

Effects of Physiological and Pathological Urea Concentrations on Human Microvascular Endothelial Cells
 ,
,  ,
,  ,
,  ,
,
Abstract
:1. Introduction
2. Results
2.1. Effect of Urea on the Growth Rate of HMEC-1
2.2. Urea-Induced Oxidative Stress Only in HMEC-1
2.3. Effect of Urea on Microfilaments Organization in HMEC-1
2.4. Urea Induces Endothelial-to-Mesenchymal Transition in HMEC-1
2.5. Effect of Urea on Protein Expression in HMEC-1
2.6. Effect of Urea on the HMEC-1 Secretome
2.7. Effect of Urea on Vasorin Expression in HMEC-1
3. Discussion
- (1)
- Vasorin is abundantly overexpressed after cell exposure to urea. This evident modulation reduces the possibility of having false positive proteins.
- (2)
- Intramembrane vasorin can be cleaved by ADAM17, a disintegrin, and metalloprotease 17, releasing the extracellular portion. This soluble and active protein binds to TGF-β and prevents its interaction with its specific receptor [26]. The solubility of released vasorin allowed us to easily verify its abundance also in plasma samples of CKD patients compared to control subjects, confirming the modulation of vasorin in human plasma.
- (3)
- Vasorin is a cell surface single-pass transmembrane glycoprotein, and it has been shown to be abundantly expressed also by vascular smooth muscle cells. Therefore, soluble vasorin could be a candidate able to cooperate and contribute to the crosstalk between endothelial cells and vascular smooth muscle cells at the systemic level [24,28].
- (4)
- Vasorin has also been demonstrated to bind TGF-β, blocking its biological activity [25]. It also regulates Notch1 signaling by interacting with Numb and preventing the degradation of Notch1 [63]. These two signaling pathways are reported to be important in the homeostasis of vascular smooth muscle cells [25,63], and vasorin could be an important signaling mediator in the crosstalk between vascular smooth muscle cells and the endothelial cell monolayer. In addition, vasorin expression is altered in several diseases; it is higher than normal in synovial fluid of patients with osteoarthritis [64], in plasma of patients with diabetic nephropathy [65], in serum of subjects with hepatocarcinoma [66], in urinary exosomes of patients with thin basement membrane nephropathy; on the contrary, vasorin expression is lower than normal in subjects with early IgA nephropathy [67,68]. Our proteomic analysis of the HMEC-1 secretome was confirmed by Western blot analysis. Vasorin downregulation was previously observed also in vivo after vascular injury. As a consequence, the expression of several cytokines, including TGF-ß, was upregulated, leading to neointimal formation, the typical fibroproliferative response to vascular injury [26]. Reverting vasorin downregulation significantly diminished injury-induced vascular lesion formation. For these reasons, vasorin has been proposed as a potential therapeutic target for vascular fibroproliferative disorders [24]. Vasorin is also linked to EMT. Vasorin is effectively cleaved by activated MMP-2 both in vitro and in vivo. We measured a significant increase in the amount of MMP-2 secreted by HMEC-1 exposed to urea. Therefore, we can hypothesize that the increased MMP-2 may have contributed to increasing the release of the soluble form of vasorin. Extracellular vasorin interferes with TGF-ß–mediated EMT, modulating E-cadherin expression and actin filament organization [25], as we have also observed in HMEC-1. In addition, vasorin modulates collagen expression [24], another EMT marker that we found among the differentially expressed proteins in HMEC-1 exposed to urea. So, vasorin downregulation, promoting TGF- ß pathway, could represent an EndMT-inducing stimulus. Since ADAM17 is the principal metalloprotease controlling vasorin cleavage, it would be interesting to investigate whether ADAM17 modulation can prevent EndMT.
4. Materials and Methods
4.1. Cell Culture
4.2. Treatment of HMEC-1 with Urea
4.3. Proliferation Assay
4.4. Quantification of Proteins Thiols
4.5. Western Blot
4.6. Immunofluorescence
4.7. Zymography
4.8. Quantitative Proteomic Analysis of HMEC-1 after a 72-h Exposure to Urea
4.9. Quantitative Proteomic Analysis of Proteins Released by HMEC-1 after a 72-h Exposure to Urea
4.10. High Resolution Mass Spectrometry Analysis (nLC-MSMS)
4.11. MS Data Analysis–Label-free Quantitative Proteomics
4.12. Validation of Proteomics Data of Proteins Released by HMEC-1 after a 72-h Exposure to Urea
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADMA | asymmetric dimethylarginine |
| AMBIC | ammonium bicarbonate |
| BCA | bicinchoninic acid |
| BSA | bovine serum albumin |
| cHDL | carbamylated high-density lipoprotein |
| cLDL | carbamylated low-density lipoprotein |
| CKD | chronic kidney disease |
| CVD | cardiovascular diseases |
| CVS | cardiovascular system |
| DAPI | 4’6-diamidino-2-phenylindole |
| DDAH | dimethylarginine dimethylaminohydrolase |
| ECL | enhanced chemiluminescence |
| EMT | epithelial-to-mesenchymal transition |
| EndMT | endothelial-to-mesenchymal transition |
| FBS | foetal bovine serum |
| FDR | false discovery rate |
| FITC | fluorescein isothiocyanate |
| GFR | glomerular filtration rate |
| HMEC-1 | human dermal microvascular endothelial cells-1 |
| LF-PVDF | low-fluorescence polyvinylidene difluoride |
| MMP | matrix metalloproteinases |
| PBS | phosphate buffered saline |
| PS | phosphatidylserine |
| SRB | sulforhodamine B |
| TFA | trifluoroacetic acid |
| TGF-β | transforming growth factor-beta |
| TRITC | tetramethylrhodamine-isothiocyanate |
| WHO | World Health Organization |
References
- Vanholder, R.; De Smet, R.; Glorieux, G.; Argilés, A.; Baurmeister, U.; Brunet, P.; Clark, W.; Cohen, G.; De Deyn, P.P.; Deppisch, R.; et al. Review on uremic toxins: Classification, concentration, and interindividual variability. Kidney Int. 2003, 63, 1934–1943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenvinkel, P.; Carrero, J.J.; Axelsson, J.; Lindholm, B.; Heimbürger, O.; Massy, Z. Emerging biomarkers for evaluating cardiovascular risk in the chronic kidney disease patient: How do new pieces fit into the uremic puzzle? Clin. J. Am. Soc. Nephrol. 2008, 3, 505–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barmore, W.; Azad, F.; Stone, W.L. Physiology, Urea Cycle. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Vanholder, R.; Argilés, A.; Baurmeister, U.; Brunet, P.; Clark, W.; Cohen, G.; De Deyn, P.P.; Deppisch, R.; Descamps-Latscha, B.; Henle, T.; et al. Uremic toxicity: Present state of the art. Int. J. Artif. Organs 2001, 24, 695–725. [Google Scholar] [CrossRef]
- Lau, W.L.; Vaziri, N.D. Urea, a true uremic toxin: The empire strikes back. Clin. Sci. 2017, 131, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Jha, V.; Garcia-Garcia, G.; Iseki, K.; Li, Z.; Naicker, S.; Plattner, B.; Saran, R.; Wang, A.Y.; Yang, C.W. Chronic kidney disease: Global dimension and perspectives. Lancet 2013, 382, 260–272. [Google Scholar] [CrossRef] [PubMed]
- Mok, Y.; Ballew, S.H.; Matsushita, K. Prognostic value of chronic kidney disease measures in patients with cardiac disease. Circ. J. 2017, 81, 1075–1084. [Google Scholar] [CrossRef] [Green Version]
- D’Apolito, M.; Du, X.; Pisanelli, D.; Pettoello-Mantovani, M.; Campanozzi, A.; Giacco, F.; Maffione, A.B.; Colia, A.L.; Brownlee, M.; Giardino, I. Urea-induced ROS cause endothelial dysfunction in chronic renal failure. Atherosclerosis 2015, 239, 393–400. [Google Scholar] [CrossRef] [Green Version]
- D’Apolito, M.; Colia, A.L.; Manca, E.; Pettoello-Mantovani, M.; Sacco, M.; Maffione, A.B.; Brownlee, M.; Giardino, I. Urea Memory: Transient cell exposure to urea causes persistent mitochondrial ros production and endothelial dysfunction. Toxins 2018, 10, 410. [Google Scholar] [CrossRef] [Green Version]
- Trécherel, E.; Godin, C.; Louandre, C.; Pettoello-Mantovani, M.; Sacco, M.; Maffione, A.B.; Brownlee, M.; Giardino, I. Upregulation of BAD, a pro-apoptotic protein of the BCL2 family, in vascular smooth muscle cells exposed to uremic conditions. Biochem. Biophys. Res. Commun. 2012, 417, 479–483. [Google Scholar] [CrossRef]
- Shroff, R.; McNair, R.; Figg, N.; Skepper, J.; Schurgers, L.; Gupta, A.; Hiorns, M.; Donald, A.E.; Deanfield, J.; Rees, L.; et al. Dialysis accelerates medial vascular calcification in part by triggering smooth muscle cell apoptosis. Circulation 2008, 118, 1748–1757. [Google Scholar] [CrossRef]
- Charreau, B. Secretome and Tunneling Nanotubes: A Multilevel Network for Long Range Intercellular Communication between Endothelial Cells and Distant Cells. Int. J. Mol. Sci. 2021, 22, 7971. [Google Scholar] [CrossRef] [PubMed]
- Dignat-George, F.; Boulanger, C.M. The many faces of endothelial microparticles. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 27–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Jong, O.G.; Verhaar, M.C.; Chen, Y.; Vader, P.; Gremmels, H.; Posthuma, G.; Schiffelers, R.M.; Gucek, M.; van Balkom, B.W. Cellular stress conditions are reflected in the protein and RNA content of endothelial cell-derived exosomes. J. Extracell. Vesicles 2012, 1, 18396. [Google Scholar] [CrossRef] [PubMed]
- Tkach, M.; Théry, C. Communication by extracellular vesicles: Where we are and where we need to go. Cell 2016, 164, 1226–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaziri, N.D. Oxidative stress in uremia: Nature, mechanisms, and potential consequences. Semin. Nephrol. 2004, 24, 469–473. [Google Scholar] [CrossRef]
- Kendrick, J.; Chonchol, M.B. Nontraditional risk factors for cardiovascular disease in patients with chronic kidney disease. Nat. Clin. Pract. Nephrol. 2008, 4, 672–681. [Google Scholar] [CrossRef]
- Hirase, T.; Node, K. Endothelial dysfunction as a cellular mechanism for vascular failure. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H499–H505. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.B.; Ren, X.; Liu, J.; Guo, X.W.; Jiang, X.P.; Zhang, D.X.; Huang, Y.S.; Zhang, J.P. HSP27 phosphorylation protects against endothelial barrier dysfunction under burn serum challenge. Biochem. Biophys. Res. Commun. 2015, 463, 377–383. [Google Scholar] [CrossRef]
- Rho, S.S.; Ando, K.; Fukuhara, S. Dynamic regulation of vascular permeability by vascular endothelial cadherin-mediated endothelial cell-cell junctions. J. Nippon Med. Sch. 2017, 84, 148–159. [Google Scholar] [CrossRef] [Green Version]
- Gong, H.; Lyu, X.; Wang, Q.; Hu, M.; Zhang, X. Endothelial to mesenchymal transition in the cardiovascular system. Life Sci. 2017, 184, 95–102. [Google Scholar] [CrossRef]
- Piera-Velazquez, S.; Jimenez, S.A. Endothelial to mesenchymal transition: Role in physiology and in the pathogenesis of human diseases. Physiol. Rev. 2019, 99, 1281–1324. [Google Scholar] [CrossRef] [PubMed]
- Toth, M.; Sohail, A.; Fridman, R. Assessment of gelatinases (MMP-2 and MMP-9) by gelatin zymography. Methods Mol. Biol. 2012, 878, 121–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, Y.; Imai, Y.; Kumagai, H.; Nosaka, T.; Morikawa, Y.; Hisaoka, T.; Manabe, I.; Maemura, K.; Nakaoka, T.; Imamura, T.; et al. Vasorin, a transforming growth factor beta-binding protein expressed in vascular smooth muscle cells, modulates the arterial response to injury in vivo. Proc. Natl. Acad. Sci. USA 2004, 101, 10732–10737. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Zhang, T.; Yang, X.; Geng, J.; Li, S.; Ding, H.; Li, H.; Huang, A.; Wang, C.; Sun, L.; et al. Identification of functional mimotopes of human vasorin ectodomain by biopanning. Int. J. Biol. Sci. 2018, 14, 461–470. [Google Scholar] [CrossRef] [Green Version]
- Malapeira, J.; Esselens, C.; Bech-Serra, J.J.; Canals, F.; Arribas, J. ADAM17 (TACE) regulates TGFβ signaling through the cleavage of vasorin. Oncogene 2011, 30, 912–1922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choksi, S.; Lin, Y.; Pobezinskaya, Y.; Chen, L.; Park, C.; Morgan, M.; Li, T.; Jitkaew, S.; Cao, X.; Kim, Y.S.; et al. A HIF-1 target, ATIA, protects cells from apoptosis by modulating the mitochondrial thioredoxin, TRX2. Mol. Cell 2011, 42, 597–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnet, A.L.; Chaussain, C.; Broutin, I.; Rochefort, G.Y.; Schrewe, H.; Gaucher, C. From vascular smooth muscle cells to folliculogenesis: What about vasorin? Front. Med. 2018, 5, 335. [Google Scholar] [CrossRef]
- Pintus, G.; Giordo, R.; Wang, Y.; Zhu, W.; Kim, S.H.; Zhang, L.; Ni, L.; Zhang, J.; Telljohann, R.; McGraw, K.R.; et al. Reduced vasorin enhances angiotensin II signaling within the aging arterial wall. Oncotarget 2018, 9, 27117–27132. [Google Scholar] [CrossRef]
- Luong, T.T.D.; Estepa, M.; Boehme, B.; Pieske, B.; Lang, F.; Eckardt, K.U.; Voelkl, J.; Alesutan, I. Inhibition of vascular smooth muscle cell calcification by vasorin through interference with TGFβ1 signaling. Cell Signal. 2019, 64, 109414. [Google Scholar] [CrossRef]
- Webster, A.C.; Nagler, E.V.; Morton, R.L.; Masson, P. Chronic Kidney Disease. Lancet 2017, 389, 1238–1252. [Google Scholar] [CrossRef]
- Evenepoel, P.; Meijers, B.K.; Bammens, B.R.; Verbeke, K. Uremic toxins originating from colonic microbial metabolism. Kidney Int. Suppl. 2009, 76, S12–S19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouré, T.; Vanholder, R. Biochemical and clinical evidence for uremic toxicity. Artif. Organs 2004, 28, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Yavuz, A.; Tetta, C.; Ersoy, F.F.; D’intini, V.; Ratanarat, R.; De Cal, M.; Bonello, M.; Bordoni, V.; Salvatori, G.; Andrikos, E.; et al. Uremic toxins: A new focus on an old subject. Semin. Dial. 2005, 18, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Apostolov, E.O.; Shah, S.V.; Ok, E.; Basnakian, A.G. Quantification of carbamylated LDL in human sera by a new sandwich ELISA. Clin. Chem. 2005, 51, 719–728. [Google Scholar] [CrossRef]
- Covic, A.; Kanbay, M.; Voroneanu, L.; Turgut, F.; Serban, D.N.; Serban, I.L.; Goldsmith, D.J. Vascular calcification in chronic kidney disease. Clin. Sci. 2010, 119, 111–121. [Google Scholar] [CrossRef]
- Lau, W.L.; Ix, J.H. Clinical detection, risk factors, and cardiovascular consequences of medial arterial calcification: A pattern of vascular injury associated with aberrant mineral metabolism. Semin. Nephrol. 2013, 33, 93–105. [Google Scholar] [CrossRef]
- Chao, C.T.; Chiang, C.K. Uremic toxins, oxidative stress, and renal fibrosis: An interwined complex. J. Ren. Nutr. 2014, 25, 55–59. [Google Scholar] [CrossRef]
- D’Apolito, M.; Du, X.; Zong, H.; Catucci, A.; Maiuri, L.; Trivisano, T.; Pettoello-Mantovani, M.; Campanozzi, A.; Raia, V.; Pessin, J.E.; et al. Urea-induced ROS generation causes insulin resistance in mice with chronic renal failure. J. Clin. Invest. 2010, 120, 203–213. [Google Scholar] [CrossRef]
- Dalle-Donne, I.; Rossi, R.; Colombo, R.; Giustarini, D.; Milzani, A. Biomarkers of oxidative damage in human disease. Clin. Chem. 2006, 52, 601–623. [Google Scholar] [CrossRef]
- Atkins, G.B.; Jain, M.K.; Hamik, A. Endothelial differentiation: Molecular mechanisms of specification and heterogeneity. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1476–1484. [Google Scholar] [CrossRef]
- Arciniegas, E.; Frid, M.G.; Douglas, I.S.; Stenmark, K.R. Perspectives on endothelial-to-mesenchymal transition: Potential contribution to vascular remodeling in chronic pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L1–L8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Duffhues, G.; García de Vinuesa, A.; Ten Dijke, P. Endothelial-to-mesenchymal transition in cardiovascular diseases: Developmental signaling pathways gone awry. Dev. Dyn. 2018, 247, 492–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, N.; Hu, M.; Khalil, R.A. Biochemical and biological attributes of matrix metalloproteinases. Prog. Mol. Biol. Transl. Sci. 2017, 147, 1–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Khalil, R.A. Matrix metalloproteinases, vascular remodeling, and vascular disease. Adv. Pharmacol. 2018, 81, 241–330. [Google Scholar] [CrossRef]
- Carbó, C.; Arderiu, G.; Escolar, G.; Fusté, B.; Cases, A.; Carrascal, M.; Abián, J.; Díaz-Ricart, M. Differential expression of proteins from cultured endothelial cells exposed to uremic versus normal serum. Am. J. Kidney Dis. 2008, 51, 603–612. [Google Scholar] [CrossRef]
- Kenis, H.; Hofstra, L.; Reutelingsperger, C.P. Annexin A5: Shifting from a diagnostic towards a therapeutic realm. Cell. Mol. Life Sci. 2007, 64, 2859–2862. [Google Scholar] [CrossRef]
- Li, G.; Ma, D.; Chen, Y. Cellular functions of programmed cell death 5. Biochim. Biophys. Acta 2016, 1863, 572–580. [Google Scholar] [CrossRef]
- Garg, S.; Hofstra, L.; Reutelingsperger, C.; Narula, J. Apoptosis as a therapeutic target in acutely ischemic myocardium. Curr. Opin. Cardiol. 2003, 18, 372–377. [Google Scholar] [CrossRef]
- Kolodgie, F.D.; Narula, J.; Burke, A.P.; Haider, N.; Farb, A.; Hui-Liang, Y.; Smialek, J.; Virmani, R. Localization of apoptotic macrophages at the site of plaque rupture in sudden coronary death. Am. J. Pathol. 2000, 157, 1259–1268. [Google Scholar] [CrossRef] [Green Version]
- Laufer, E.M.; Reutelingsperger, C.P.; Narula, J.; Hofstra, L. Annexin A5: An imaging biomarker of cardiovascular risk. Basic Res. Cardiol. 2008, 103, 95–104. [Google Scholar] [CrossRef]
- Roth, W.; Hatzfeld, M.; Friedrich, M.; Thiering, S.; Magin, T.M. Keratin function and regulation in tissue homeostasis and pathogenesis. Biomol. Concepts 2012, 3, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Choi, W.J.; Lee, C.H. Phosphorylation and reorganization of keratin networks: Implications for carcinogenesis and epithelial mesenchymal transition. Biomol. Ther. 2015, 23, 301–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez, F.; Sakai, L.Y. Biogenesis and function of fibrillin assemblies. Cell Tissue Res. 2010, 339, 71–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyer, A.S.; Erickson, C.P.; Runyan, R.B. Epithelial-mesenchymal transformation in the embryonic heart is mediated through distinct pertussis toxin-sensitive and TGFbeta signal transduction mechanisms. Dev. Dyn. 1999, 214, 81–91. [Google Scholar] [CrossRef]
- Revert, F.; Revert-Ros, F.; Blasco, R.; Artigot, A.; López-Pascual, E.; Gozalbo-Rovira, R.; Ventura, I.; Gutiérrez-Carbonell, E.; Roda, N.; Ruíz-Sanchis, D.; et al. Selective targeting of collagen IV in the cancer cell microenvironment reduces tumor burden. Oncotarget 2018, 9, 11020–11045. [Google Scholar] [CrossRef] [Green Version]
- Smith, B.N.; Bhowmick, N.A. Role of EMT in metastasis and therapy resistance. J. Clin. Med. 2016, 5, 17. [Google Scholar] [CrossRef] [Green Version]
- Anderssohn, M.; Schwedhelm, E.; Lüneburg, N.; Vasan, R.S.; Böger, R.H. Asymmetric dimethylarginine as a mediator of vascular dysfunction and a marker of cardiovascular disease and mortality: An intriguing interaction with diabetes mellitus. Diab. Vasc. Dis. Res. 2010, 7, 105–118. [Google Scholar] [CrossRef] [Green Version]
- Leiper, J.; Nandi, M.; Torondel, B.; Murray-Rust, J.; Malaki, M.; O′Hara, B.; Rossiter, S.; Anthony, S.; Madhani, M.; Selwood, D.; et al. Disruption of methylarginine metabolism impairs vascular homeostasis. Nat. Med. 2007, 13, 198–203. [Google Scholar] [CrossRef]
- Palm, F.; Onozato, M.L.; Luo, Z.; Wilcox, C.S. Dimethylarginine dimethylaminohydrolase (DDAH): Expression, regulation, and function in the cardiovascular and renal systems. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H3227–H3245. [Google Scholar] [CrossRef] [Green Version]
- Tripepi, G.; Mattace Raso, F.; Sijbrands, E.; Seck, M.S.; Maas, R.; Boger, R.; Witteman, J.; Rapisarda, F.; Malatino, L.; Mallamaci, F.; et al. Inflammation and asymmetric dimethylarginine for predicting death and cardiovascular events in ESRD patients. Clin. J. Am. Soc. Nephrol. 2011, 6, 1714–1721. [Google Scholar] [CrossRef]
- Jacobi, J.; Tsao, P.S. Asymmetrical dimethylarginine in renal disease: Limits of variation or variation limits? A systematic review. Am. J. Nephrol. 2008, 28, 224–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Man, J.; Yu, X.; Huang, H.; Zhou, W.; Xiang, C.; Huang, H.; Miele, L.; Liu, Z.; Bebek, G.; Bao, S.; et al. Hypoxic induction of vasorin regulates Notch1 turnover to maintain glioma stem-like cells. Cell Stem Cell 2018, 22, 104–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, W.; Li, Z.; Li, T.; Zhang, Q.; Zhang, H.; Wang, X. Proteomic analysis of synovial fluid in osteoarthritis using SWATH-mass spectrometry. Mol. Med. Rep. 2018, 17, 2827–2836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, J.M.; Kim, B.G.; Yu, M.H.; Lee, I.K.; Cho, J.Y. Identification of diabetic nephropathy-selective proteins in human plasma by multi-lectin affinity chromatography and LC-MS/MS. Proteomics Clin. Appl. 2010, 4, 644–653. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, H.; Yang, X.; Wang, W.; Huang, A.; Li, J.; Qin, X.; Li, F.; Lu, G.; Ding, H.; et al. Vasorin is a potential serum biomarker and drug target of hepatocarcinoma screened by subtractive-EMSA-SELEX to clinic patient serum. Oncotarget 2015, 6, 10045–10059. [Google Scholar] [CrossRef] [Green Version]
- Moon, P.G.; Lee, J.E.; You, S.; Kim, T.K.; Cho, J.H.; Kim, I.S.; Kwon, T.H.; Kim, C.D.; Park, S.H.; Hwang, D.; et al. Proteomic analysis of urinary exosomes from patients of early IgA nephropathy and thin basement membrane nephropathy. Proteomics 2011, 11, 2459–2475. [Google Scholar] [CrossRef]
- Samavat, S.; Kalantari, S.; Nafar, M.; Rutishauser, D.; Rezaei-Tavirani, M.; Parvin, M.; Zubarev, R.A. Diagnostic urinary proteome profile for immunoglobulin a nephropathy. Iran. J. Kidney Dis. 2015, 9, 239–248. [Google Scholar]
- Orellana, E.A.; Kasinski, A.L. Sulforhodamine B (SRB) assay in cell culture to investigate cell proliferation. Bio Protoc. 2016, 6, e1984. [Google Scholar] [CrossRef] [Green Version]
- Hill, B.G.; Reily, C.; Oh, J.Y.; Johnson, M.S.; Landar, A. Methods for the determination and quantification of the reactive thiol proteome. Free Radic. Biol. Med. 2009, 47, 675–683. [Google Scholar] [CrossRef] [Green Version]
- Rivero-Gutiérrez, B.; Anzola, A.; Martínez-Augustin, O.; de Medina, F.S. Stain-free detection as loading control alternative to Ponceau and housekeeping protein immunodetection in Western blotting. Anal. Biochem. 2014, 467, 1–3. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
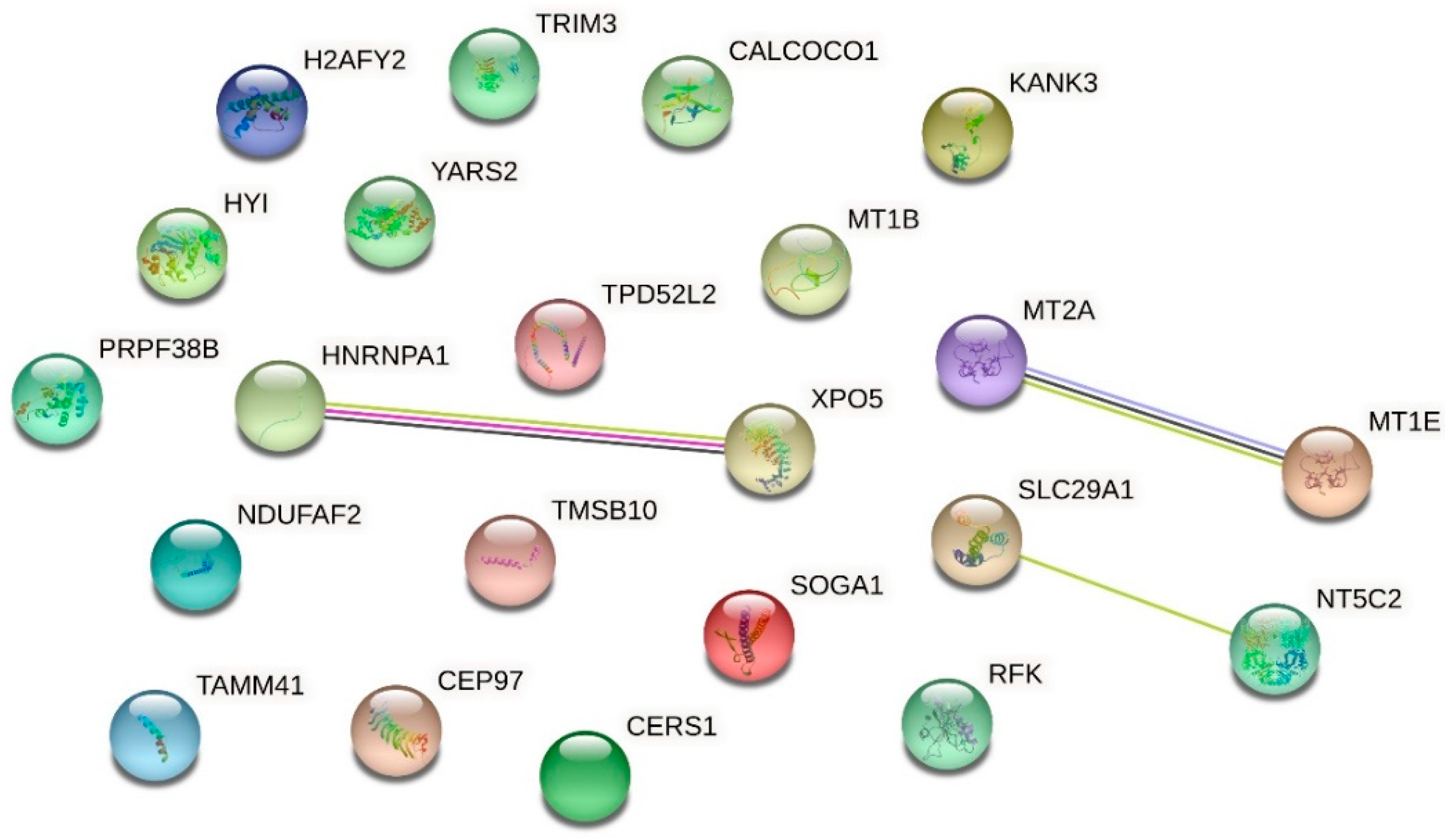
| Up-Regulated Proteins | ||||
|---|---|---|---|---|
| Accession | Description | Abundance Ratio | p-Value | |
| 5 g/L vs. 0.25 g/L | ||||
| O94964 | Isoform 2 of Protein SOGA1 | 2.267 | 0.000000020868 | |
| Q5VTL8 | Pre-mRNA-splicing factor 38B | 2.156 | 0.000000000010 | |
| Q8IW35 | Centrosomal protein of 97 kDa | 1.953 | 0.000004938470 | |
| P09651 | Isoform A1-A of Heterogeneous nuclear ribonucleoprotein A1 | 1.949 | 0.000001279087 | |
| Q9HAV4 | Exportin-5 | 1.944 | 0.000000008737 | |
| P04732 | Metallothionein-1E | 1.865 | 0.000033970136 | |
| P07438 | Metallothionein-1B | 1.865 | 0.000033970136 | |
| Q9P0M6 | Core histone macro-H2A.2 | 1.830 | 0.000073230796 | |
| Q9Y2Z4 | Tyrosine--tRNA ligase, mitochondrial | 1.739 | 0.000365094872 | |
| Q8N183 | NADH dehydrogenase [ubiquinone] 1 alpha subcomplex factor 2 | 1.722 | 0.000007391038 | |
| P49902 | Cytosolic purine 5′-nucleotidase | 1.719 | 0.000237839101 | |
| Q6NY19 | KN motif and ankyrin repeat domain-containing protein 3 | 1.669 | 0.013682890309 | |
| O43399 | Isoform 3 of Tumor protein D54 | 1.594 | 0.006287115623 | |
| Q5T013 | Putative hydroxypyruvate isomerase | 1.584 | 0.003957519742 | |
| Q9P1Z2 | Calcium-binding and coiled-coil domain-containing protein 1 | 1.547 | 0.014584529276 | |
| P63313 | Thymosin beta-10 | 1.508 | 0.000000312149 | |
| Q969G6 | Riboflavin kinase | 1.506 | 0.025817204876 | |
| O75382 | Tripartite motif-containing protein 3 | 1.499 | 0.036583959883 | |
| Q99808 | Equilibrative nucleoside transporter 1 | 1.489 | 0.032611308850 | |
| Q96BW9 | Phosphatidate cytidylyltransferase, mitochondrial | 1.464 | 0.006245305736 | |
| P27544 | Ceramide synthase 1 | 1.445 | 0.017775493602 | |
| P02795 | Metallothionein-2 | 1.418 | 0.000036164289 | |
| Down-regulated proteins | ||||
| Accession | Description | Abundance Ratio | p-Value | |
| 5 g/L vs. 0.25 g/L | 0.25 g/L vs. 5 g/L | |||
| Q12800 | Alpha-globin transcription factor CP2 | 0.265 | 3.773 | 0.0000000000000015 |
| O75962 | Triple functional domain protein | 0.454 | 2.202 | 0.0002507027368594 |
| Q04837 | Single-stranded DNA-binding protein, mitochondrial | 0.474 | 2.109 | 0.0000000000000015 |
| Q9NQG1 | Protein MANBAL | 0.495 | 2.020 | 0.0027647462862580 |
| Q13416 | Origin recognition complex subunit 2 | 0.503 | 1.988 | 0.0000031299038550 |
| P04818 | Thymidylate synthase | 0.558 | 1.792 | 0.0101710429818778 |
| Q92542 | Nicastrin | 0.564 | 1.773 | 0.0031084727012270 |
| Q9GZM8 | Nuclear distribution protein nudE-like 1 | 0.575 | 1.739 | 0.0000538096333800 |
| P24821 | Isoform 4 of Tenascin | 0.593 | 1.686 | 0.0002257365561964 |
| Q9Y5U9 | Immediate early response 3-interacting protein 1 | 0.606 | 1.650 | 0.0035816066338457 |
| P01892 | HLA class I histocompatibility antigen, A-2 alpha chain | 0.622 | 1.607 | 0.0074612565194133 |
| Q53H12 | Acylglycerol kinase, mitochondrial | 0.629 | 1.589 | 0.0136828903087287 |
| P40306 | Proteasome subunit beta type-10 | 0.644 | 1.552 | 0.0064794736331468 |
| Q99988 | Growth/differentiation factor 15 | 0.649 | 1.540 | 0.0000143152780648 |
| Q9UHF1 | Epidermal growth factor-like protein 7 | 0.654 | 1.529 | 0.0308946734033236 |
| P24821 | Tenascin | 0.666 | 1.501 | 0.0000035594104464 |
| Q6NXE6 | Armadillo repeat-containing protein 6 | 0.674 | 1.483 | 0.0340911967228681 |
| Q15149 | Isoform 3 of Plectin | 0.691 | 1.447 | 0.0496321589542498 |
| Q92743 | Serine protease HTRA1 | 0.693 | 1.443 | 0.0001037941772726 |
| P00505 | Aspartate aminotransferase, mitochondrial | 0.700 | 1.428 | 0.0001349131929805 |
| Q6PIU2 | Neutral cholesterol ester hydrolase 1 | 0.707 | 1.414 | 0.0045281605174441 |
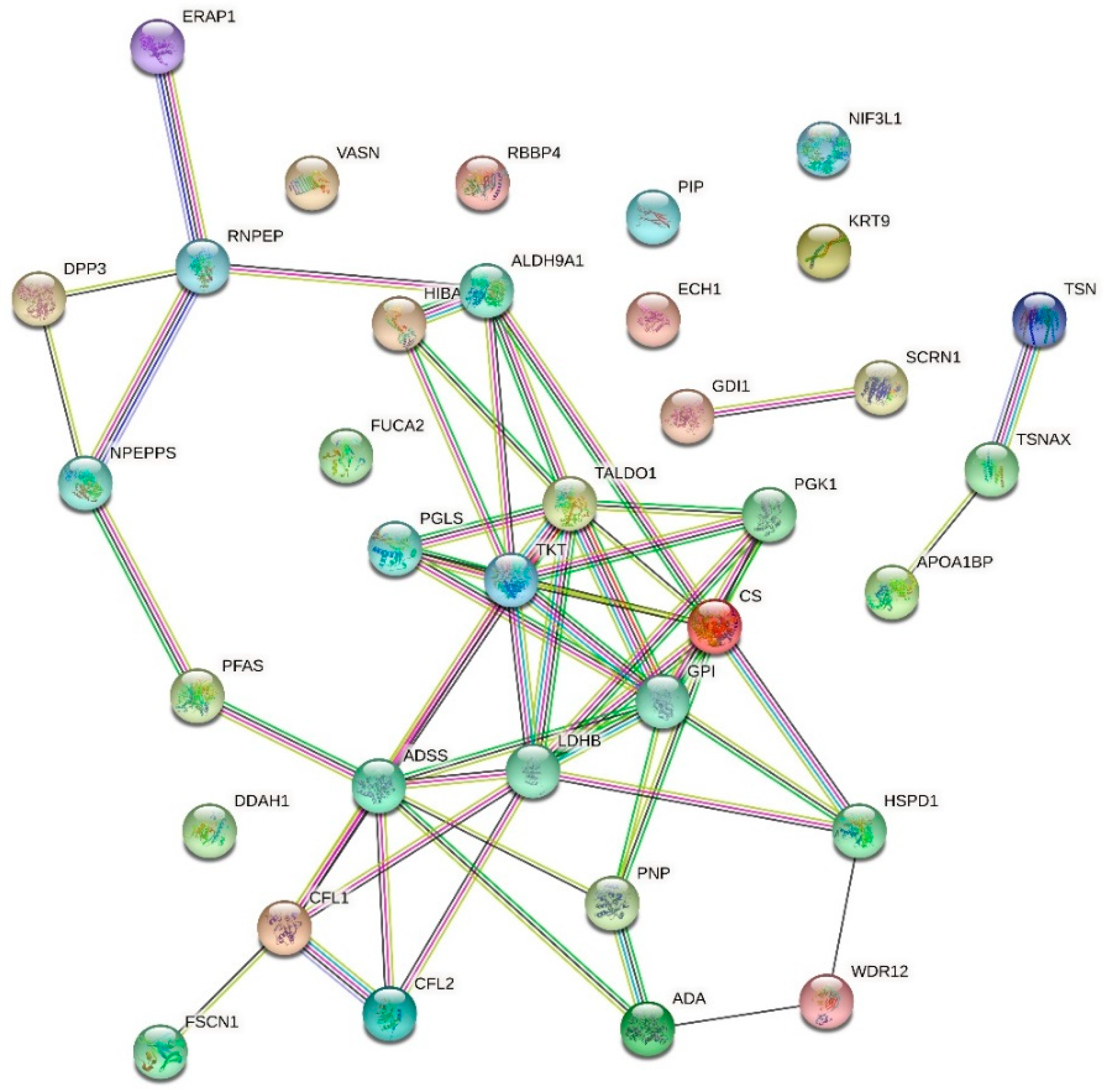
| Up-Regulated Proteins | ||||
|---|---|---|---|---|
| Accession | Description | Abundance Ratio | p-Value | |
| Control/Urea (5 g/L) | ||||
| APOA1BP | NAD(P)H-hydrate epimerase | 49.966 | 0.000000000000022 | |
| O94760 | N(G),N(G)-dimethylarginine dimethylaminohydrolase 1 | 45.754 | 0.000000000000091 | |
| A0A024R3X4 | Epididymis secretory sperm binding protein | 30.065 | 0.000000000046539 | |
| P30520 | Adenylosuccinate synthetase isozyme 2 | 26.540 | 0.000000000936884 | |
| A0A024R3V7 | NIF3-like protein 1 | 14.996 | 0.000009726416165 | |
| A0A024RB75 | Citrate synthase | 14.257 | 0.000000573618455 | |
| P12273 | Prolactin-inducible protein | 14.216 | 0.000000591547361 | |
| P55786 | Puromycin-sensitive aminopeptidase | 14.108 | 0.000000641574461 | |
| Q549N0 | Cofilin 2 (Muscle), isoform CRA_a | 12.992 | 0.000001555188153 | |
| Q8N7G1 | Purine nucleoside phosphorylase | 12.660 | 0.000001995520621 | |
| P29401 | Transketolase | 11.977 | 0.000003566403024 | |
| Q16658 | Fascin | 11.654 | 0.000004723432374 | |
| Q9BTY2 | Plasma alpha-L-fucosidase | 11.477 | 0.000018168658036 | |
| Q13011 | Delta(3,5)-Delta(2,4)-dienoyl-CoA isomerase, mitochondrial | 11.337 | 0.000007886470783 | |
| PFAS | Phosphoribosylformylglycinamidine synthase, isoform CRA_b | 11.234 | 0.000006831656478 | |
| Q53T99 | Ribosome biogenesis protein WDR12 | 9.173 | 0.000048900585780 | |
| Q6EMK4 | Vasorin | 9.026 | 0.000641197347120 | |
| V9HWF4 | Phosphoglycerate kinase | 8.109 | 0.000151428360979 | |
| G3V180 | Dipeptidyl peptidase 3 | 7.275 | 0.002621057250876 | |
| GDI1 | Rab GDP dissociation inhibitor | 6.991 | 0.000533863830113 | |
| P49189 | 4-trimethylaminobutyraldehyde dehydrogenase | 6.658 | 0.000770649443919 | |
| P00813 | Adenosine deaminase | 6.281 | 0.001233369407633 | |
| P31937 | 3-hydroxyisobutyrate dehydrogenase, mitochondrial | 5.937 | 0.008777288491631 | |
| E9PK25 | Cofilin-1 | 5.435 | 0.003602441604062 | |
| SCRN1 | Secernin 1 | 5.201 | 0.020151555976657 | |
| GPI | Glucose-6-phosphate isomerase | 5.044 | 0.006722750720656 | |
| P37837 | Transaldolase | 5.013 | 0.006387658270684 | |
| P07195 | L-lactate dehydrogenase B chain | 4.842 | 0.008086580808851 | |
| A0A024R3V8 | Translin-associated factor X, isoform CRA_c | 4.789 | 0.030367926630212 | |
| Q09028 | Histone-binding protein RBBP4 | 4.762 | 0.021788773185764 | |
| HEL-S-304 | 6-phosphogluconolactonase | 4.740 | 0.027747024650690 | |
| Q15631 | Translin | 4.612 | 0.011041331019512 | |
| RNPEP | Aminopeptidase B | 4.580 | 0.030367926630212 | |
| Q9NZ08 | Endoplasmic reticulum aminopeptidase 1 | 3.997 | 0.025908828760954 | |
| P35527 | Keratin, type I cytoskeletal 9 | 3.948 | 0.027944488189618 | |
| Down-regulated proteins | ||||
| Accession | Description | Abundance Ratio | p-Value: | |
| Control/Urea (5 g/L) | Urea (5g/L)/Control | |||
| P35556 | Fibrillin-2 | 0.068 | 14.705 | 0.000001550 |
| P06899 | Histone H2B type 1-J | 0.082 | 12.195 | 0.000010319 |
| P02462 | Collagen alpha-1(IV) chain | 0.127 | 7.874 | 0.002994145 |
| P08758 | Annexin A5 | 0.127 | 7.874 | 0.000533864 |
| Q5TEC6 | Histone H3 | 0.128 | 7.812 | 0.000563535 |
| P18065 | Insulin-like growth factor-binding protein 2 | 0.129 | 7.751 | 0.000601115 |
| Q01844 | RNA-binding protein EWS | 0.131 | 7.633 | 0.005687063 |
| P46783 | 40S ribosomal protein S10 | 0.136 | 7.352 | 0.000918185 |
| P62857 | 40S ribosomal protein S28 | 0.157 | 6.369 | 0.009562979 |
| O00273 | DNA fragmentation factor subunit alpha | 0.164 | 6.097 | 0.020667882 |
| B2R4R0 | Histone H4 | 0.171 | 5.847 | 0.005017696 |
| O14737 | Programmed cell death protein 5 | 0.182 | 5.494 | 0.033480893 |
| EFEMP1 | EGF containing fibulin-like extracellular matrix protein 1 isoform 1 | 0.189 | 5.291 | 0.010064879 |
| P52943 | Cysteine-rich protein 2 | 0.189 | 5.291 | 0.044393972 |
| Q01995 | Transgelin | 0.208 | 4.807 | 0.017871069 |
| O00233 | 26S proteasome non-ATPase regulatory subunit 9 | 0.209 | 4.784 | 0.042721853 |
| Q14847 | LIM and SH3 domain protein 1 | 0.210 | 4.761 | 0.019123177 |
| E5RIM7 | Copper transport protein ATOX1 | 0.218 | 4.587 | 0.023827383 |
| P62750 | 60S ribosomal protein L23a | 0.231 | 4.329 | 0.032476814 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Colombo, G.; Altomare, A.; Astori, E.; Landoni, L.; Garavaglia, M.L.; Rossi, R.; Giustarini, D.; Lionetti, M.C.; Gagliano, N.; Milzani, A.; et al. Effects of Physiological and Pathological Urea Concentrations on Human Microvascular Endothelial Cells. Int. J. Mol. Sci. 2023, 24, 691. https://doi.org/10.3390/ijms24010691
Colombo G, Altomare A, Astori E, Landoni L, Garavaglia ML, Rossi R, Giustarini D, Lionetti MC, Gagliano N, Milzani A, et al. Effects of Physiological and Pathological Urea Concentrations on Human Microvascular Endothelial Cells. International Journal of Molecular Sciences. 2023; 24(1):691. https://doi.org/10.3390/ijms24010691
Chicago/Turabian StyleColombo, Graziano, Alessandra Altomare, Emanuela Astori, Lucia Landoni, Maria Lisa Garavaglia, Ranieri Rossi, Daniela Giustarini, Maria Chiara Lionetti, Nicoletta Gagliano, Aldo Milzani, and et al. 2023. "Effects of Physiological and Pathological Urea Concentrations on Human Microvascular Endothelial Cells" International Journal of Molecular Sciences 24, no. 1: 691. https://doi.org/10.3390/ijms24010691
APA StyleColombo, G., Altomare, A., Astori, E., Landoni, L., Garavaglia, M. L., Rossi, R., Giustarini, D., Lionetti, M. C., Gagliano, N., Milzani, A., & Dalle-Donne, I. (2023). Effects of Physiological and Pathological Urea Concentrations on Human Microvascular Endothelial Cells. International Journal of Molecular Sciences, 24(1), 691. https://doi.org/10.3390/ijms24010691