
p53 Family in Resistance to Targeted Therapy of Melanoma




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Metastatic Melanoma—Progress, but Still no Cure
2. Molecular Mechanisms of Resistance to Targeted Therapy
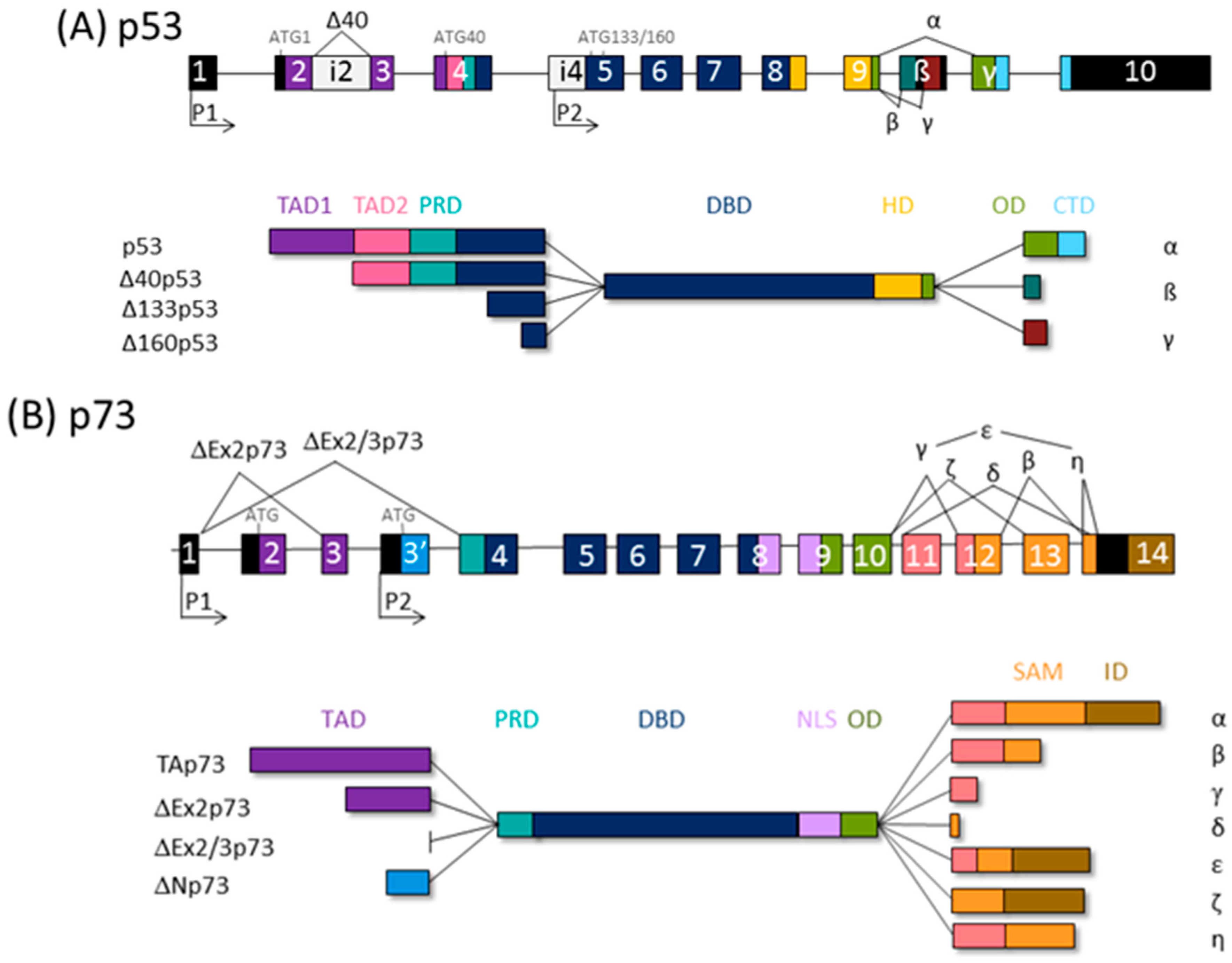
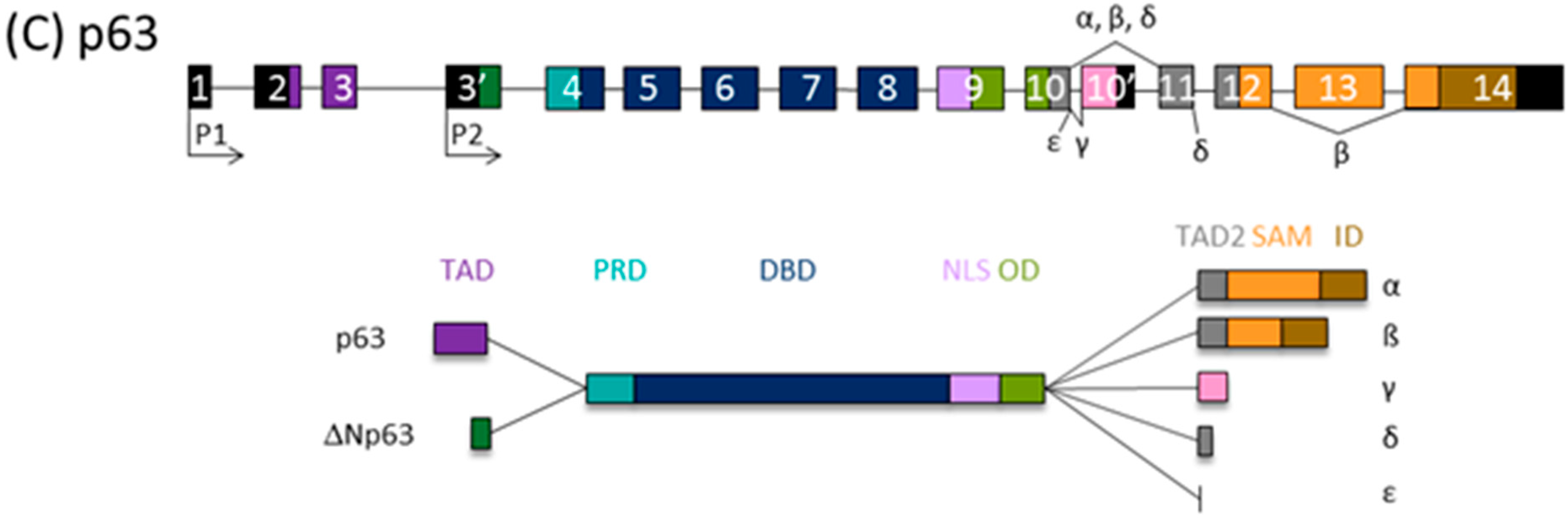
3. The p53 Family Isoforms
4. The Role of p53 Family Isoforms in Melanoma
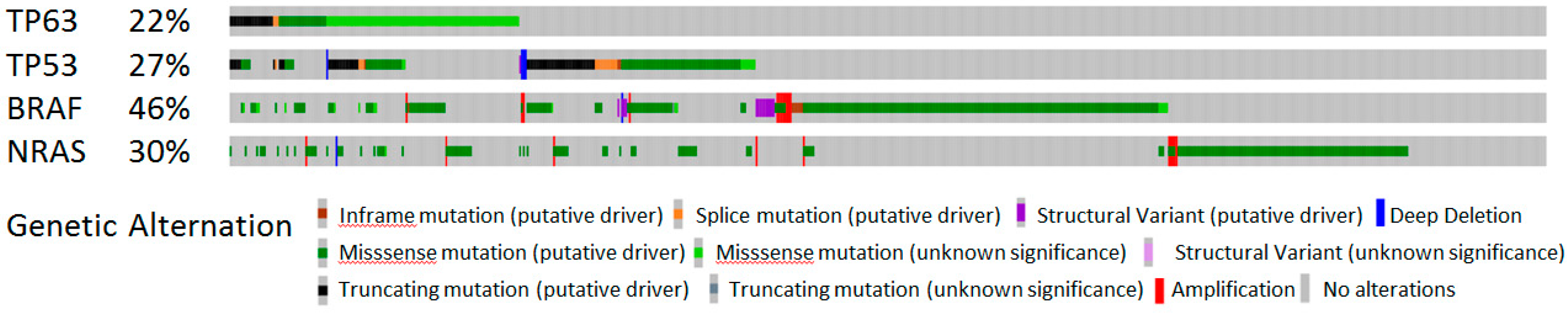
4.1. The Expression and Activities of p53 Isoforms in Melanoma
4.2. The Expression and Activities of p73 Isoforms in Melanoma
4.3. The Expression and Activities of p63 Isoforms in Melanoma
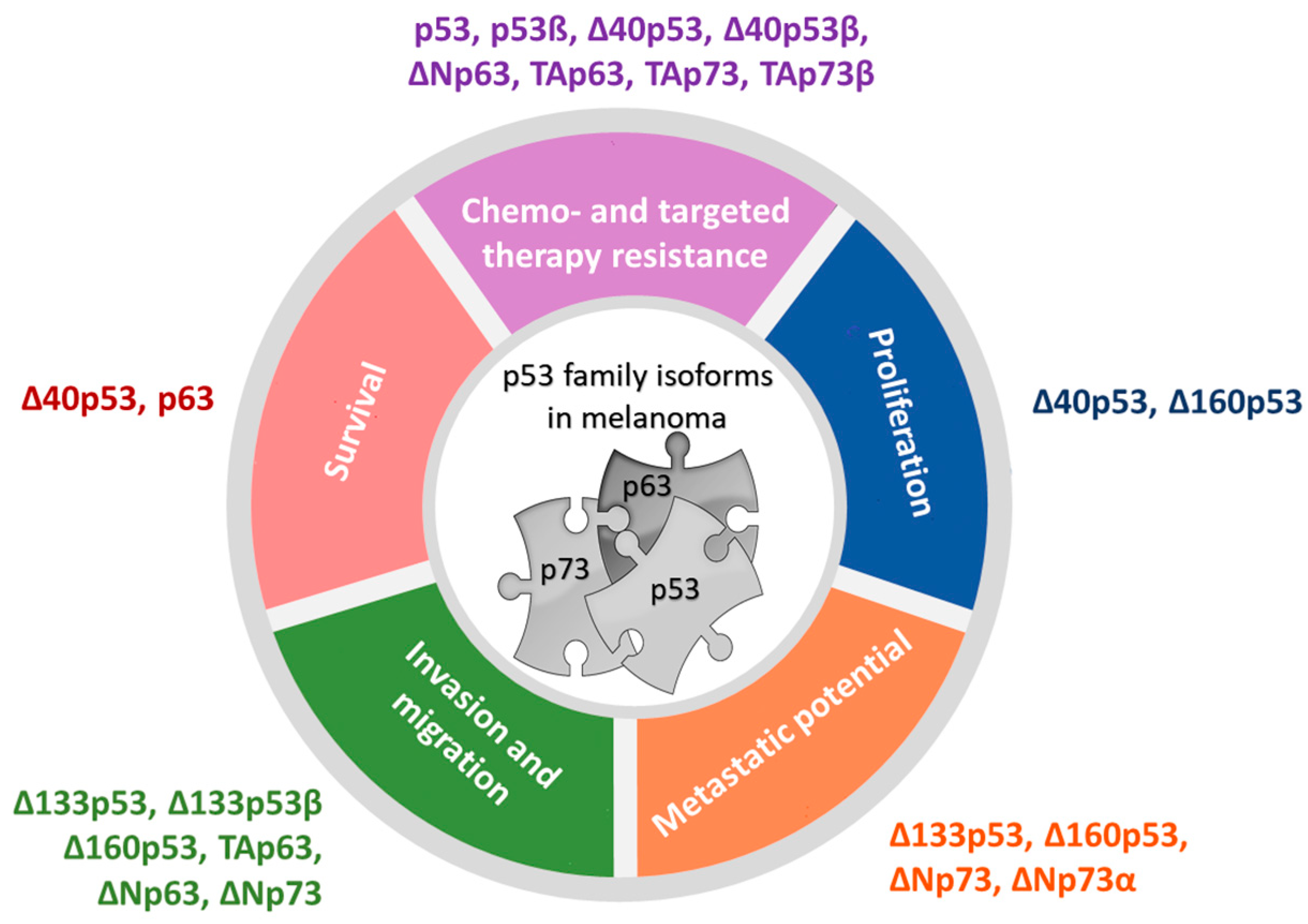
5. The Role of p53 Family Isoforms in Resistance to Targeted Therapy
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Saginala, K.; Barsouk, A.; Aluru, J.; Rawla, P.; Barsouk, A. Epidemiology of Melanoma. Med. Sci. 2021, 9, 63. [Google Scholar] [CrossRef] [PubMed]
- Reddy, B.Y.; Miller, D.M.; Tsao, H. Somatic Driver Mutations in Melanoma. Cancer 2017, 123, 2104–2117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawryluk, E.B.; Tsao, H. Melanoma: Clinical Features and Genomic Insights. Cold Spring Harb. Perspect. Med. 2014, 4, a015388. [Google Scholar] [CrossRef] [Green Version]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A Landscape of Driver Mutations in Melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [Green Version]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF Gene in Human Cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of Mutated, Activated BRAF in Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sosman, J.A.; Kim, K.B.; Schuchter, L.; Gonzalez, R.; Pavlick, A.C.; Weber, J.S.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; et al. Survival in BRAF V600-Mutant Advanced Melanoma Treated with Vemurafenib. N. Engl. J. Med. 2012, 366, 707–714. [Google Scholar] [CrossRef] [Green Version]
- Flaherty, K.T.; Infante, J.R.; Daud, A.; Gonzalez, R.; Kefford, R.F.; Sosman, J.; Hamid, O.; Schuchter, L.; Cebon, J.; Ibrahim, N.; et al. Combined BRAF and MEK Inhibition in Melanoma with BRAF V600 Mutations. N. Engl. J. Med. 2012, 367, 1694–1703. [Google Scholar] [CrossRef] [Green Version]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK Inhibition versus BRAF Inhibition Alone in Melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef]
- Kakadia, S.; Yarlagadda, N.; Awad, R.; Kundranda, M.; Niu, J.; Naraev, B.; Mina, L.; Dragovich, T.; Gimbel, M.; Mahmoud, F. Mechanisms of Resistance to BRAF and MEK Inhibitors and Clinical Update of US Food and Drug Administration-Approved Targeted Therapy in Advanced Melanoma. Onco. Targets. Ther. 2018, 11, 7095–7107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimaldi, A.M.; Simeone, E.; Festino, L.; Vanella, V.; Ascierto, P.A. Combined BRAF and MEK Inhibition with Vemurafenib and Cobimetinib for Patients with Advanced Melanoma. Eur. Oncol. Haematol. 2017, 13, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Larkin, J.; Ascierto, P.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Combined Vemurafenib and Cobimetinib in BRAF-Mutated Melanoma. N. Engl. J. Med. 2014, 371, 1867–1876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willsmore, Z.N.; Coumbe, B.G.T.; Crescioli, S.; Reci, S.; Gupta, A.; Harris, R.J.; Chenoweth, A.; Chauhan, J.; Bax, H.J.; McCraw, A.; et al. Combined Anti-PD-1 and Anti-CTLA-4 Checkpoint Blockade: Treatment of Melanoma and Immune Mechanisms of Action. Eur. J. Immunol. 2021, 51, 544–556. [Google Scholar] [CrossRef]
- Yearley, J.H.; Gibson, C.; Yu, N.; Moon, C.; Murphy, E.; Juco, J.; Lunceford, J.; Cheng, J.; Chow, L.Q.M.; Seiwert, T.Y.; et al. PD-L2 Expression in Human Tumors: Relevance to Anti-PD-1 Therapy in Cancer. Clin. Cancer Res. 2017, 23, 3158–3167. [Google Scholar] [CrossRef] [Green Version]
- Ng, G.; Xu, W.; Atkinson, V. Treatment Approaches for Melanomas That Relapse After Adjuvant or Neoadjuvant Therapy. Curr. Oncol. Rep. 2022, 24, 1–8. [Google Scholar] [CrossRef]
- Martin, C.A.; Cullinane, C.; Kirby, L.; Abuhammad, S.; Lelliott, E.J.; Waldeck, K.; Young, R.J.; Brajanovski, N.; Cameron, D.P.; Walker, R.; et al. Palbociclib Synergizes with BRAF and MEK Inhibitors in Treatment Naïve Melanoma but Not after the Development of BRAF Inhibitor Resistance. Int. J. Cancer 2018, 142, 2139–2152. [Google Scholar] [CrossRef] [Green Version]
- AbuHammad, S.; Cullinane, C.; Martin, C.; Bacolas, Z.; Ward, T.; Chen, H.; Slater, A.; Ardley, K.; Kirby, L.; Chan, K.T.; et al. Regulation of PRMT5-MDM4 Axis Is Critical in the Response to CDK4/6 Inhibitors in Melanoma. Proc. Natl. Acad. Sci. USA 2019, 116, 17990–18000. [Google Scholar] [CrossRef]
- Patel, H.; Yacoub, N.; Mishra, R.; White, A.; Yuan, L.; Alanazi, S.; Garrett, J.T. Current Advances in the Treatment of BRAF-Mutant Melanoma. Cancers 2020, 12, 482. [Google Scholar] [CrossRef] [Green Version]
- Tangella, L.P.; Clark, M.E.; Gray, E.S. Resistance Mechanisms to Targeted Therapy in BRAF-Mutant Melanoma—A Mini Review. Biochim. Biophys. Acta-Gen. Subj. 2021, 1865, 129736. [Google Scholar] [CrossRef]
- Lim, S.Y.; Menzies, A.M.; Rizos, H. Mechanisms and Strategies to Overcome Resistance to Molecularly Targeted Therapy for Melanoma. Cancer 2017, 123, 2118–2129. [Google Scholar] [CrossRef] [Green Version]
- Rizos, H.; Menzies, A.M.; Pupo, G.M.; Carlino, M.S.; Fung, C.; Hyman, J.; Haydu, L.E.; Mijatov, B.; Becker, T.M.; Boyd, S.C.; et al. BRAF Inhibitor Resistance Mechanisms in Metastatic Melanoma: Spectrum and Clinical Impact. Clin. Cancer Res. 2014, 20, 1965–1977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zecena, H.; Tveit, D.; Wang, Z.; Farhat, A.; Panchal, P.; Liu, J.; Singh, S.J.; Sanghera, A.; Bainiwal, A.; Teo, S.Y.; et al. Systems Biology Analysis of Mitogen Activated Protein Kinase Inhibitor Resistance in Malignant Melanoma. BMC Syst. Biol. 2018, 12, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najem, A.; Krayem, M.; Salès, F.; Hussein, N.; Badran, B.; Robert, C.; Awada, A.; Journe, F.; Ghanem, G.E. P53 and MITF/Bcl-2 Identified as Key Pathways in the Acquired Resistance of NRAS-Mutant Melanoma to MEK Inhibition. Eur. J. Cancer 2017, 83, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.P.; Brunton, H.; Rowling, E.J.; Ferguson, J.; Arozarena, I.; Miskolczi, Z.; Lee, J.L.; Girotti, M.R.; Marais, R.; Levesque, M.P.; et al. Inhibiting Drivers of Non-Mutational Drug Tolerance Is a Salvage Strategy for Targeted Melanoma Therapy. Cancer Cell 2016, 29, 270–284. [Google Scholar] [CrossRef] [Green Version]
- Fallahi-Sichani, M.; Becker, V.; Izar, B.; Baker, G.J.; Lin, J.; Boswell, S.A.; Shah, P.; Rotem, A.; Garraway, L.A.; Sorger, P.K. Adaptive Resistance of Melanoma Cells to RAF Inhibition via Reversible Induction of a Slowly Dividing De-Differentiated State. Mol. Syst. Biol. 2017, 13, 905. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Wei, W.; Robert, L.; Xue, M.; Tsoi, J.; Garcia-Diaz, A.; Homet Moreno, B.; Kim, J.; Ng, R.H.; Lee, J.W.; et al. Single-Cell Analysis Resolves the Cell State Transition and Signaling Dynamics Associated with Melanoma Drug-Induced Resistance. Proc. Natl. Acad. Sci. USA 2017, 114, 13679–13684. [Google Scholar] [CrossRef] [Green Version]
- Webster, M.R.; Fane, M.E.; Alicea, G.M.; Basu, S.; Kossenkov, A.V.; Marino, G.E.; Douglass, S.M.; Kaur, A.; Ecker, B.L.; Gnanapradeepan, K.; et al. Paradoxical Role for Wild-Type P53 in Driving Therapy Resistance in Melanoma. Mol. Cell 2020, 77, 681. [Google Scholar] [CrossRef] [Green Version]
- Restivo, G.; Diener, J.; Cheng, P.F.; Kiowski, G.; Bonalli, M.; Biedermann, T.; Reichmann, E.; Levesque, M.P.; Dummer, R.; Sommer, L. Low Neurotrophin Receptor CD271 Regulates Phenotype Switching in Melanoma. Nat. Commun. 2017, 8, 1988. [Google Scholar] [CrossRef] [Green Version]
- Filipp, F.V.; Li, C.; Boiko, A.D. CD271 Is a Molecular Switch with Divergent Roles in Melanoma and Melanocyte Development. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Jiang, L.; Huang, S.; Wang, J.; Zhang, Y.; Xiong, Y.; Zeng, S.X.; Lu, H. Inactivating P53 Is Essential for Nerve Growth Factor Receptor to Promote Melanoma-Initiating Cell-Stemmed Tumorigenesis. Cell Death Dis. 2020, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Saltari, A.; Dzung, A.; Quadri, M.; Tiso, N.; Facchinello, N.; Hernandez-Barranco, A.; Garcia-Silva, S.; Nogues, L.; Stoffel, C.I.; Cheng, P.F.; et al. Specific Activation of the CD271 Intracellular Domain in Combination with Chemotherapy or Targeted Therapy Inhibits Melanoma Progression. Cancer Res. 2021, 81, 6044–6057. [Google Scholar] [CrossRef] [PubMed]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Auman, J.T.; Ayala, B.; et al. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Dutton-Regester, K.; Brown, K.M.; Hayward, N.K. The Genomic Landscape of Cutaneous Melanoma. Pigment Cell Melanoma Res. 2016, 29, 266–283. [Google Scholar] [CrossRef] [PubMed]
- Moll, U.M.; Slade, N. P63 and P73: Roles in Development and Tumor Formation. Mol. Cancer Res. 2004, 2, 371–386. [Google Scholar] [CrossRef] [PubMed]
- Murray-Zmijewski, F.; Lane, D.P.; Bourdon, J.C. P53/P63/P73 Isoforms: An Orchestra of Isoforms to Harmonise Cell Differentiation and Response to Stress. Cell Death Differ. 2006, 13, 962–972. [Google Scholar] [CrossRef]
- Joruiz, S.M.; Bourdon, J.C. P53 Isoforms: Key Regulators of the Cell Fate Decision. Cold Spring Harb. Perspect. Med. 2016, 6, a026039. [Google Scholar] [CrossRef] [Green Version]
- Fisher, M.L.; Balinth, S.; Mills, A.A. P63-Related Signaling At a Glance. J. Cell Sci. 2020, 133, jcs228015. [Google Scholar] [CrossRef]
- Horvat, A.; Tadijan, A.; Vlašić, I.; Slade, N. P53/P73 Protein Network in Colorectal Cancer and Other Human Malignancies. Cancers 2021, 13, 2885. [Google Scholar] [CrossRef]
- Anbarasan, T.; Bourdon, J.C. The Emerging Landscape of P53 Isoforms in Physiology, Cancer and Degenerative Diseases. Int. J. Mol. Sci. 2019, 20, 6257. [Google Scholar] [CrossRef]
- Vikhreva, P.; Melino, G.; Amelio, I. P73 Alternative Splicing: Exploring a Biological Role for the C-Terminal Isoforms. J. Mol. Biol. 2018, 430, 1829–1838. [Google Scholar] [CrossRef] [PubMed]
- Ishimoto, O.; Kawahara, C.; Enjo, K.; Obinata, M.; Nukiwa, T.; Ikawa, S. Possible Oncogenic Potential of ΔNp73: A Newly Identified Isoform of Human P73. Cancer Res. 2002, 62, 636–641. [Google Scholar] [PubMed]
- Stiewe, T.; Pützer, B.M. Role of P73 in Malignancy: Tumor Suppressor or Oncogene? Cell Death Differ. 2002, 9, 237–245. [Google Scholar] [CrossRef] [Green Version]
- Fillippovich, I.; Sorokina, N.; Gatei, M.; Haupt, Y.; Hobson, K.; Moallem, E.; Spring, K.; Mould, M.; Mcguckin, M.A.; Lavin, M.F.; et al. Transactivation-Deficient P73α (P73Δexon2) Inhibits Apoptosis and Competes with P53. Oncogene 2001, 20, 514–522. [Google Scholar] [CrossRef] [Green Version]
- McLure, K.G.; Lee, P.W.K. How P53 Binds DNA as a Tetramer. EMBO J. 1998, 17, 3342–3350. [Google Scholar] [CrossRef]
- Bourdon, J.-C.; Fernandes, K.; Murray-zmijewski, F.; Liu, G.; Diot, A.; Xirodimas, D.P.; Saville, M.K.; Lane, D.P. P53 Isoforms Can Regulate P53 Transcriptional Activity. Genes Dev. 2005, 19, 2122–2137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slade, N.; Zaika, A.I.; Erster, S.; Moll, U.M. ΔNp73 Stabilises TAp73 Proteins but Compromises Their Function Due to Inhibitory Hetero-Oligomer Formation. Cell Death Differ. 2004, 11, 357–360. [Google Scholar] [CrossRef]
- Zorić, A.; Horvat, A.A.; Slade, N. Differential Effects of Diverse P53 Isoforms on TAp73 Transcriptional Activity and Apoptosis. Carcinogenesis 2013, 34, 522–529. [Google Scholar] [CrossRef] [Green Version]
- Zaika, A.I.; Slade, N.; Erster, S.H.; Sansome, C.; Joseph, T.W.; Pearl, M.; Chalas, E.; Moll, U.M. DeltaNp73, a Dominant-Negative Inhibitor of Wild-Type P53 and TAp73, Is up-Regulated in Human Tumors. J. Exp. Med. 2002, 196, 765–780. [Google Scholar] [CrossRef]
- Stiewe, T.; Zimmermann, S.; Frilling, A.; Esche, H.; Pützer, B.M. Transactivation-Deficient ΔTA-P73 Acts as an Oncogene. Cancer Res. 2002, 62, 3598–3602. [Google Scholar]
- Avery-Kiejda, K.A.; Bowden, N.A.; Croft, A.J.; Scurr, L.L.; Kairupan, C.F.; Ashton, K.A.; Talseth-Palmer, B.A.; Rizos, H.; Zhang, X.D.; Scott, R.J.; et al. P53 in Human Melanoma Fails to Regulate Target Genes Associated with Apoptosis and the Cell Cycle and May Contribute to Proliferation. BMC Cancer 2011, 11, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Du, N.; Huang, T.; Guo, J.; Mo, X.; Yuan, T.; Chen, Y.; Ye, T.; Xu, C.; Wang, W.; et al. TP53 Mutation as Potential Negative Predictor for Response of Anti-CTLA-4 Therapy in Metastatic Melanoma. EBioMedicine 2018, 32, 119–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, G.; Pan, C.; Bei, J.X.; Li, B.; Liang, C.; Xu, Y.; Fu, X. Mutant P53 in Cancer Progression and Targeted Therapies. Front. Oncol. 2020, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.; Heine, M.; Arden, K.C.; Körner, B.; Pilch, H.; Herbst, R.A.; Jung, E.G. Mutation and Expression of TP53 in Malignant Melanomas. Recent Results Cancer Res. 1995, 139, 137–154. [Google Scholar] [CrossRef]
- Palmieri, G.; Ombra, M.N.; Colombino, M.; Casula, M.; Sini, M.C.; Manca, A.; Paliogiannis, P.; Ascierto, P.A.; Cossu, A. Multiple Molecular Pathways in Melanomagenesis: Characterization of Therapeutic Targets. Front. Oncol. 2015, 5, 183. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The CBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Shoushtari, A.N.; Chatila, W.K.; Arora, A.; Sanchez-Vega, F.; Kantheti, H.S.; Zamalloa, J.A.R.; Krieger, P.; Callahan, M.K.; Warner, A.B.; Postow, M.A.; et al. Therapeutic Implications of Detecting MAPK-Activating Alterations in Cutaneous and Unknown Primary Melanomas. Clin. Cancer Res. 2021, 27, 2226–2235. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the CBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, G.; Capone, M.; Ascierto, M.L.; Gentilcore, G.; Stroncek, D.F.; Casula, M.; Sini, M.C.; Palla, M.; Mozzillo, N.; Ascierto, P.A. Main Roads to Melanoma. J. Transl. Med. 2009, 7, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Box, N.F.; Vukmer, T.O.; Terzian, T. Targeting P53 in Melanoma. Pigment Cell Melanoma Res. 2014, 27, 8–10. [Google Scholar] [CrossRef] [Green Version]
- Gembarska, A.; Luciani, F.; Fedele, C.; Russell, E.A.; Dewaele, M.; Villar, S.; Zwolinska, A.; Haupt, S.; De Lange, J.; Yip, D.; et al. MDM4 Is a Key Therapeutic Target in Cutaneous Melanoma. Nat. Med. 2012, 18, 1239–1247. [Google Scholar] [CrossRef] [PubMed]
- Potu, H.; Peterson, L.F.; Pal, A.; Verhaegen, M.; Cao, J.; Talpaz, M.; Donato, N.J. Usp5 Links Suppression of P53 and FAS Levels in Melanoma to the BRAF Pathway. Oncotarget 2014, 5, 5559–5569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, R.; Giannini, C.; Sarkaria, J.N.; Schroeder, M.; Rogers, J.; Mastroeni, D.; Scrable, H. P53 Isoform Profiling in Glioblastoma and Injured Brain. Oncogene 2013, 32, 3165–3174. [Google Scholar] [CrossRef] [Green Version]
- Hofstetter, G.; Berger, A.; Fiegl, H.; Slade, N.; Zori, A.; Holzer, B.; Schuster, E.; Mobus, V.J.; Reimer, D.; Daxenbichler, G.; et al. Alternative Splicing of P53 and P73: The Novel P53 Splice Variant P53delta Is an Independent Prognostic Marker in Ovarian Cancer. Oncogene 2010, 29, 1997–2004. [Google Scholar] [CrossRef]
- Hofstetter, G.; Berger, A.; Schuster, E.; Wolf, A.; Hager, G.; Vergote, I.; Cadron, I.; Sehouli, J.; Braicu, E.I.; Mahner, S.; et al. Δ133p53 Is an Independent Prognostic Marker in P53 Mutant Advanced Serous Ovarian Cancer. Br. J. Cancer 2011, 105, 1593–1599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nutthasirikul, N.; Limpaiboon, T.; Leelayuwat, C.; Patrakitkomjorn, S.; Jearanaikoon, P. Ratio Disruption of the ∆133p53 and TAp53 Isoform Equilibrium Correlates with Poor Clinical Outcome in Intrahepatic Cholangiocarcinoma. Int. J. Oncol. 2013, 42, 1181–1188. [Google Scholar] [CrossRef] [Green Version]
- Anensen, N.; Oyan, A.M.; Bourdon, J.C.; Kalland, K.H.; Bruserud, O.; Gjertsen, B.T. A Distinct P53 Protein Isoform Signature Reflects the Onset of Induction Chemotherapy for Acute Myeloid Leukemia. Clin. Cancer Res. 2006, 12, 3985–3992. [Google Scholar] [CrossRef] [Green Version]
- Bourdon, J.C.; Khoury, M.P.; Diot, A.; Baker, L.; Fernandes, K.; Aoubala, M.; Quinlan, P.; Purdie, C.A.; Jordan, L.B.; Prats, A.C.; et al. P53 Mutant Breast Cancer Patients Expressing P53γ Have as Good a Prognosis as Wild-Type P53 Breast Cancer Patients. Breast Cancer Res. 2011, 13, 1–10. [Google Scholar] [CrossRef]
- Avery-Kiejda, K.A.; Xu, D.Z.; Adams, L.J.; Scott, R.J.; Vojtesek, B.; Lane, D.P.; Hersey, P. Small Molecular Weight Variants of P53 Are Expressed in Human Melanoma Cells and Are Induced by the DNA-Damaging Agent Cisplatin. Clin. Cancer Res. 2008, 14, 1659–1668. [Google Scholar] [CrossRef] [Green Version]
- Knezović Florijan, M.; Ozretić, P.; Bujak, M.; Pezzè, L.; Ciribilli, Y.; Kaštelan, Ž.; Slade, N.; Hudolin, T. The Role of P53 Isoforms’ Expression and P53 Mutation Status in Renal Cell Cancer Prognosis. Urol. Oncol. 2019, 37, e1–e578. [Google Scholar] [CrossRef]
- Tuve, S.; Racek, T.; Niemetz, A.; Schultz, J.; Soengas, M.S.; Pützer, B.M. Adenovirus-Mediated TA-P73beta Gene Transfer Increases Chemosensitivity of Human Malignant Melanomas. Apoptosis 2006, 11, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Tuve, S.; Wagner, S.N.; Schitrek, B.; Pützer, B.M. Alterations of DeltaTA-p 73 Splice Transcripts during Melanoma Development and Progression. Int. J. Cancer 2004, 108, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Ozretić, P.; Hanžić, N.; Proust, B.; Sabol, M.; Trnski, D.; Radić, M.; Musani, V.; Ciribilli, Y.; Milas, I.; Puljiz, Z.; et al. Expression Profiles of P53/P73, NME and GLI Families in Metastatic Melanoma Tissue and Cell Lines. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roth, I.; Campbell, H.; Rubio, C.; Vennin, C.; Wilson, M.; Wiles, A.; Williams, G.; Woolley, A.; Timpson, P.; Berridge, M.V.; et al. The Δ133p53 Isoform and Its Mouse Analogue Δ122p53 Promote Invasion and Metastasis Involving Pro-Inflammatory Molecules Interleukin-6 and CCL2. Oncogene 2016, 35, 4981–4989. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Alonso, R.; Martin-Lopez, M.; Gonzalez-Cano, L.; Garcia, S.; Castrillo, F.; Diez-Prieto, I.; Fernandez-Corona, A.; Lorenzo-Marcos, M.E.; Li, X.; Claesson-Welsh, L.; et al. P73 Is Required for Endothelial Cell Differentiation, Migration and the Formation of Vascular Networks Regulating VEGF and TGFβ Signaling. Cell Death Differ. 2015, 22, 1287–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, R.; Markovic, S.N.; Scrable, H.J. Dominant Effects of Δ40p53 on P53 Function and Melanoma Cell Fate. J. Investig. Dermatol. 2014, 134, 791–800. [Google Scholar] [CrossRef] [Green Version]
- Matin, R.N.; Chikh, A.; Chong, S.L.P.; Mesher, D.; Graf, M.; Sanza, P.; Senatore, V.; Scatolini, M.; Moretti, F.; Leigh, I.M.; et al. P63 Is an Alternative P53 Repressor in Melanoma That Confers Chemoresistance and a Poor Prognosis. J. Exp. Med. 2013, 210, 581–603. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.; Garcia, L.F.; Mannella, V.; Gammon, L.; Borg, T.M.; Maffucci, T.; Scatolini, M.; Chiorino, G.; Vergani, E.; Rodolfo, M.; et al. Targeting P63 Upregulation Abrogates Resistance to MAPK Inhibitors in Melanoma. Cancer Res. 2020, 80, 2676–2688. [Google Scholar] [CrossRef] [Green Version]
- Tadijan, A.; Precazzini, F.; Hanžić, N.; Radić, M.; Gavioli, N.; Vlašić, I.; Ozretić, P.; Pinto, L.; Škreblin, L.; Barban, G.; et al. Altered Expression of Shorter P53 Family Isoforms Can Impact Melanoma Aggressiveness. Cancers 2021, 13, 5231. [Google Scholar] [CrossRef]
- Makino, E.; Gutmann, V.; Kosnopfel, C.; Niessner, H.; Forschner, A.; Garbe, C.; Sinnberg, T.; Schittek, B. Melanoma Cells Resistant towards MAPK Inhibitors Exhibit Reduced TAp73 Expression Mediating Enhanced Sensitivity to Platinum-Based Drugs. Cell Death Dis. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Candeias, M.M.; Hagiwara, M.; Matsuda, M. Cancer-specific Mutations in P53 Induce the Translation of Δ160p53 Promoting Tumorigenesis. EMBO Rep. 2016, 17, 1542–1551. [Google Scholar] [CrossRef] [PubMed]
- Arsic, N.; Gadea, G.; Lagerqvist, E.L.; Bußon, M.; Cahuzac, N.; Brock, C.; Hollande, F.; Gire, V.; Pannequin, J.; Roux, P. The P53 Isoform Δ133p53β Promotes Cancer Stem Cell Potential. Stem Cell Rep. 2015, 4, 531–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gadea, G.; Arsic, N.; Fernandes, K.; Diot, A.; Joruiz, S.M.; Abdallah, S.; Meuray, V.; Vinot, S.; Anguille, C.; Remenyi, J.; et al. TP53 Drives Invasion through Expression of Its Δ133p53β Variant. Elife 2016, 5, e14734. [Google Scholar] [CrossRef]
- Kazantseva, M.; Mehta, S.; Eiholzer, R.A.; Gimenez, G.; Bowie, S.; Campbell, H.; Reily-Bell, A.L.; Roth, I.; Ray, S.; Drummond, C.J.; et al. The Δ133p53β Isoform Promotes an Immunosuppressive Environment Leading to Aggressive Prostate Cancer. Cell Death Dis. 2019, 10, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazantseva, M.; Eiholzer, R.A.; Mehta, S.; Taha, A.; Bowie, S.; Roth, I.; Zhou, J.; Joruiz, S.M.; Royds, J.A.; Hung, N.A.; et al. Elevation of the TP53 Isoform Δ133p53β in Glioblastomas: An Alternative to Mutant P53 in Promoting Tumor Development. J. Pathol. 2018, 246, 77–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arsic, N.; Slatter, T.; Gadea, G.; Villain, E.; Fournet, A.; Kazantseva, M.; Allemand, F.; Sibille, N.; Seveno, M.; de Rossi, S.; et al. Δ133p53β Isoform Pro-Invasive Activity Is Regulated through an Aggregation-Dependent Mechanism in Cancer Cells. Nat. Commun. 2021, 12, 1–18. [Google Scholar] [CrossRef]
- Sun, Y.; Manceau, A.; Frydman, L.; Cappuccio, L.; Neves, D.; Basso, V.; Wang, H.; Fombonne, J.; Maisse, C.; Mehlen, P.; et al. Δ40p53 Isoform Up-Regulates Netrin-1/UNC5B Expression and Potentiates Netrin-1 pro-Oncogenic Activity. Proc. Natl. Acad. Sci. USA 2021, 118, e2103319118. [Google Scholar] [CrossRef]
- Slade, N.; Horvat, A. Targeting P73--a Potential Approach in Cancer Treatment. Curr. Pharm. Des. 2011, 17, 591–602. [Google Scholar] [CrossRef]
- Zhang, H.; Schneider, J.; Rosdahl, I. Expression of P16, P27, P53, P73 and Nup88 Proteins in Matched Primary and Metastatic Melanoma Cells. Int. J. Oncol. 2002, 21, 43–48. [Google Scholar] [CrossRef]
- Steder, M.; Alla, V.; Meier, C.; Spitschak, A.; Pahnke, J.; Fürst, K.; Kowtharapu, B.S.; Engelmann, D.; Petigk, J.; Egberts, F.; et al. DNp73 Exerts Function in Metastasis Initiation by Disconnecting the Inhibitory Role of EPLIN on IGF1R-AKT/STAT3 Signaling. Cancer Cell 2013, 24, 512–527. [Google Scholar] [CrossRef] [Green Version]
- Nyman, U.; Sobczak-Pluta, A.; Vlachos, P.; Perlmann, T.; Zhivotovsky, B.; Joseph, B. Full-Length P73alpha Represses Drug-Induced Apoptosis in Small Cell Lung Carcinoma Cells. J. Biol. Chem. 2005, 280, 34159–34169. [Google Scholar] [CrossRef] [PubMed]
- Radić, M.; Šoštar, M.; Weber, I.; Ćetković, H.; Slade, N.; Bosnar, M.H. The Subcellular Localization and Oligomerization Preferences of NME1/NME2 upon Radiation-Induced DNA Damage. Int. J. Mol. Sci. 2020, 21, 2363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smirnov, A.; Anemona, L.; Novelli, F.; Piro, C.M.; Annicchiarico-Petruzzelli, M.; Melino, G.; Candi, E. P63 Is a Promising Marker in the Diagnosis of Unusual Skin Cancer. Int. J. Mol. Sci. 2019, 20, 5781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truong, A.B.; Kretz, M.; Ridky, T.W.; Kimmel, R.; Khavari, P.A. P63 Regulates Proliferation and Differentiation of Developmentally Mature Keratinocytes. Genes Dev. 2006, 20, 3185–3197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, B.C.; Lefort, K.; Mandinova, A.; Antonini, D.; Devgan, V.; Della Gatta, G.; Koster, M.I.; Zhang, Z.; Wang, J.; Di Vignano, A.T.; et al. Cross-Regulation between Notch and P63 in Keratinocyte Commitment to Differentiation. Genes Dev. 2006, 20, 1028. [Google Scholar] [CrossRef] [Green Version]
- Osada, M.; Nagakawa, Y.; Park, H.L.; Yamashita, K.; Wu, G.; Kim, M.S.; Fomenkov, A.; Trink, B.; Sidransky, D. P63-Specific Activation of the BPAG-1e Promoter. J. Investig. Dermatol. 2005, 125, 52–60. [Google Scholar] [CrossRef] [Green Version]
- Ihrie, R.A.; Marques, M.R.; Nguyen, B.T.; Horner, J.S.; Papazoglu, C.; Bronson, R.T.; Mills, A.A.; Attardi, L.D. Perp Is a P63-Regulated Gene Essential for Epithelial Integrity. Cell 2005, 120, 843–856. [Google Scholar] [CrossRef] [Green Version]
- Carroll, D.K.; Carroll, J.S.; Leong, C.O.; Cheng, F.; Brown, M.; Mills, A.A.; Brugge, J.S.; Ellisen, L.W. P63 Regulates an Adhesion Programme and Cell Survival in Epithelial Cells. Nat. Cell Biol. 2006, 8, 551–561. [Google Scholar] [CrossRef]
- Shimomura, Y.; Wajid, M.; Shapiro, L.; Christiano, A.M. P-Cadherin Is a P63 Target Gene with a Crucial Role in the Developing Human Limb Bud and Hair Follicle. Development 2008, 135, 743–753. [Google Scholar] [CrossRef] [Green Version]
- Candi, E.; Rufini, A.; Terrinoni, A.; Dinsdale, D.; Ranalli, M.; Paradisi, A.; De Laurenzi, V.; Spagnoli, L.G.; Catani, M.V.; Ramadan, S.; et al. Differential Roles of P63 Isoforms in Epidermal Development: Selective Genetic Complementation in P63 Null Mice. Cell Death Differ. 2006, 13, 1037–1047. [Google Scholar] [CrossRef] [Green Version]
- Koster, M.I.; Dai, D.; Marinari, B.; Sano, Y.; Costanzo, A.; Karin, M.; Roop, D.R. P63 Induces Key Target Genes Required for Epidermal Morphogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 3255–3260. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, Y.; Tamura, A.; Kamiya, M.; Fukuda, T.; Ishikawa, O. Immunohistochemical Analyses of P63 Expression in Cutaneous Tumours. Br. J. Dermatol. 2005, 153, 1230–1232. [Google Scholar] [CrossRef] [PubMed]
- Sakiz, D.; Turkmenoglu, T.T.; Kabukcuoglu, F. The Expression of P63 and P53 in Keratoacanthoma and Intraepidermal and Invasive Neoplasms of the Skin. Pathol. Res. Pract. 2009, 205, 589–594. [Google Scholar] [CrossRef]
- Kanner, W.A.; Brill, L.B.; Patterson, J.W.; Wick, M.R. CD10, P63 and CD99 Expression in the Differential Diagnosis of Atypical Fibroxanthoma, Spindle Cell Squamous Cell Carcinoma and Desmoplastic Melanoma. J. Cutan. Pathol. 2010, 37, 744–750. [Google Scholar] [CrossRef] [PubMed]
- Monti, P.; Ghiorzo, P.; Menichini, P.; Foggetti, G.; Queirolo, P.; Izzotti, A.; Fronza, G. TP63 Mutations Are Frequent in Cutaneous Melanoma, Support UV Etiology, but Their Role in Melanomagenesis Is Unclear. Oncol. Rep. 2017, 38, 1985–1994. [Google Scholar] [CrossRef] [Green Version]
- Radić, M.; Vlašić, I.; Jazvinšćak Jembrek, M.; Horvat, A.; Tadijan, A.; Sabol, M.; Dužević, M.; Herak Bosnar, M.; Slade, N. Characterization of Vemurafenib-Resistant Melanoma Cell Lines Reveals Novel Hallmarks of Targeted Therapy Resistance. Int. J. Mol. Sci. 2022, 23, 9910. [Google Scholar] [CrossRef]
- Bálint, E.; Bates, S.; Vousden, K.H. Mdm2 Binds P73α without Targeting Degradation. Oncogene 1999, 18, 3923–3929. [Google Scholar] [CrossRef] [Green Version]
- Dobbelstein, M.; Wienzek, S.; König, C.; Roth, J. Inactivation of the P53-Homologue P73 by the Mdm2-Oncoprotein. Oncogene 1999, 18, 2101–2106. [Google Scholar] [CrossRef] [Green Version]
- Ongkeko, W.M.; Wang, X.Q.; Siu, W.Y.; Lau, A.W.S.; Yamashita, K.; Harris, A.L.; Cox, L.S.; Poon, R.Y.C. MDM2 and MDMX Bind and Stabilize the P53-Related Protein P73. Curr. Biol. 1999, 9, 829–832. [Google Scholar] [CrossRef] [Green Version]
- Zeng, X.; Chen, L.; Jost, C.A.; Maya, R.; Keller, D.; Wang, X.; Kaelin, W.G.; Oren, M.; Chen, J.; Lu, H. MDM2 Suppresses P73 Function without Promoting P73 Degradation. Mol. Cell. Biol. 1999, 19, 3257–3266. [Google Scholar] [CrossRef] [Green Version]
- Camus, S.; Menéndez, S.; Fernandes, K.; Kua, N.; Liu, G.; Xirodimas, D.P.; Lane, D.P.; Bourdon, J. The P53 Isoforms Are Differentially Modified by Mdm2 Do Not Distribute. © 2012 Landes Bioscience. Cell Cycle 2012, 11, 1646–1655. [Google Scholar] [CrossRef] [PubMed]
- Honda, R.; Tanaka, H.; Yasuda, H. Oncoprotein MDM2 Is a Ubiquitin Ligase E3 for Tumor Suppressor P53. FEBS Lett. 1997, 420, 25–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoude, L.G.; Gartside, M.; Johansson, P.; Palmer, J.M.; Symmons, J.; Martin, N.G.; Montgomery, G.W.; Hayward, N.K. Prevalence of Germline BAP1, CDKN2A, and CDK4 Mutations in an Australian Population-Based Sample of Cutaneous Melanoma Cases. Twin Res. Hum. Genet. 2015, 18, 126–133. [Google Scholar] [CrossRef] [Green Version]
- Luo, L.; Shen, R.; Arora, A.; Orlow, I.; Busam, K.J.; Lezcano, C.; Lee, T.K.; Hernando, E.; Gorlov, I.; Amos, C.; et al. Landscape of Mutations in Early Stage Primary Cutaneous Melanoma: An InterMEL Study. Pigment Cell Melanoma Res. 2022, 35, 605–612. [Google Scholar] [CrossRef]
- Marcel, V.; Fernandes, K.; Terrier, O.; Lane, D.P.; Bourdon, J.C. Modulation of P53β and P53γ Expression by Regulating the Alternative Splicing of TP53 Gene Modifies Cellular Response. Cell Death Differ. 2014, 21, 1377. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vlašić, I.; Horvat, A.; Tadijan, A.; Slade, N. p53 Family in Resistance to Targeted Therapy of Melanoma. Int. J. Mol. Sci. 2023, 24, 65. https://doi.org/10.3390/ijms24010065
Vlašić I, Horvat A, Tadijan A, Slade N. p53 Family in Resistance to Targeted Therapy of Melanoma. International Journal of Molecular Sciences. 2023; 24(1):65. https://doi.org/10.3390/ijms24010065
Chicago/Turabian StyleVlašić, Ignacija, Anđela Horvat, Ana Tadijan, and Neda Slade. 2023. "p53 Family in Resistance to Targeted Therapy of Melanoma" International Journal of Molecular Sciences 24, no. 1: 65. https://doi.org/10.3390/ijms24010065
APA StyleVlašić, I., Horvat, A., Tadijan, A., & Slade, N. (2023). p53 Family in Resistance to Targeted Therapy of Melanoma. International Journal of Molecular Sciences, 24(1), 65. https://doi.org/10.3390/ijms24010065