

Regulation of Transcriptional Activity of Merkel Cell Polyomavirus Large T-Antigen by PKA-Mediated Phosphorylation

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
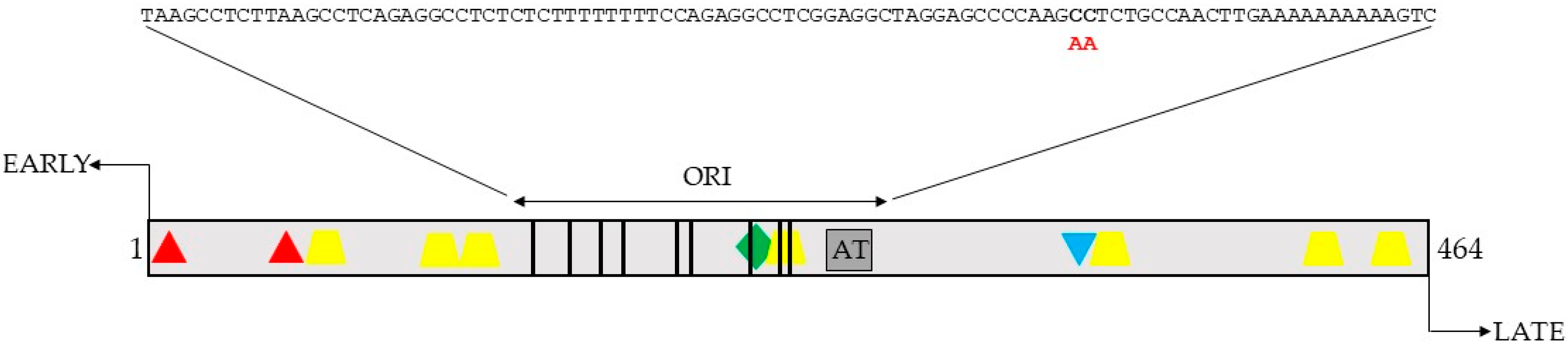
2.1. Identification of Putative PKA Phosphoacceptor Sites
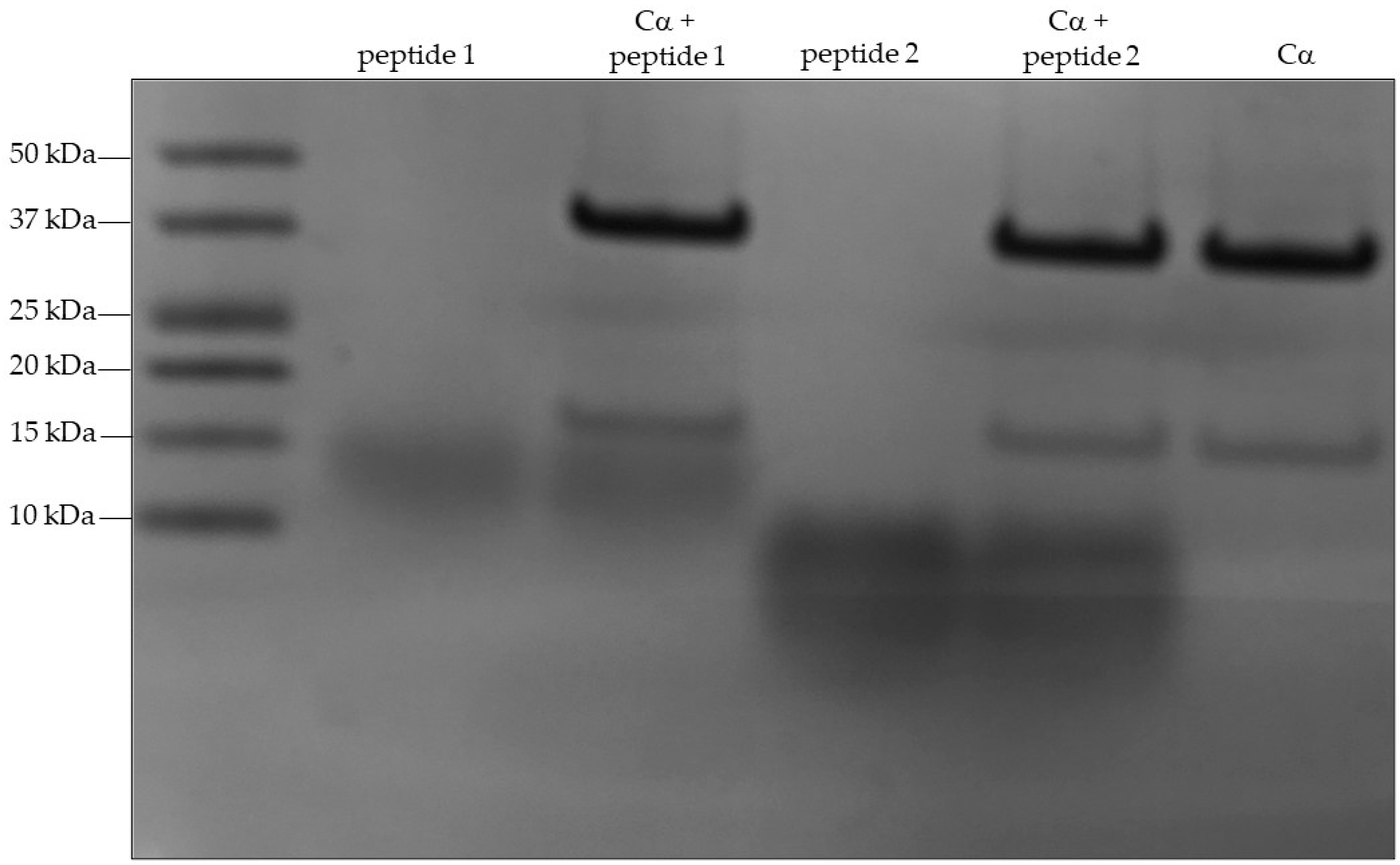
2.2. Mass Spectrometry of In Vitro Phosphorylated Peptides Suggest S203 and S265 as PKA Phosphoacceptor Residues
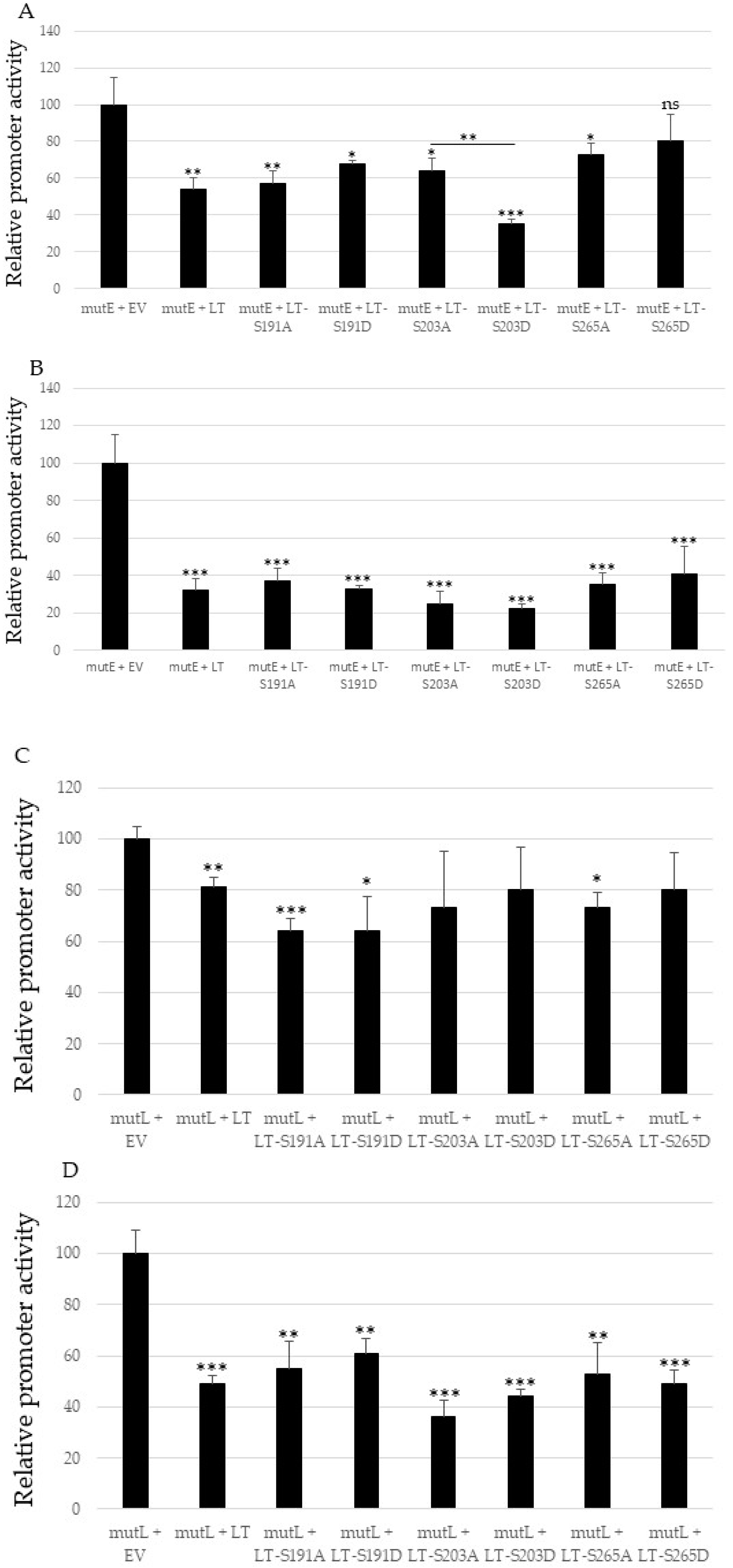
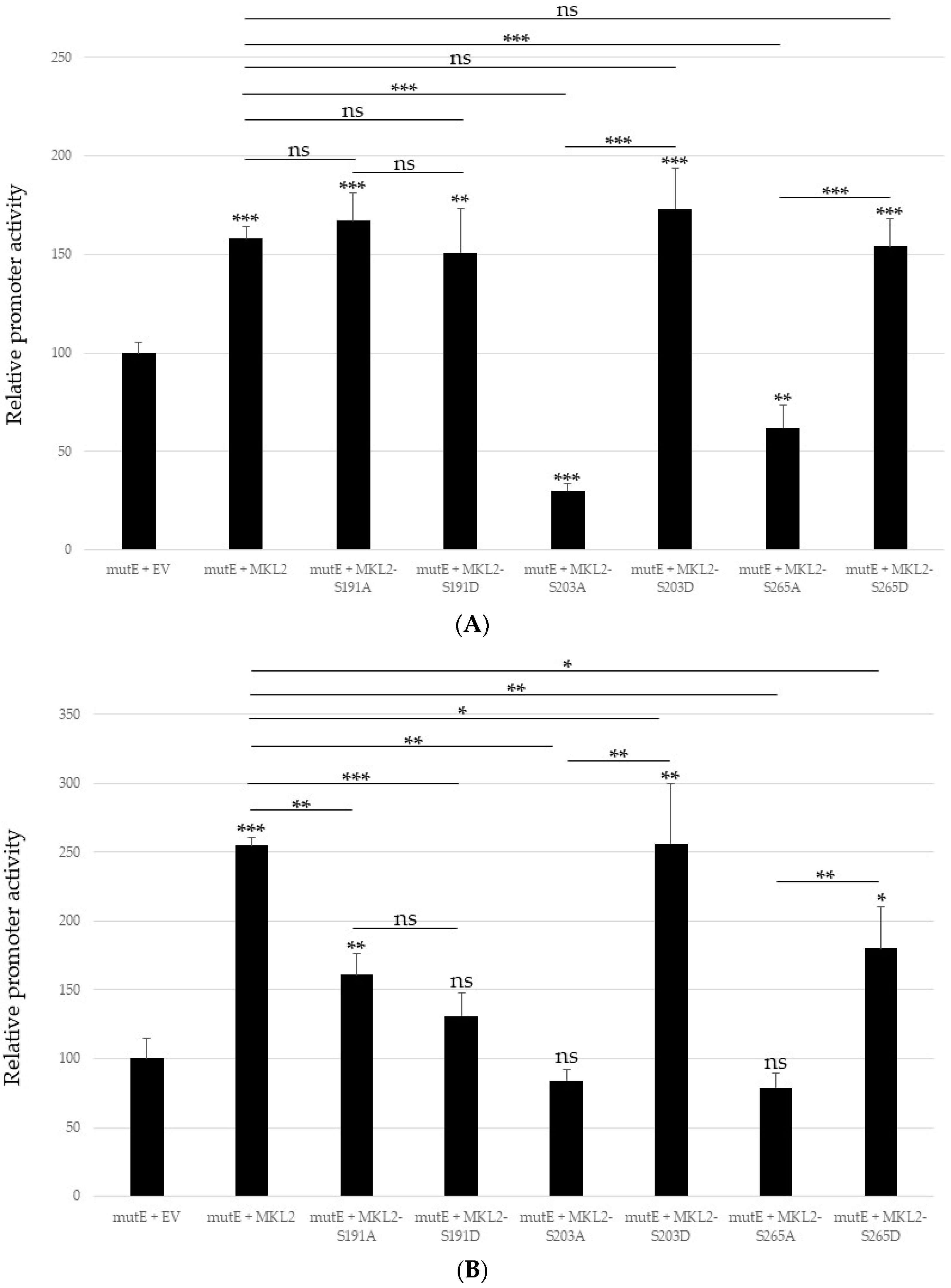
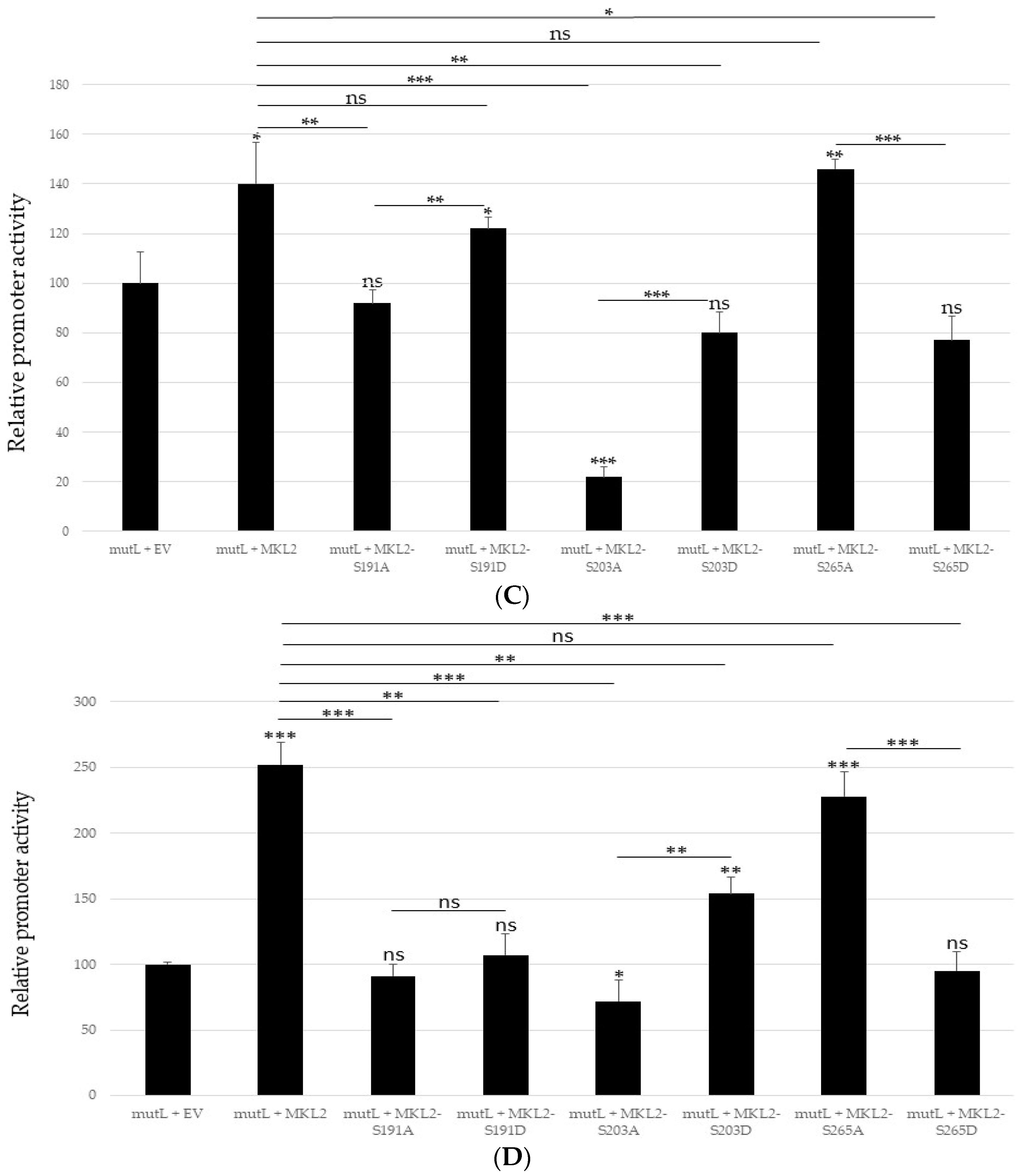
2.3. Mutating S191, S203 and S265 Has No Significant Effect the Inhibitory Effect ofMCPyV Full-Length LTag on the Viral Early and Late Promoter
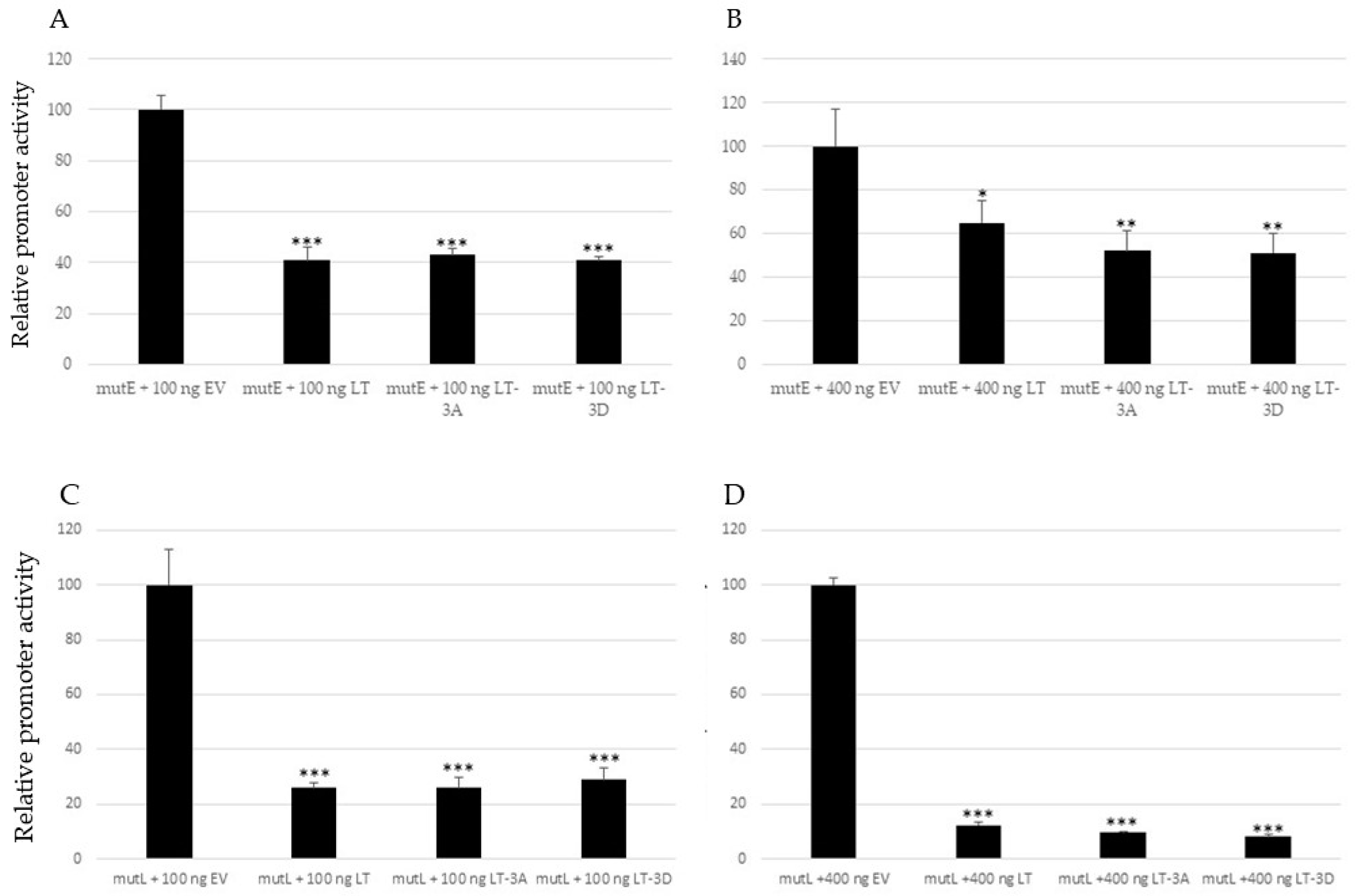
2.4. Non-Phosphorylable and Phosphomimicking Substitutions of the PKA Phosphoacceptor Sites Have Opposite Effect on MKL2 LTag Induced Activation of the MCPyV Promoters
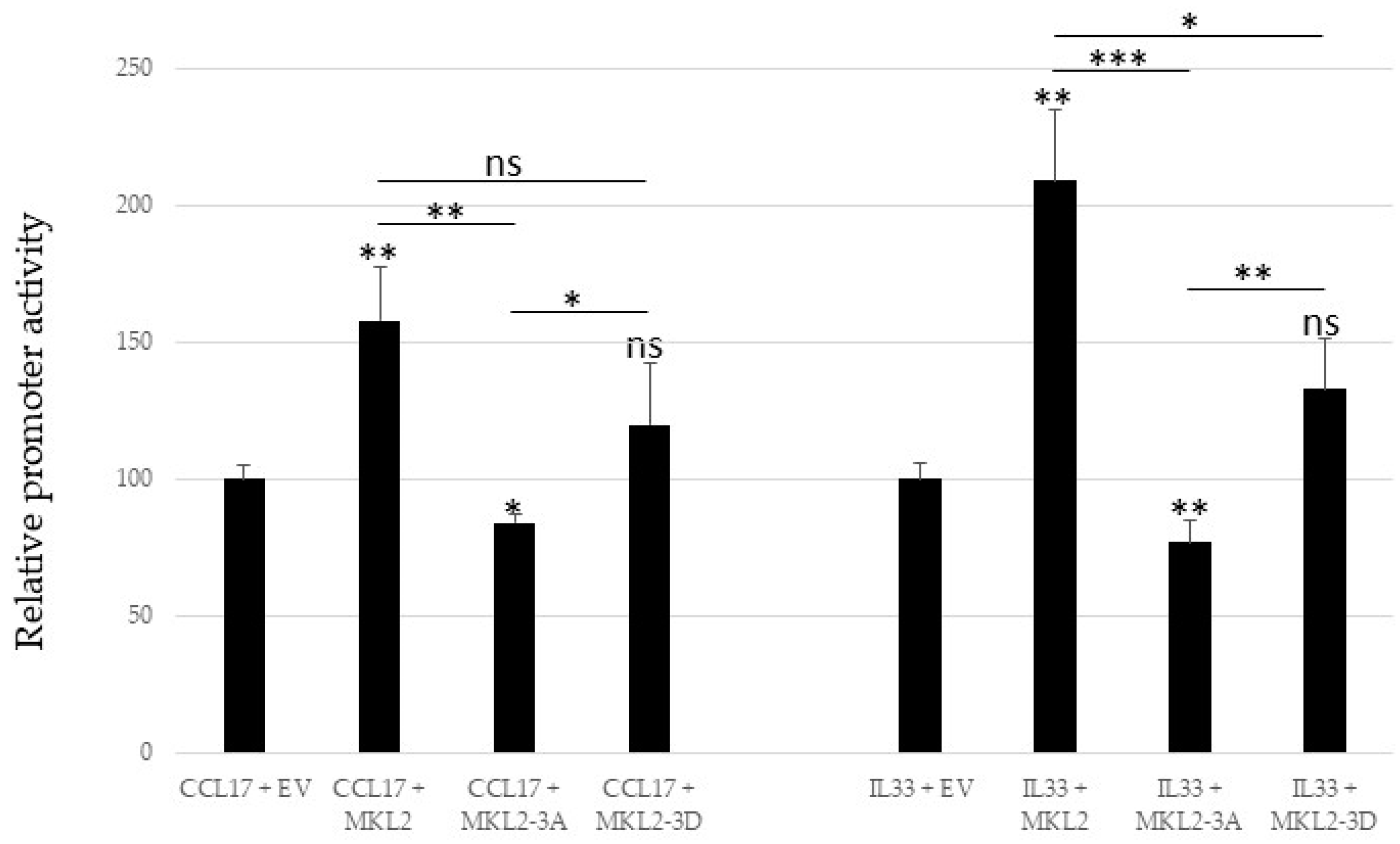
2.5. Mutations in PKA Phosphoacceptor Sites of MKL2 LTag Affect Transactivation of the Cellular Promoters CCL17 and IL33
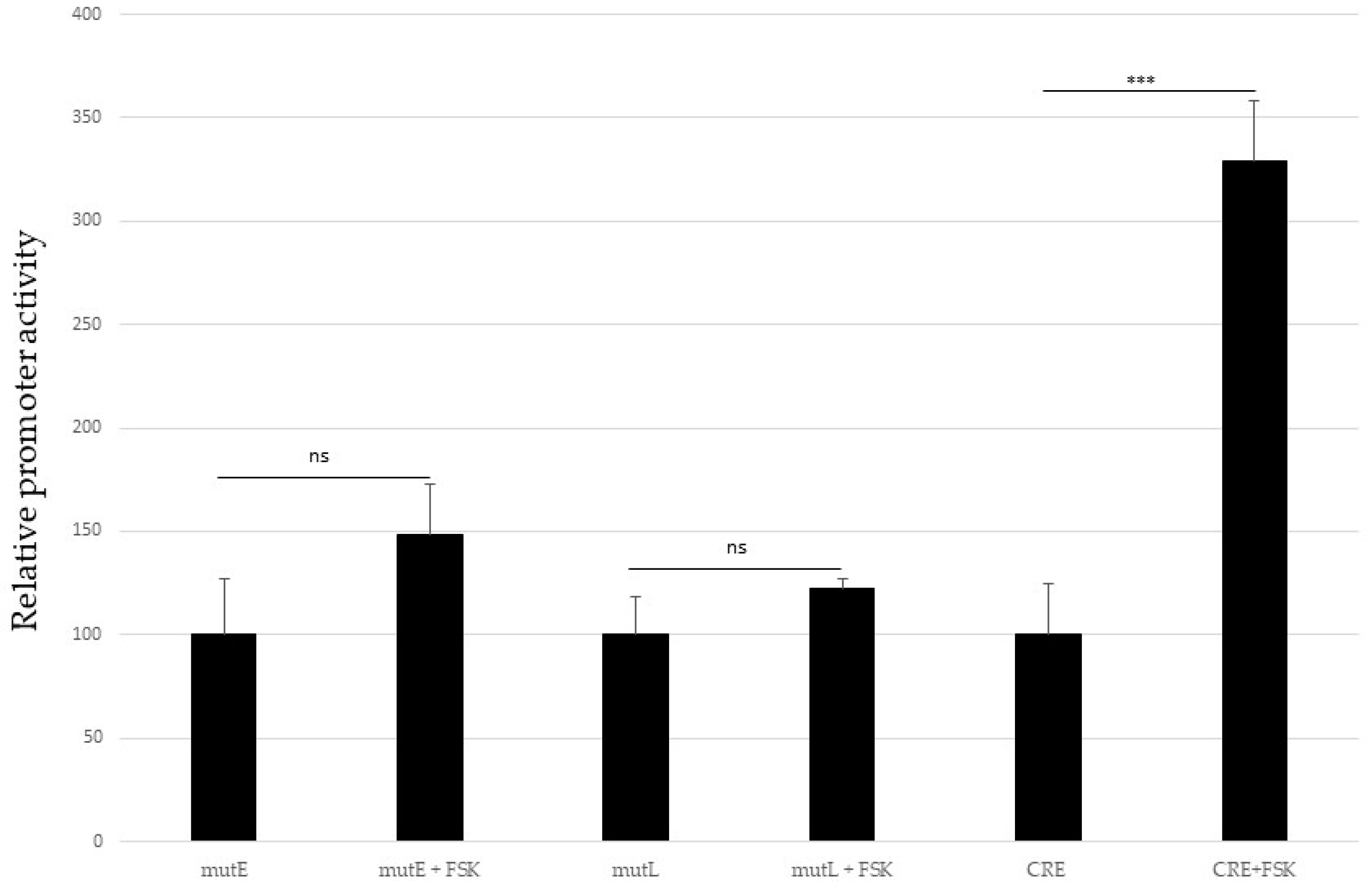
2.6. Activation of the cAMP-Dependent Protein Kinase Pathway Does Not Affect MCPyV Promoter Activity
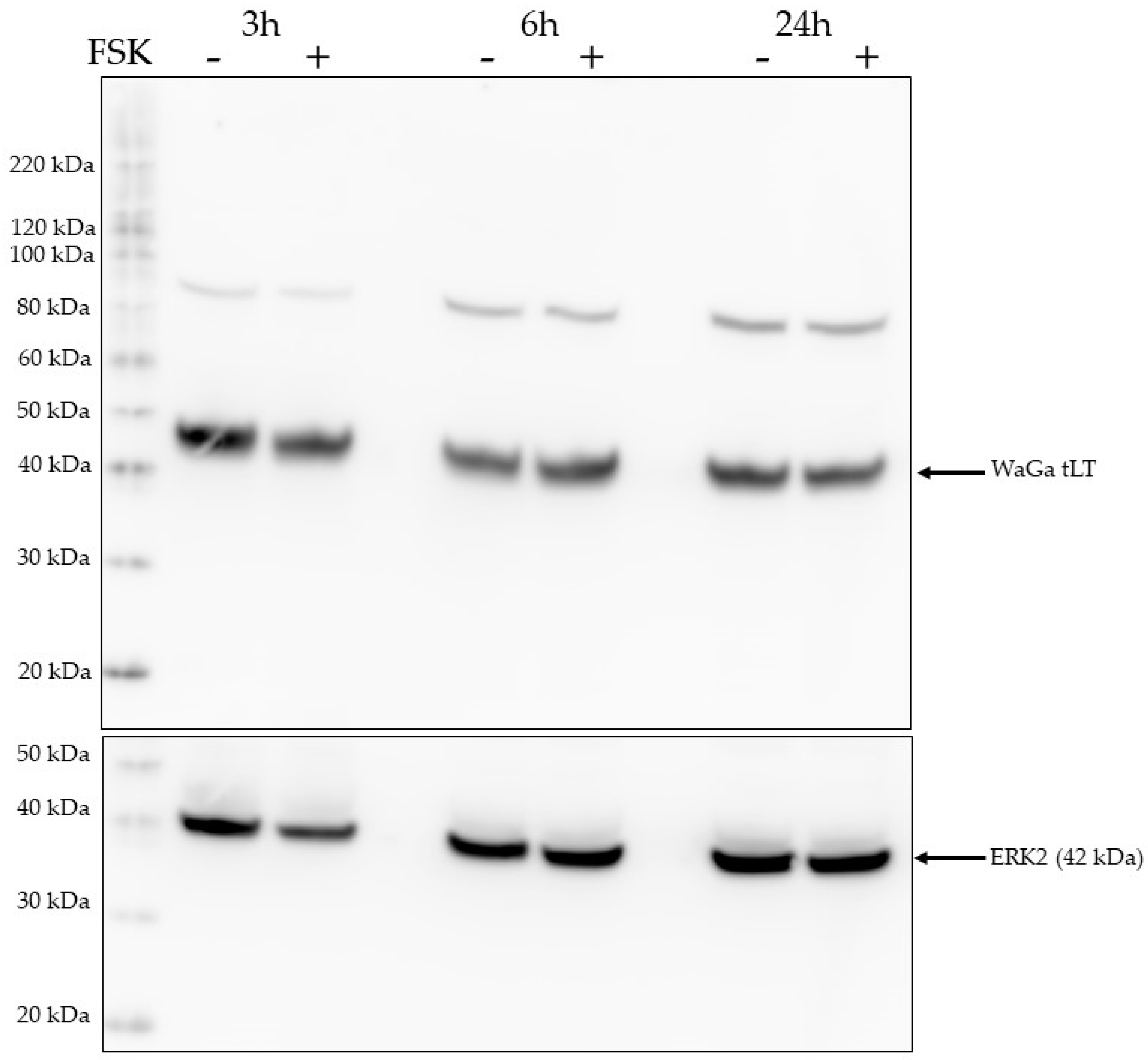
2.7. Activation of the PKA Pathway Does Not Affect LTag Expression Levels
3. Discussion
4. Materials and Methods
4.1. Cells
4.2. Plasmids
4.3. Forskolin Stimulation
4.4. Transfection and Luciferase Assay
4.5. Western Blot
4.6. In Vitro Kinase Assay
4.7. Mass Spectrometry
4.8. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, J.C.; Stang, A.; DeCaprio, J.A.; Cerroni, L.; Lebbé, C.; Veness, M.; Nghiem, P. Merkel cell carcinoma. Nat. Rev. Dis. Prim. 2017, 3, 17077. [Google Scholar] [CrossRef] [PubMed]
- Moens, U.; Krumbholz, A.; Ehlers, B.; Zell, R.; Johne, R.; Calvignac-Spencer, S.; Lauber, C. Biology, evolution, and medical importance of polyomaviruses: An update. Infect. Genet. Evol. 2017, 54, 18–38. [Google Scholar] [CrossRef] [PubMed]
- Schowalter, R.M.; Buck, C.B. The Merkel cell polyomavirus minor capsid protein. PLoS Pathog. 2013, 9, e1003558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeCaprio, J.A.; Garcea, R.L. A cornucopia of human polyomaviruses. Nat. Rev. Microbiol. 2013, 11, 264–276. [Google Scholar] [CrossRef]
- Chang, Y.; Moore, P.S. Merkel cell carcinoma: A virus-induced human cancer. Annu. Rev. Pathol. 2012, 7, 123–144. [Google Scholar] [CrossRef] [Green Version]
- Pietropaolo, V.; Prezioso, C.; Moens, U. Merkel Cell Polyomavirus and Merkel Cell Carcinoma. Cancers 2020, 12, 1774. [Google Scholar] [CrossRef]
- De Caprio, J.A. Molecular Pathogenesis of Merkel Cell Carcinoma. Annu. Rev. Pathol. 2021, 16, 69–91. [Google Scholar] [CrossRef]
- Schlemeyer, T.; Ohnezeit, D.; Virdi, S.; Körner, C.; Weißelberg, S.; Starzonek, S.; Schumacher, U.; Grundhoff, A.; Indenbirken, D.; Albertini, S.; et al. Merkel cell carcinoma and immune evasion: Merkel cell polyomavirus small T-antigen induced surface changes can be reverted by therapeutic intervention. J. Investig. Dermatol. 2022, 142, 3071–3081.e13. [Google Scholar] [CrossRef]
- Kwun, H.J.; Chang, Y.; Moore, P.S. Protein-mediated viral latency is a novel mechanism for Merkel cell polyomavirus persistence. Proc. Natl. Acad. Sci. USA 2017, 114, E4040–E4047. [Google Scholar] [CrossRef]
- Nwogu, N.; Ortiz, L.E.; Kwun, H.J. Merkel Cell Polyomavirus Large T Antigen Unique Domain Regulates Its Own Protein Stability and Cell Growth. Viruses 2020, 12, 1043. [Google Scholar] [CrossRef] [PubMed]
- Schrama, D.; Hesbacher, S.; Angermeyer, S.; Schlosser, A.; Haferkamp, S.; Aue, A.; Adam, C.; Weber, A.; Schmidt, M.; Houben, R. Serine 220 phosphorylation of the Merkel cell polyomavirus large T antigen crucially supports growth of Merkel cell carcinoma cells. Int. J. Cancer 2016, 138, 1153–1162. [Google Scholar] [CrossRef] [PubMed]
- Diaz, J.; Wang, X.; Tsang, S.H.; Jiao, J.; You, J. Phosphorylation of large T antigen regulates merkel cell polyomavirus replication. Cancers 2014, 6, 1464–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Diaz, J.; Wang, X.; Tsang, S.H.; You, J. Phosphorylation of Merkel cell polyomavirus large tumor antigen at serine 816 by ATM kinase induces apoptosis in host cells. J. Biol. Chem. 2015, 290, 1874–1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez Orellana, J.; Kwun, H.J.; Artusi, S.; Chang, Y.; Moore, P.S. Sirolimus and Other Mechanistic Target of Rapamycin Inhibitors Directly Activate Latent Pathogenic Human Polyomavirus Replication. J. Infect. Dis. 2021, 224, 1160–1169. [Google Scholar] [CrossRef] [Green Version]
- Cohen, P. The regulation of protein function by multisite phosphorylation—A 25 year update. Trends Biochem. Sci. 2000, 25, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Pawson, T.; Scott, J.D. Protein phosphorylation in signaling—50 years and counting. Trends Biochem. Sci. 2005, 30, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Ardito, F.; Giuliani, M.; Perrone, D.; Troiano, G.; Lo Muzio, L. The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (Review). Int. J. Mol. Med. 2017, 40, 271–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases [published correction appears in Microbiol. Mol. Biol. Rev. 2012, 76, 496]. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [Green Version]
- Trenker, R.; Jura, N. Receptor tyrosine kinase activation: From the ligand perspective. Curr. Opin. Cell Biol. 2020, 63, 174–185. [Google Scholar] [CrossRef]
- Pinna, L.A.; Ruzzene, M. How do protein kinases recognize their substrates? Biochim. Biophys. Acta 1996, 1314, 191–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayr, B.; Montminy, M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell Biol. 2001, 2, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, M.; Delghandi, M.P.; Moens, U. What turns CREB on? Cell Signal 2004, 16, 1211–1227. [Google Scholar] [CrossRef] [PubMed]
- Moens, U.; Sundsfjord, A.; Flaegstad, T.; Traavik, T. BK virus early RNA transcripts in stably transformed cells: Enhanced levels induced by dibutyryl cyclic AMP, forskolin and 12-O-tetradecanoylphorbol-13-acetate treatment. J. Gen. Virol. 1990, 71, 1461–1471. [Google Scholar] [CrossRef]
- Love, T.M.; de Jesus, R.; Kean, J.A.; Sheng, Q.; Leger, A.; Schaffhausen, B. Activation of CREB/ATF sites by polyomavirus large T antigen. J. Virol. 2005, 79, 4180–4190. [Google Scholar] [CrossRef] [Green Version]
- Olsen, J.V.; Blagoev, B.; Gnad, F.; Macek, B.; Kumar, C.; Mortensen, P.; Mann, M. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 2006, 127, 635–648. [Google Scholar] [CrossRef] [Green Version]
- Smith, F.D.; Samelson, B.K.; Scott, J.D. Discovery of cellular substrates for protein kinase A using a peptide array screening protocol. Biochem. J. 2011, 438, 103–110. [Google Scholar] [CrossRef] [Green Version]
- Imamura, H.; Wagih, O.; Niinae, T.; Sugiyama, N.; Beltrao, P.; Ishihama, Y. Identifications of Putative PKA Substrates with Quantitative Phosphoproteomics and Primary-Sequence-Based Scoring. J. Proteome Res. 2017, 16, 1825–1830. [Google Scholar] [CrossRef]
- Isobe, K.; Jung, H.J.; Yang, C.R.; Claxton, J.; Sandoval, P.; Burg, M.B.; Raghuram, V.; Knepper, M.A. Systems-level identification of PKA-dependent signaling in epithelial cells. Proc. Natl. Acad. Sci. USA 2017, 114, E8875–E8884. [Google Scholar] [CrossRef] [Green Version]
- Karamafrooz, A.; Brennan, J.; Thomas, D.D.; Parker, L.L. Integrated Phosphoproteomics for Identifying Substrates of Human Protein Kinase A (PRKACA) and Its Oncogenic Mutant DNAJB1-PRKACA. J. Proteome Res. 2021, 20, 4815–4830. [Google Scholar] [CrossRef]
- Blom, N.; Gammeltoft, S.; Brunak, S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J. Mol. Biol. 1999, 294, 1351–1362. [Google Scholar] [CrossRef] [PubMed]
- Hashida, Y.; Imajoh, M.; Kamioka, M.; Taniguchi, A.; Kuroda, N.; Hayashi, K.; Nakajima, H.; Sano, S.; Daibata, M. Phylogenetic analysis of Merkel cell polyomavirus based on full-length LT and VP1 gene sequences derived from neoplastic tumours in Japanese patients. J. Gen. Virol. 2014, 95, 135–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsushita, M.; Iwasaki, T.; Kuwamoto, S.; Kato, M.; Nagata, K.; Murakami, I.; Kitamura, Y.; Hayashi, K. Merkel cell polyomavirus (MCPyV) strains in Japanese merkel cell carcinomas (MCC) are distinct from Caucasian type MCPyVs: Genetic variability and phylogeny of MCPyV genomes obtained from Japanese MCPyV-infected MCCs. Virus Genes 2014, 48, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Jin, Y.; Chen, B.; Gugger, M.K.; Wilkinson-Johnson, C.L.; Tiambeng, T.N.; Jin, S.; Ge, Y. Comprehensive Characterization of the Recombinant Catalytic Subunit of cAMP-Dependent Protein Kinase by Top-Down Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2019, 30, 2561–2570. [Google Scholar] [CrossRef] [PubMed]
- Abdulsalam, I.; Rasheed, K.; Sveinbjørnsson, B.; Ehlers, B.; Moens, U. Promoter activity of Merkel cell Polyomavirus variants in human dermal fibroblasts and a Merkel cell carcinoma cell line. Virol. J. 2020, 17, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boukamp, P.; Petrussevska, R.T.; Breitkreutz, D.; Hornung, J.; Markham, A.; Fusenig, N.E. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 1988, 106, 761–771. [Google Scholar] [CrossRef] [Green Version]
- Sunshine, J.C.; Jahchan, N.S.; Sage, J.; Choi, J. Are there multiple cells of origin of Merkel cell carcinoma? Oncogene 2018, 37, 1409–1416. [Google Scholar] [CrossRef]
- Kervarrec, T.; Samimi, M.; Guyétant, S.; Sarma, B.; Chéret, J.; Blanchard, E.; Berthon, P.; Schrama, D.; Houben, R.; Touzé, A. Histogenesis of Merkel Cell Carcinoma: A Comprehensive Review. Front. Oncol. 2019, 9, 451. [Google Scholar] [CrossRef] [Green Version]
- Farmerie, W.G.; Folk, W.R. Regulation of polyomavirus transcription by large tumor antigen. Proc. Natl. Acad. Sci. USA 1984, 81, 6919–6923. [Google Scholar] [CrossRef] [Green Version]
- Rasheed, K.; Sveinbjørnsson, B.; Moens, U. Reciprocal transactivation of Merkel cell polyomavirus and high-risk human papillomavirus promoter activities and increased expression of their oncoproteins. Virol. J. 2021, 18, 139. [Google Scholar] [CrossRef]
- Rasheed, K.; Moens, U.; Policastro, B.; Johnsen, J.I.; Koljonen, V.; Sihto, H.; Lui, W.O.; Sveinbjørnsson, B. The Merkel Cell Polyomavirus T-Antigens and IL-33/ST2-IL1RAcP Axis: Possible Role in Merkel Cell Carcinoma. Int. J. Mol. Sci. 2022, 23, 3702. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, K.; Abdulsalam, I.; Fismen, S.; Grimstad, Ø.; Sveinbjørnsson, B.; Moens, U. CCL17/TARC and CCR4 expression in Merkel cell carcinoma. Oncotarget 2018, 9, 31432–31447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seamon, K.B.; Padgett, W.; Daly, J.W. Forskolin: Unique diterpene activator of adenylate cyclase in membranes and in intact cells. Proc. Natl. Acad. Sci. USA 1981, 78, 3363–3367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanassi, P.; Paolillo, M.; Feliciello, A.; Avvedimento, E.V.; Gallo, V.; Schinelli, S. cAMP-dependent protein kinase induces cAMP-response element-binding protein phosphorylation via an intracellular calcium release/ERK-dependent pathway in striatal neurons. J. Biol. Chem. 2001, 276, 11487–11495. [Google Scholar] [CrossRef] [Green Version]
- Dittmer, A.; Dittmer, J. Beta-actin is not a reliable loading control in Western blot analysis. Electrophoresis 2006, 27, 2844–2845. [Google Scholar] [CrossRef] [PubMed]
- Vuchak, L.A.; Tsygankova, O.M.; Prendergast, G.V.; Meinkoth, J.L. Protein kinase A and B-Raf mediate extracellular signal-regulated kinase activation by thyrotropin. Mol. Pharmacol. 2009, 76, 1123–1129. [Google Scholar] [CrossRef] [Green Version]
- Mohr, I.J.; Stillman, B.; Gluzman, Y. Regulation of SV40 DNA replication by phosphorylation of T antigen. EMBO J. 1987, 6, 153–160. [Google Scholar] [CrossRef]
- Bockus, B.J.; Schaffhausen, B. Phosphorylation of polyomavirus large T antigen: Effects of viral mutations and cell growth state. J. Virol. 1987, 61, 1147–1154. [Google Scholar] [CrossRef] [Green Version]
- Swenson, J.J.; Frisque, R.J. Biochemical characterization and localization of JC virus large T antigen phosphorylation domains. Virology 1995, 212, 295–308. [Google Scholar] [CrossRef] [Green Version]
- Swenson, J.J.; Trowbridge, P.W.; Frisque, R.J. Replication activity of JC virus large T antigen phosphorylation and zinc finger domain mutants. J. Neurovirol. 1996, 2, 78–86. [Google Scholar] [CrossRef]
- Larsen, K.M.; Minaya, M.K.; Vaish, V.; Peña, M.M.O. The Role of IL-33/ST2 Pathway in Tumorigenesis. Int. J. Mol. Sci. 2018, 19, 2676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, J.; Kim, S.; Lin, P.C. Interleukin-33 and ST2 Signaling in Tumor Microenvironment. J. Interferon Cytokine Res. 2019, 39, 61–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korbecki, J.; Kojder, K.; Simińska, D.; Bohatyrewicz, R.; Gutowska, I.; Chlubek, D.; Baranowska-Bosiacka, I. CC Chemokines in a Tumor: A Review of Pro-Cancer and Anti-Cancer Properties of the Ligands of Receptors CCR1, CCR2, CCR3, and CCR4. Int. J. Mol. Sci. 2020, 21, 8412. [Google Scholar] [CrossRef] [PubMed]
- Schneider-Yin, X.; Gouya, L.; Dorsey, M.; Rüfenacht, U.; Deybach, J.C.; Ferreira, G.C. Mutations in the iron-sulfur cluster ligands of the human ferrochelatase lead to erythropoietic protoporphyria. Blood 2000, 96, 1545–1549. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.Q.; Grønborg, M.; Huang, H.; Kim, J.W.; Otto, T.C.; Pandey, A.; Lane, M.D. Sequential phosphorylation of CCAAT enhancer-binding protein beta by MAPK and glycogen synthase kinase 3beta is required for adipogenesis. Proc. Natl. Acad. Sci. USA 2005, 102, 9766–9771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, G.; Chen, S.; Li, S.; Cha, J.; Long, C.; Li, L.; He, Q.; Liu, Y. Protein kinase A and casein kinases mediate sequential phosphorylation events in the circadian negative feedback loop. Genes Dev. 2007, 21, 3283–3295. [Google Scholar] [CrossRef] [Green Version]
- Woo, Y.; Kim, S.J.; Suh, B.K.; Kwak, Y.; Jung, H.J.; Nhung, T.T.M.; Mun, D.J.; Hong, J.H.; Noh, S.J.; Kim, S.; et al. Sequential phosphorylation of NDEL1 by the DYRK2-GSK3β complex is critical for neuronal morphogenesis. eLife 2019, 8, e50850. [Google Scholar] [CrossRef]
- Xiao, C.Y.; Hübner, S.; Elliot, R.M.; Caon, A.; Jans, D.A. A consensus cAMP-dependent protein kinase (PK-A) site in place of the CcN motif casein kinase II site simian virus 40 large T-antigen confers PK-A-mediated regulation of nuclear import. J. Biol. Chem. 1996, 271, 6451–6457. [Google Scholar] [CrossRef] [Green Version]
- Timney, B.L.; Raveh, B.; Mironska, R.; Trivedi, J.M.; Kim, S.J.; Russel, D.; Wente, S.R.; Sali, A.; Rout, M.P. Simple rules for passive diffusion through the nuclear pore complex. J. Cell Biol. 2016, 215, 57–76. [Google Scholar] [CrossRef] [Green Version]
- Houben, R.; Shuda, M.; Weinkam, R.; Schrama, D.; Feng, H.; Chang, Y.; Moore, P.S.; Becker, J.C. Merkel cell polyomavirus-infected Merkel cell carcinoma cells require expression of viral T antigens. J. Virol. 2010, 84, 7064–7072. [Google Scholar] [CrossRef]
- Guastafierro, A.; Feng, H.; Thant, M.; Kirkwood, J.M.; Chang, Y.; Moore, P.S.; Shuda, M. Characterization of an early passage Merkel cell polyomavirus-positive Merkel cell carcinoma cell line, MS-1, and its growth in NOD scid gamma mice. J. Virol. Methods 2013, 187, 6–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moens, U.; Van Ghelue, M.; Ludvigsen, M.; Korup-Schulz, S.; Ehlers, B. Early and late promoters of BK polyomavirus, Merkel cell polyomavirus, Trichodysplasia spinulosa-associated polyomavirus and human polyomavirus 12 are among the strongest of all known human polyomaviruses in 10 different cell lines. J. Gen. Virol. 2015, 96, 2293–2303. [Google Scholar] [CrossRef] [PubMed]
- Van Lint, J.; Ni, Y.; Valius, M.; Merlevede, W.; Vandenheede, J.R. Platelet-derived growth factor stimulates protein kinase D through the activation of phospholipase Cgamma and protein kinase C. J. Biol. Chem. 1998, 273, 7038–7043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Hein, J.; Richardson, S.C.; Basse, P.H.; Toptan, T.; Moore, P.S.; Gjoerup, O.V.; Chang, Y. Merkel cell polyomavirus large T antigen disrupts lysosome clustering by translocating human Vam6p from the cytoplasm to the nucleus. J. Biol. Chem. 2011, 286, 17079–17090. [Google Scholar] [CrossRef] [PubMed]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Falquet, M.; Prezioso, C.; Ludvigsen, M.; Bruun, J.-A.; Passerini, S.; Sveinbjørnsson, B.; Pietropaolo, V.; Moens, U. Regulation of Transcriptional Activity of Merkel Cell Polyomavirus Large T-Antigen by PKA-Mediated Phosphorylation. Int. J. Mol. Sci. 2023, 24, 895. https://doi.org/10.3390/ijms24010895
Falquet M, Prezioso C, Ludvigsen M, Bruun J-A, Passerini S, Sveinbjørnsson B, Pietropaolo V, Moens U. Regulation of Transcriptional Activity of Merkel Cell Polyomavirus Large T-Antigen by PKA-Mediated Phosphorylation. International Journal of Molecular Sciences. 2023; 24(1):895. https://doi.org/10.3390/ijms24010895
Chicago/Turabian StyleFalquet, Mar, Carla Prezioso, Maria Ludvigsen, Jack-Ansgar Bruun, Sara Passerini, Baldur Sveinbjørnsson, Valeria Pietropaolo, and Ugo Moens. 2023. "Regulation of Transcriptional Activity of Merkel Cell Polyomavirus Large T-Antigen by PKA-Mediated Phosphorylation" International Journal of Molecular Sciences 24, no. 1: 895. https://doi.org/10.3390/ijms24010895
APA StyleFalquet, M., Prezioso, C., Ludvigsen, M., Bruun, J. -A., Passerini, S., Sveinbjørnsson, B., Pietropaolo, V., & Moens, U. (2023). Regulation of Transcriptional Activity of Merkel Cell Polyomavirus Large T-Antigen by PKA-Mediated Phosphorylation. International Journal of Molecular Sciences, 24(1), 895. https://doi.org/10.3390/ijms24010895