory Properties of Two Akkermansia muciniphila Strains on Chronic Colitis in Mice. Front. Cell. Infect. Microbiol. 2019, 9, 239. [Google Scholar] [CrossRef] [Green Version]
- Hagag, A.A.; Amin, S.M.; El-Fiky, R.B.; El-Sayad, M.E. Study of Serum Levels of Some Oxidative Stress Markers in Children with Helicobacter pylori Infection. Infect. Disord.-Drug Targets 2018, 18, 52–59. [Google Scholar] [CrossRef]
- Bhattacharyya, A.; Chattopadhyay, R.; Mitra, S.; Crowe, S.E. Oxidative Stress: An Essential Factor in the Pathogenesis of Gastrointestinal Mucosal Diseases. Physiol. Rev. 2014, 94, 329–354. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Yao, H.; Gao, X.; Zhang, Z.; Wang, J.-F.; Xu, S.-W. The Role of Nitric Oxide and Oxidative Stress in Intestinal Damage Induced by Selenium Deficiency in Chickens. Biol. Trace Elem. Res. 2015, 163, 144–153. [Google Scholar] [CrossRef]
- Tako, E.A.; Hassimi, M.F.; Li, E.; Singer, S.M.; Miller, L.H. Transcriptomic Analysis of the Host Response to Giardia duodenalis Infection Reveals Redundant Mechanisms for Parasite Control. MBio 2013, 4, e00660-13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- AL-Hasnawy, M.H.M. Host Cell Response Analysis during Giardia duodenalis Infection. Ph.D. Thesis, Monash University, Melbourne, Australia, 2018. [Google Scholar]
- Kerr, S.C.; Carrington, S.D.; Oscarson, S.; Gallagher, M.E.; Solon, M.; Yuan, S.; Ahn, J.N.; Dougherty, R.H.; Finkbeiner, W.E.; Peters, M.C.; et al. Intelectin-1 Is a Prominent Protein Constituent of Pathologic Mucus Associated with Eosinophilic Airway Inflammation in Asthma. Am. J. Respir. Crit. Care Med. 2014, 189, 1005–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, T.; Chibana, K.; Shiobara, T.; Tei, R.; Koike, R.; Nakamura, Y.; Arai, R.; Horigane, Y.; Shimizu, Y.; Takemasa, A.; et al. Expression of intelectin-1 in bronchial epithelial cells of asthma is correlated with T-helper 2 (Type-2) related parameters and its function. Allergy Asthma Clin. Immunol. 2017, 13, 35. [Google Scholar] [CrossRef] [Green Version]
- Okada, T.; Kanda, T.; Ueda, N.; Ikebuchi, Y.; Hashiguchi, K.; Nakao, K.; Isomoto, H. IL-8 and LYPD8 expression levels are associated with the inflammatory response in the colon of patients with ulcerative colitis. Biomed. Rep. 2020, 12, 193–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dkhil, M.A. Sex-determined susceptibility and differential MUC2 mRNA expression during the course of murine intestinal eimeriosis. Parasitol. Res. 2015, 114, 283–288. [Google Scholar] [CrossRef]
- Rao, Y.; Kuang, Z.; Li, C.; Guo, S.; Xu, Y.; Zhao, D.; Hu, Y.; Song, B.; Jiang, Z.; Ge, Z.; et al. Gut Akkermansia muciniphila ameliorates metabolic dysfunction-associated fatty liver disease by regulating the metabolism of L-aspartate via gut-liver axis. Gut Microbes 2021, 13, 1927633. [Google Scholar] [CrossRef]
- Toro-Londono, M.A.; Bedoya-Urrego, K.; Garcia-Montoya, G.M.; Galvan-Diaz, A.L.; Alzate, J.F. Intestinal parasitic infection alters bacterial gut microbiota in children. PeerJ 2019, 7, e6200. [Google Scholar] [CrossRef] [Green Version]
- Karpe, A.; Hutton, M.; Mileto, S.; James, M.; Evans, C.; Shah, R.; Ghodke, A.; Hillyer, K.; Metcalfe, S.; Liu, J. Cryptosporidiosis modulates gut microbiome metabolism and the immune response in an infected host. Metabolites 2020, 11, 380. [Google Scholar] [CrossRef]
- Brandl, K.; Schnabl, B. Intestinal microbiota and nonalcoholic steatohepatitis. Curr. Opin. Gastroenterol. 2017, 33, 128–133. [Google Scholar] [CrossRef] [Green Version]
- Hage, R.E.; Hernandez-Sanabria, E.; Arroyo, M.C.; Van de Wiele, T. Supplementation of a propionate-producing consortium improves markers of insulin resistance in an in vitro model of gut-liver axis. Am. J. Physiol.-Endocrinol. Metab. 2020, 318, E742–E749. [Google Scholar] [CrossRef]
- Perry, R.J.; Borders, C.B.; Cline, G.W.; Zhang, X.-M.; Alves, T.C.; Petersen, K.F.; Rothman, D.L.; Kibbey, R.G.; Shulman, G.I. Propionate Increases Hepatic Pyruvate Cycling and Anaplerosis and Alters Mitochondrial Metabolism. J. Biol. Chem. 2016, 291, 12161–12170. [Google Scholar] [CrossRef] [Green Version]
- Trend, S.; Leffler, J.; Jones, A.P.; Cha, L.; Gorman, S.; Brown, D.A.; Breit, S.N.; Kermode, A.G.; French, M.A.; Ward, N.C.; et al. Associations of serum short-chain fatty acids with circulating immune cells and serum biomarkers in patients with multiple sclerosis. Sci. Rep. 2021, 11, 5244. [Google Scholar] [CrossRef]
- Just, S. Impact of the Interplay between Bile Acids, Lipids, Intestinal Coriobacteriaceae and Diet on Host Metabolism. Ph.D. Thesis, Technische Universität München, Munich, Germany, 2017. [Google Scholar]
- Ducarmon, Q.R.; Terveer, E.M.; Nooij, S.; Bloem, M.N.; Vendrik, K.E.W.; Caljouw, M.A.A.; Sanders, I.M.J.G.; van Dorp, S.M.; Wong, M.C.; Zwittink, R.D.; et al. Microbiota-associated risk factors for asymptomatic gut colonisation with multi-drug-resistant organisms in a Dutch nursing home. Genome Med. 2021, 13, 54. [Google Scholar] [CrossRef]
- Clavel, T.; Lepage, P.; Charrier, C. The Family Coriobacteriaceae. In The Prokaryotes: Actinobacteria; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 201–238. [Google Scholar]
- Kilkenny, C.; Browne, W.J.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Improving Bioscience Research Reporting: The ARRIVE Guidelines for Reporting Animal Research. PLOS Biol. 2010, 8, e1000412. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef] [Green Version]
- Azizan, K.A.; Baharum, S.N.; Ressom, H.W.; Noor, N.M. GC-MS analysis and PLS-DA validation of the trimethyl silyl-derivatization techniques. Am. J. Appl. Sci. 2012, 9, 1124–1136. [Google Scholar] [CrossRef]
- Noecker, C.; Eng, A.; Srinivasan, S.; Theriot, C.M.; Young, V.B.; Jansson, J.K.; Fredricks, D.N.; Borenstein, E. Metabolic model-based integration of microbiome taxonomic and metabolomic profiles elucidates mechanistic links between ecological and metabolic variation. MSystems 2016, 1, e00013-15. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway | Duodenum | Jejunum | Ileum | Caecum | Colon | Faeces | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Impact | FDR | Impact | FDR | Impact | FDR | Impact | FDR | Impact | FDR | Impact | FDR | |
| Aminoacyl-tRNA biosynthesis | 0.38 | 6.3 × 10−6 | 0.41 | 6.62 × 10−6 | 0.41 | 8.37 × 10−6 | 0.41 | 2.06 × 10−5 | 0.44 | 9.07 × 10−6 | 0.33 | 0.0001 |
| Glyoxylate, dicarboxylate metabolism | 1.05 | 4.3 × 10−5 | 1.16 | 5.64 × 10−7 | 0.91 | 7 × 10−5 | 1.16 | 1.33 × 10−6 | 1.35 | 4.18 × 10−9 | 1.13 | 4.85 × 10−5 |
| Alanine, aspartate, and glutamate metabolism | 1.17 | 0.0038 | 1.10 | 0.0024 | 1.07 | 0.0010 | 1.20 | 2.06 × 10−5 | 1.15 | 0.0008 | 0.82 | 0.0012 |
| Arginine biosynthesis | 0.88 | 0.0039 | 1.12 | 1.11 × 10−5 | 1.12 | 0.0006 | 1.31 | 1.33 × 10−6 | 1.35 | 1.69 × 10−5 | 0.85 | 0.0010 |
| Glutathione metabolism | 0.95 | 4.26 × 10−7 | 0.84 | 3.43 × 10−6 | 1.00 | 1.99 × 10−6 | 1.16 | 1.44 × 10−5 | 0.96 | 7.43 × 10−6 | 0.87 | 0.0001 |
| Citrate cycle (TCA cycle) | 1.63 | 0.0027 | 2.44 | 2.07 × 10−7 | 1.95 | 0.0007 | 2.07 | 0.0002 | 2.66 | 3.89 × 10−6 | 1.56 | 0.0022 |
| Glycolysis or gluconeogenesis | 1.17 | 0.0446 | 1.95 | 3.92 × 10−13 | 1.60 | 0.0006 | 1.55 | 0.0001 | 1.68 | 0.0004 | 1.48 | 0.0010 |
| Pantothenate and CoA biosynthesis | 0.73 | 0.0378 | 0.79 | 0.0398 | 0.79 | 0.0210 | 0.79 | 0.0084 | 0.73 | 0.0233 | 0.36 | 0.0878 |
| Glycine, serine, and threonine metabolism | 1.46 | 0.0275 | 1.63 | 0.0001 | 1.44 | 0.0027 | 1.66 | 0.0003 | 1.63 | 0.0028 | 1.48 | 0.0010 |
| Phenylalanine metabolism | 0.87 | 0.1507 | 0.96 | 0.0708 | 0.61 | 0.2560 | 1.22 | 0.0032 | 1.22 | 0.0099 | 0.78 | 0.0350 |
| Pentose phosphate pathway | 0.78 | 0.5734 | 1.48 | 0.0315 | 1.26 | 0.0021 | 1.22 | 0.0044 | 1.13 | 0.0197 | 1.13 | 0.0126 |
| Butanoate metabolism | 1.18 | 0.0027 | 0.93 | 0.0509 | 1.14 | 0.0027 | 1.11 | 0.0303 | 1.18 | 0.0016 | 0.25 | 0.2035 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karpe, A.V.; Hutton, M.L.; Mileto, S.J.; James, M.L.; Evans, C.; Ghodke, A.B.; Shah, R.M.; Metcalfe, S.S.; Liu, J.-W.; Walsh, T.; et al. Gut Microbial Perturbation and Host Response Induce Redox Pathway Upregulation along the Gut–Liver Axis during Giardiasis in C57BL/6J Mouse Model. Int. J. Mol. Sci. 2023, 24, 1636. https://doi.org/10.3390/ijms24021636
Karpe AV, Hutton ML, Mileto SJ, James ML, Evans C, Ghodke AB, Shah RM, Metcalfe SS, Liu J-W, Walsh T, et al. Gut Microbial Perturbation and Host Response Induce Redox Pathway Upregulation along the Gut–Liver Axis during Giardiasis in C57BL/6J Mouse Model. International Journal of Molecular Sciences. 2023; 24(2):1636. https://doi.org/10.3390/ijms24021636
Chicago/Turabian StyleKarpe, Avinash V., Melanie L. Hutton, Steven J. Mileto, Meagan L. James, Chris Evans, Amol B. Ghodke, Rohan M. Shah, Suzanne S. Metcalfe, Jian-Wei Liu, Tom Walsh, and et al. 2023. "Gut Microbial Perturbation and Host Response Induce Redox Pathway Upregulation along the Gut–Liver Axis during Giardiasis in C57BL/6J Mouse Model" International Journal of Molecular Sciences 24, no. 2: 1636. https://doi.org/10.3390/ijms24021636
APA StyleKarpe, A. V., Hutton, M. L., Mileto, S. J., James, M. L., Evans, C., Ghodke, A. B., Shah, R. M., Metcalfe, S. S., Liu, J. -W., Walsh, T., Lyras, D., Palombo, E. A., & Beale, D. J. (2023). Gut Microbial Perturbation and Host Response Induce Redox Pathway Upregulation along the Gut–Liver Axis during Giardiasis in C57BL/6J Mouse Model. International Journal of Molecular Sciences, 24(2), 1636. https://doi.org/10.3390/ijms24021636