

Meta-Inflammation and New Anti-Diabetic Drugs: A New Chance to Knock Down Residual Cardiovascular Risk

 , ,
, ,
Abstract
:1. Introduction
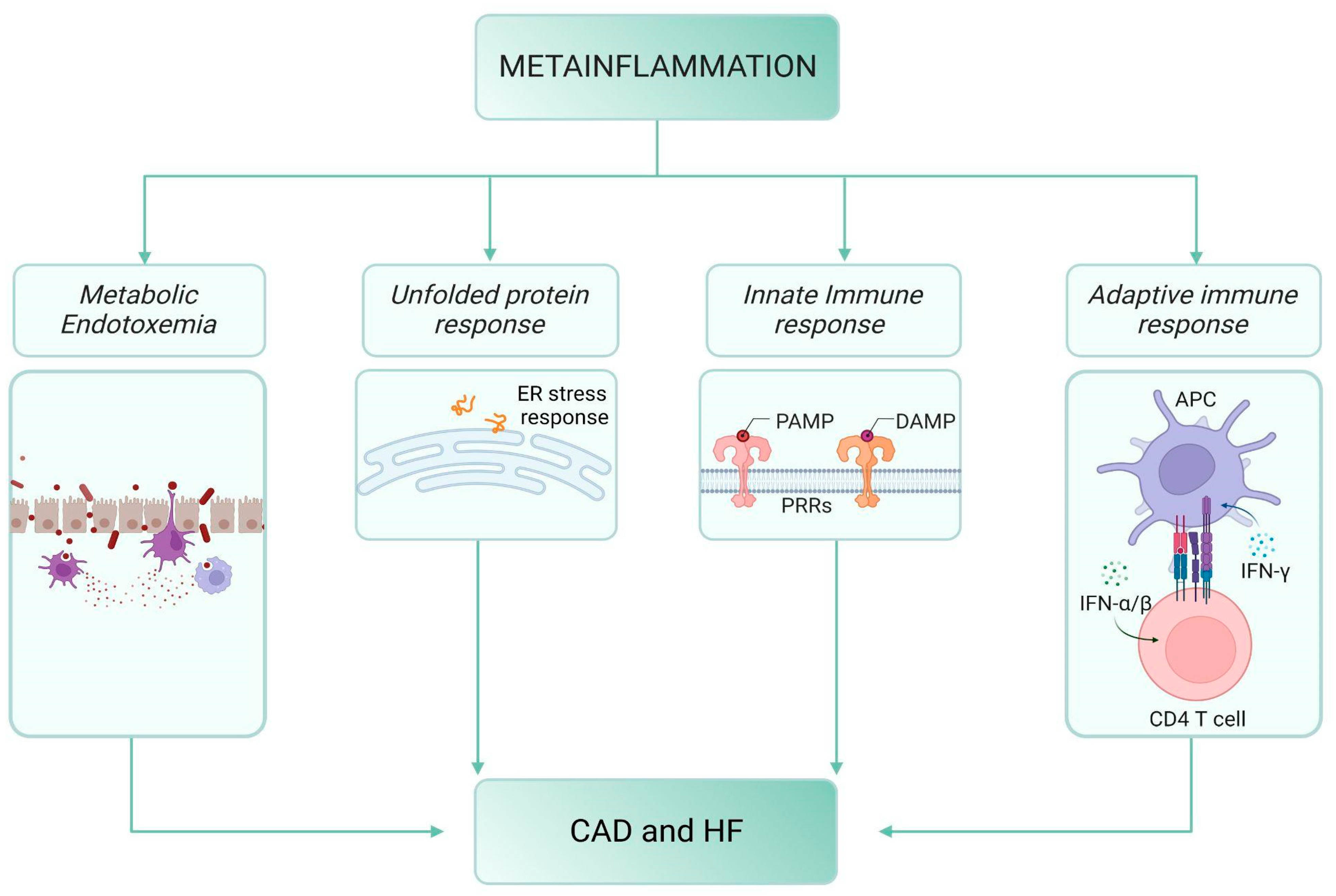
2. Meta-Inflammation, Diabetes and Cardiovascular Disease
2.1. Meta-Inflammation and Metabolic Endotoxemia
2.2. Meta-Inflammation and the “Unfolded Protein Response” (UPR)
2.3. The Innate Immune Response
2.4. The Adaptive Immune Response
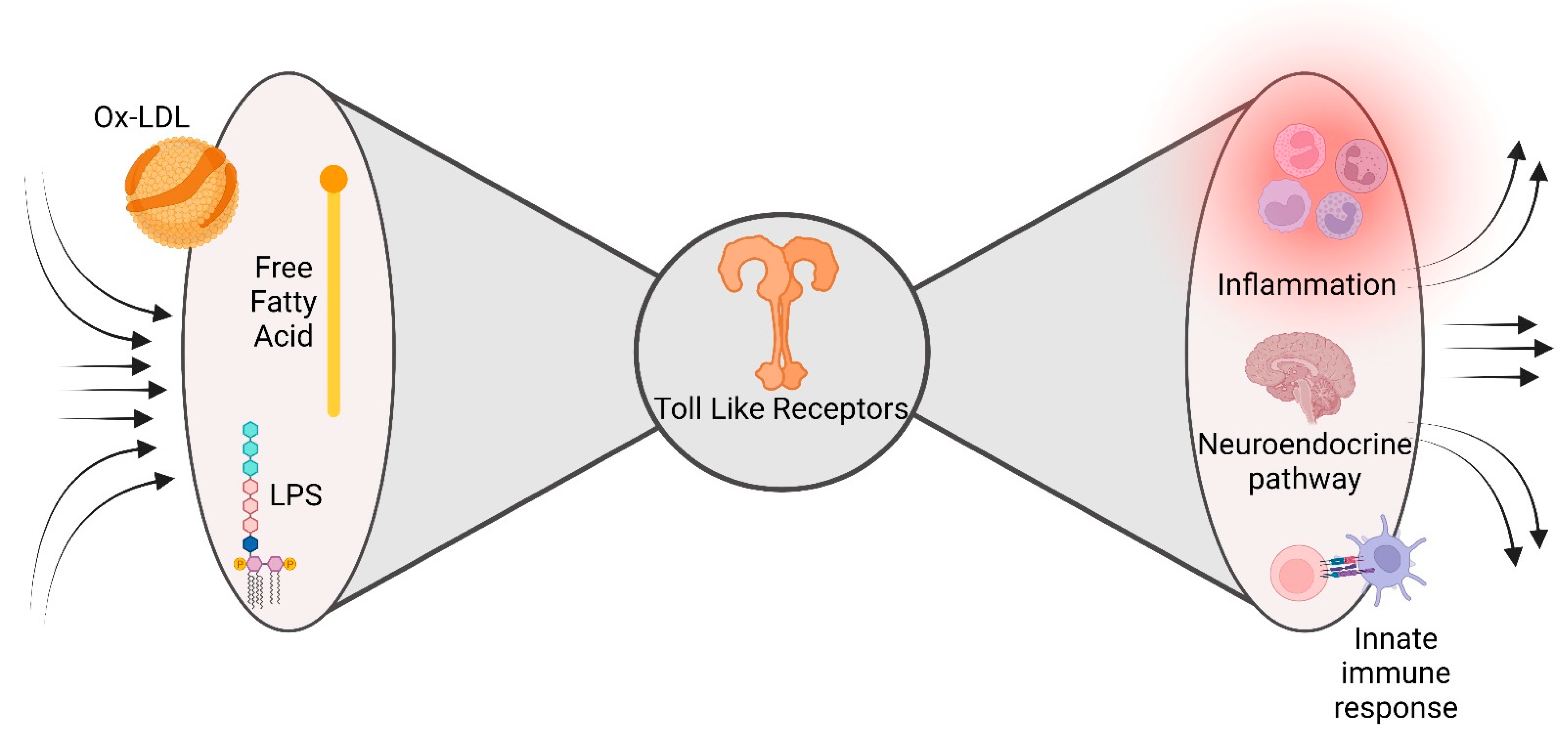
2.5. The Bow Tie Model
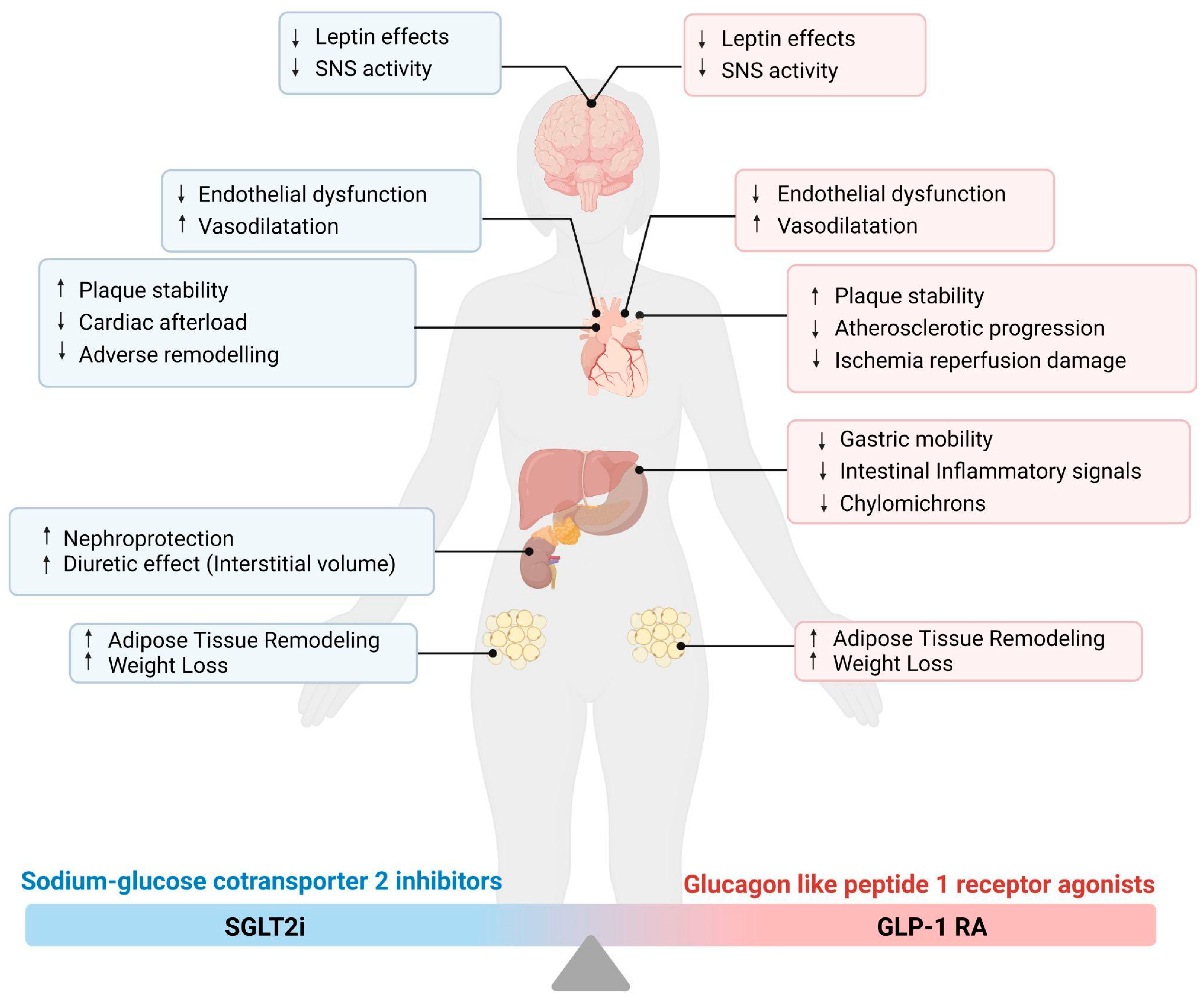
3. Newer Glucose-Lowering Medications and Cardiovascular Effects
3.1. Sodium–Glucose Cotransporter 2 Inhibitors (SGLT2i)
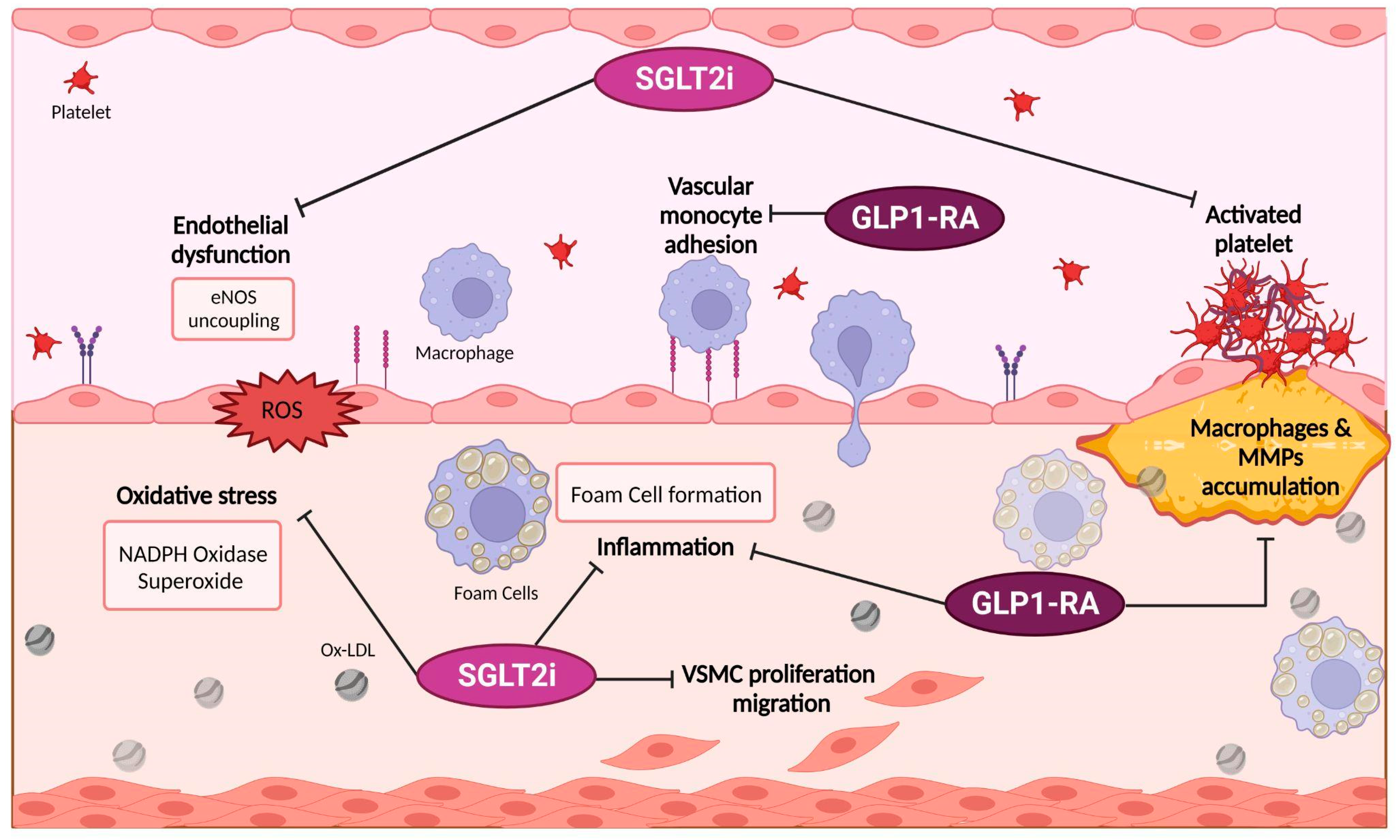
3.1.1. Possible Targets of SGLT2i Pharmacodynamics
3.1.2. SGLT2i and Meta-inflammation
3.2. Glucagon-like Peptide 1 Receptor Agonists (GLP-1 RA)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GLP-1RA Name | Clinical Trial Name (CVOT) | Brief Reference | Number of Patients | Definition of CV Outcomes | Median Follow Up (Months) | Principal Findings |
|---|---|---|---|---|---|---|
| Lixisenatide | ELIXA | Holman RR et al., New England Journal of Medicine, 2017 [78] | 6068 | CV death, myocardial infarction, stroke, or hospitalization for unstable angina | 25 | The primary endpoint (a composite of the first occurrence of any of the following: death from CV causes, non-fatal myocardial infarction, non-fatal stroke, or hospitalization for unstable angina) event occurred in 13.4% of the lixisenatide group and in 13.2% of the placebo group (HR, 1.02; 95% CI, 0.89 to 1.17), which showed the non-inferiority of lixisenatide to placebo (p < 0.001) but did not show superiority (p = 0.81). |
| Liraglutide | LEADER | Marso SP et al., New England Journal of Medicine, 2016 [79] | 9340 | The first occurrence of death from CV causes non-fatal myocardial infarction or non-fatal stroke | 15.8 | The primary outcome (the first occurrence of death from CV causes, non-fatal myocardial infarction, or non-fatal stroke) occurred in significantly fewer patients in the liraglutide group (13.0%) than in the placebo group (14.9%) (HR, 0.87; 95% CI, 0.78 to 0.97; p < 0.001 for non-inferiority; p = 0.01 for superiority). Fewer patients died from cardiovascular causes in the liraglutide group (4.7%) than in the placebo group (6.0%) (HR, 0.78; 95% CI, 0.66 to 0.93; p = 0.007). |
| Mann JFE et al., Circulation, 2018 [87] | 9340 | Composite of CV death, non-fatal myocardial infarction, or non-fatal stroke | 36 | In patients with eGFR < 60 mL/min/1.73 m2, risk reduction for the primary composite CV outcome with liraglutide was greater (HR, 0.69; 95% CI, 0.57–0.85) versus those with eGFR ≥ 60 mL/min/1.73 m2 (HR, 0.94; 95% CI, 0.83–1.07; interaction p = 0.01). | ||
| Semaglutide | SUSTAIN-6 | Marso SP et al., New England Journal of Medicine, 2016 [80] | 3297 | The first occurrence of CV death, non-fatal myocardial infarction, or non-fatal stroke | 24 | The primary outcome (composite of first occurrence of death from CV causes, non-fatal myocardial infarction, or non-fatal stroke. Occurred in 6.6% of the semaglutide group and in 8.9% of the placebo group (HR, 0.74; 95% CI, 0.58 to 0.95; p < 0.001 for non-inferiority). Non-fatal myocardial infarction occurred in 2.9% of the patients receiving semaglutide and in 3.9% of those receiving placebo (HR, 0.74; 95% CI, 0.51 to 1.08; p = 0.12); non-fatal stroke occurred in 1.6% and 2.7%, respectively (HR, 0.61; 95% CI, 0.38 to 0.99; p = 0.04). |
| PIONEER-6 | Husain M et al., New England Journal of Medicine, 2019 [84] | 3183 | The first occurrence of CV death, non-fatal myocardial infarction, or non-fatal stroke | 15.9 | Major adverse CV events occurred in 3.8% of the oral semaglutide group and in 4.8% of the placebo group (hazard ratio, 0.79; 95% CI, 0.57 to 1.11; p < 0.001 for non-inferiority). | |
| Exenatide | EXSCEL | Holman RR et al., New England Journal of Medicine, 2016 [78] | 14,752 | The first occurrence of death from CV causes non-fatal myocardial infarction or non-fatal stroke | 38.4 | A primary outcome (composite of the first occurrence of any component of the composite outcome of death from CV causes, non-fatal myocardial infarction, or non-fatal stroke) event occurred in 11.4% in the exenatide group and in 12.2% in the placebo group (HR, 0.91; 95% CI, 0.83 to 1.00) |
| Dulaglutide | REWIND | Gerstein HC et al., Lancet, 2019 [83] | 9901 | The first occurrence of the composite endpoint of non-fatal myocardial infarction, non-fatal stroke, or CV death | 64.8 | The primary outcome (composite of first occurrence of non-fatal myocardial infarction, non-fatal stroke, and death from CV causes or unknown causes) occurred in 12.0% in the dulaglutide group and in 13.4% in the placebo group (HR 0.88, 95% CI 0.79–0.99; p = 0.026). |
Possible Target of GLP-1 RA Pharmacodynamics and Their Link with Meta-Inflammation
4. From the Bed to the Bench Side: The New Glucose-Lowering Drugs and Related Cardiovascular Benefits
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sun, H.; Saeedi, P.; Karuranga, S.; Pinkepank, M.; Ogurtsova, K.; Duncan, B.B.; Stein, C.; Basit, A.; Chan, J.C.; Mbanya, J.C.; et al. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res. Clin. Pract. 2022, 183, 109119. [Google Scholar] [CrossRef] [PubMed]
- Zelniker, T.A.; Wiviott, S.D.; Raz, I.; Im, K.; Goodrich, E.; Bonaca, M.P.; Mosenzon, O.; Kato, E.; Cahn, A.; Furtado, R.H.M.; et al. SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: A systematic review and meta-analysis of cardiovascular outcome trials. Lancet 2019, 393, 31–39, Erratum in: Lancet 2019, 393, 30. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, S.L.; Rørth, R.; Jhund, P.S.; Docherty, K.F.; Sattar, N.; Preiss, D.; Køber, L.; Petrie, M.C.; McMurray, J.J.V. Cardiovascular, mortality, and kidney outcomes with GLP-1 receptor agonists in patients with type 2 diabetes: A systematic review and meta-analysis of cardiovascular outcome trials. Lancet Diabetes Endocrinol. 2019, 7, 776–785, Erratum in: Lancet Diabetes Endocrinol. 2020, 8, e2. [Google Scholar] [CrossRef]
- Wilcox, T.; De Block, C.; Schwartzbard, A.Z.; Newman, J.D. Diabetic Agents, From Metformin to SGLT2 Inhibitors and GLP1 Receptor Agonists: JACC Focus Seminar. J. Am. Coll. Cardiol. 2020, 75, 1956–1974, Erratum in: J. Am. Coll. Cardiol. 2020, 76, 1719–1722. [Google Scholar] [CrossRef]
- Schiattarella, G.G.; Rodolico, D.; A Hill, J. Metabolic inflammation in heart failure with preserved ejection fraction. Cardiovasc. Res. 2021, 117, 423–434. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: Developed by the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) With the special contribution of the Heart Failure Association (HFA) of the ESC. Rev. Esp. Cardiol. 2022, 75, 523. [Google Scholar] [CrossRef]
- Aronson, D.; Edelman, E.R. Coronary Artery Disease and Diabetes Mellitus. Heart Fail. Clin. 2016, 12, 117–133. [Google Scholar] [CrossRef] [PubMed]
- Gregor, M.F.; Hotamisligil, G.S. Inflammatory Mechanisms in Obesity. Annu. Rev. Immunol. 2011, 29, 415–445. [Google Scholar] [CrossRef]
- Pedicino, D.; Liuzzo, G.; Trotta, F.; Giglio, A.F.; Giubilato, S.; Martini, F.; Zaccardi, F.; Scavone, G.; Previtero, M.; Massaro, G.; et al. Adaptive Immunity, Inflammation, and Cardiovascular Complications in Type 1 and Type 2 Diabetes Mellitus. J. Diabetes Res. 2013, 2013, 184258. [Google Scholar] [CrossRef]
- Neri, S.; Calvagno, S.; Mauceri, B.; Misseri, M.; Tsami, A.; Vecchio, C.; Mastrosimone, G.; Di Pino, A.; Maiorca, D.; Judica, A.; et al. Effects of antioxidants on postprandial oxidative stress and endothelial dysfunction in subjects with impaired glucose tolerance and Type 2 diabetes. Eur. J. Nutr. 2010, 49, 409–416. [Google Scholar] [CrossRef]
- Saxena, R.; Madhu, S.V.; Shukla, R.; Prabhu, K.M.; Gambhir, J.K. Postprandial hypertriglyceridemia and oxidative stress in patients of type 2 diabetes mellitus with macrovascular complications. Clin. Chim. Acta 2005, 359, 101–108, Erratum in: Clin. Chim. Acta. 2006, 368, 203. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.I.; Bettaieb, A.; Sun, C.; DeVerse, J.S.; Radecke, C.E.; Mathew, S.; Edwards, C.M.; Haj, F.G.; Passerini, A.G.; Simon, S.I. Triglyceride-Rich Lipoprotein Modulates Endothelial Vascular Cell Adhesion Molecule (VCAM)-1 Expression via Differential Regulation of Endoplasmic Reticulum Stress. PLoS ONE 2013, 8, e78322. [Google Scholar] [CrossRef]
- van Oostrom, A.J.H.H.M.; Sijmonsma, T.P.; Verseyden, C.; Jansen, E.H.J.M.; de Koning, E.J.P.; Rabelink, T.; Cabezas, M.C. Postprandial recruitment of neutrophils may contribute to endothelial dysfunction. J. Lipid Res. 2003, 44, 576–583. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, Y.; Saito, T.; Ogihara, T.; Ishigaki, Y.; Yamada, T.; Imai, J.; Uno, K.; Gao, J.; Kaneko, K.; Shimosawa, T.; et al. Blockade of the Nuclear Factor-κB Pathway in the Endothelium Prevents Insulin Resistance and Prolongs Life Spans. Circulation 2012, 125, 1122–1133. [Google Scholar] [CrossRef]
- Solinas, G.; Becattini, B. JNK at the crossroad of obesity, insulin resistance, and cell stress response. Mol. Metab. 2016, 6, 174–184. [Google Scholar] [CrossRef]
- Erridge, C.; Attina, T.; Spickett, C.M.; Webb, D.J. A high-fat meal induces low-grade endotoxemia: Evidence of a novel mechanism of postprandial inflammation. Am. J. Clin. Nutr. 2007, 86, 1286–1292. [Google Scholar] [CrossRef]
- Pahwa, R.; Devaraj, S.; Jialal, I. The effect of the accessory proteins, soluble CD14 and lipopolysaccharide-binding protein on Toll-like receptor 4 activity in human monocytes and adipocytes. Int. J. Obes. 2016, 40, 907–911. [Google Scholar] [CrossRef] [PubMed]
- Vandanmagsar, B.; Youm, Y.-H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Biasucci, L.M.; La Rosa, G.; Pedicino, D.; D’aiello, A.; Galli, M.; Liuzzo, G. Where Does Inflammation Fit? Curr. Cardiol. Rep. 2017, 19, 84. [Google Scholar] [CrossRef]
- Levine, B.; Kalman, J.; Mayer, L.; Fillit, H.M.; Packer, M. Elevated Circulating Levels of Tumor Necrosis Factor in Severe Chronic Heart Failure. N. Engl. J. Med. 1990, 323, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, S.L.; Spengler, M.; May, M.A.; Spengler, R.; Larrick, J.; Remick, D. Prostaglandin E2 regulates macrophage-derived tumor necrosis factor gene expression. J. Biol. Chem. 1988, 263, 5380–5384. [Google Scholar] [CrossRef] [PubMed]
- Giordano, A.; Murano, I.; Mondini, E.; Perugini, J.; Smorlesi, A.; Severi, I.; Barazzoni, R.; Scherer, P.E.; Cinti, S. Obese adipocytes show ultrastructural features of stressed cells and die of pyroptosis. J. Lipid Res. 2013, 54, 2423–2436. [Google Scholar] [CrossRef] [PubMed]
- Gregor, M.F.; Hotamisligil, G.S. Thematic review series: Adipocyte Biology. Adipocyte stress: The endoplasmic reticulum and metabolic disease. J. Lipid Res. 2007, 48, 1905–1914. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Todd, D.J.; Lee, A.-H.; Glimcher, L.H. The endoplasmic reticulum stress response in immunity and autoimmunity. Nat. Rev. Immunol. 2008, 8, 663–674. [Google Scholar] [CrossRef]
- Hu, P.; Han, Z.; Couvillon, A.D.; Kaufman, R.J.; Exton, J.H. Autocrine Tumor Necrosis Factor Alpha Links Endoplasmic Reticulum Stress to the Membrane Death Receptor Pathway through IRE1α-Mediated NF-κB Activation and Down-Regulation of TRAF2 Expression. Mol. Cell. Biol. 2006, 26, 3071–3084. [Google Scholar] [CrossRef]
- Zhang, K.; Shen, X.; Wu, J.; Sakaki, K.; Saunders, T.; Rutkowski, D.T.; Back, S.H.; Kaufman, R.J. Endoplasmic Reticulum Stress Activates Cleavage of CREBH to Induce a Systemic Inflammatory Response. Cell 2006, 124, 587–599. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int. Immunol. 2009, 21, 317–337. [Google Scholar] [CrossRef]
- Pedicino, D.; Giglio, A.F.; Galiffa, V.A.; Cialdella, P.; Trotta, F.; Graziani, F.; Liuzzo, G. Infections, immunity and atherosclerosis: Pathogenic mechanisms and unsolved questions. Int. J. Cardiol. 2013, 166, 572–583. [Google Scholar] [CrossRef]
- Eguchi, K.; Nagai, R. Islet inflammation in type 2 diabetes and physiology. J. Clin. Investig. 2017, 127, 14–23. [Google Scholar] [CrossRef]
- Shioi, T.; Matsumori, A.; Kihara, Y.; Inoko, M.; Ono, K.; Iwanaga, Y.; Yamada, T.; Iwasaki, A.; Matsushima, K.; Sasayama, S. Increased Expression of Interleukin-1β and Monocyte Chemotactic and Activating Factor/Monocyte Chemoattractant Protein-1 in the Hypertrophied and Failing Heart With Pressure Overload. Circ. Res. 1997, 81, 664–671. [Google Scholar] [CrossRef] [PubMed]
- Pedicino, D.; Severino, A.; Ucci, S.; Bugli, F.; Flego, D.; Giglio, A.F.; Trotta, F.; Ruggio, A.; Lucci, C.; Iaconelli, A.; et al. Epicardial adipose tissue microbial colonization and inflammasome activation in acute coronary syndrome. Int. J. Cardiol. 2017, 236, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Liuzzo, G.; Montone, R.A.; Gabriele, M.; Pedicino, D.; Giglio, A.F.; Trotta, F.; Galiffa, V.A.; Previtero, M.; Severino, A.; Biasucci, L.M.; et al. Identification of unique adaptive immune system signature in acute coronary syndromes. Int. J. Cardiol. 2013, 168, 564–567. [Google Scholar] [CrossRef]
- Kintscher, U.; Hartge, M.; Hess, K.; Foryst-Ludwig, A.; Clemenz, M.; Wabitsch, M.; Fischer-Posovszky, P.; Barth, T.F.; Dragun, D.; Skurk, T.; et al. T-lymphocyte infiltration in visceral adipose tissue: A primary event in adipose tissue inflammation and the development of obesity-mediated insulin resistance. Arter. Thromb. Vasc. Biol. 2008, 28, 1304–1310. [Google Scholar] [CrossRef] [PubMed]
- Madhumitha, H.; Mohan, V.; Deepa, M.; Babu, S.; Aravindhan, V. Increased Th1 and suppressed Th2 serum cytokine levels in subjects with diabetic coronary artery disease. Cardiovasc. Diabetol. 2014, 13, 1. [Google Scholar] [CrossRef]
- McGillicuddy, F.C.; Chiquoine, E.H.; Hinkle, C.C.; Kim, R.J.; Shah, R.; Roche, H.M.; Smyth, E.M.; Reilly, M.P. Interferon γ Attenuates Insulin Signaling, Lipid Storage, and Differentiation in Human Adipocytes via Activation of the JAK/STAT Pathway. J. Biol. Chem. 2009, 284, 31936–31944. [Google Scholar] [CrossRef] [PubMed]
- Ruggio, A.; Pedicino, D.; Flego, D.; Vergallo, R.; Severino, A.; Lucci, C.; Niccoli, G.; Trani, C.; Burzotta, F.; Aurigemma, C.; et al. Correlation between CD4+CD28null T lymphocytes, regulatory T cells and plaque rupture: An Optical Coherence Tomography study in Acute Coronary Syndromes. Int. J. Cardiol. 2019, 276, 289–292. [Google Scholar] [CrossRef]
- Canonico, F.; Pedicino, D.; Severino, A.; Vinci, R.; Flego, D.; Pisano, E.; D’aiello, A.; Ciampi, P.; Ponzo, M.; Bonanni, A.; et al. GLUT-1/PKM2 loop dysregulation in patients with non-ST-segment elevation myocardial infarction promotes metainflammation. Cardiovasc. Res. 2022; Online ahead of print. [Google Scholar] [CrossRef]
- Bansal, S.S.; Ismahil, M.A.; Goel, M.; Patel, B.; Hamid, T.; Rokosh, G.; Prabhu, S.D. Activated T Lymphocytes are Essential Drivers of Pathological Remodeling in Ischemic Heart Failure. Circ. Heart Fail. 2017, 10, e003688. [Google Scholar] [CrossRef]
- Tieri, P.; Grignolio, A.; Zaikin, A.; Mishto, M.; Remondini, D.; Castellani, G.C.; Franceschi, C. Network, degeneracy and bow tie. Integrating paradigms and architectures to grasp the complexity of the immune system. Theor. Biol. Med. Model. 2010, 7, 32. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune–metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Zhao, L.; Youn, H.S.; Weatherill, A.R.; Tapping, R.; Feng, L.; Lee, W.H.; Fitzgerald, K.A.; Hwang, D.H. Saturated Fatty Acid Activates but Polyunsaturated Fatty Acid Inhibits Toll-like Receptor 2 Dimerized with Toll-like Receptor 6 or 1. J. Biol. Chem. 2004, 279, 16971–16979. [Google Scholar] [CrossRef] [PubMed]
- Gallo, L.A.; Wright, E.M.; Vallon, V. Probing SGLT2 as a therapeutic target for diabetes: Basic physiology and consequences. Diabetes Vasc. Dis. Res. 2015, 12, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R.; et al. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.L.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; et al. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2019, 380, 347–357. [Google Scholar] [CrossRef]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef]
- Cannon, C.P.; Pratley, R.; Dagogo-Jack, S.; Mancuso, J.; Huyck, S.; Masiukiewicz, U.; Charbonnel, B.; Frederich, R.; Gallo, S.; Cosentino, F.; et al. Cardiovascular Outcomes with Ertugliflozin in Type 2 Diabetes. N. Engl. J. Med. 2020, 383, 1425–1435. [Google Scholar] [CrossRef]
- Bhatt, D.L.; Szarek, M.; Pitt, B.; Cannon, C.P.; Leiter, L.A.; McGuire, D.K.; Lewis, J.B.; Riddle, M.C.; Inzucchi, S.E.; Kosiborod, M.N.; et al. Sotagliflozin in Patients with Diabetes and Chronic Kidney Disease. N. Engl. J. Med. 2021, 384, 129–139. [Google Scholar] [CrossRef]
- Mazidi, M.; Rezaie, P.; Gao, H.-K.; Kengne, A.P. Effect of Sodium-Glucose Cotransport-2 Inhibitors on Blood Pressure in People With Type 2 Diabetes Mellitus: A Systematic Review and Meta-Analysis of 43 Randomized Control Trials With 22 528 Patients. J. Am. Heart Assoc. 2017, 6, e004007. [Google Scholar] [CrossRef] [PubMed]
- Hallow, K.M.; Helmlinger, G.; Greasley, P.J.; McMurray, J.J.V.; Boulton, D.W. Why do SGLT2 inhibitors reduce heart failure hospitalization? A differential volume regulation hypothesis. Diabetes Obes. Metab. 2018, 20, 479–487. [Google Scholar] [CrossRef]
- Neubauer, S. The Failing Heart—An Engine Out of Fuel. N. Engl. J. Med. 2007, 356, 1140–1151. [Google Scholar] [CrossRef] [PubMed]
- Ferrannini, E.; Mark, M.; Mayoux, E. CV Protection in the EMPA-REG OUTCOME Trial: A “Thrifty Substrate” Hypothesis. Diabetes Care 2016, 39, 1108–1114. [Google Scholar] [CrossRef] [PubMed]
- Jordan, J.; Tank, J.; Heusser, K.; Heise, T.; Wanner, C.; Heer, M.; Macha, S.; Mattheus, M.; Lund, S.S.; Woerle, H.J.; et al. The effect of empagliflozin on muscle sympathetic nerve activity in patients with type II diabetes mellitus. J. Am. Soc. Hypertens. 2017, 11, 604–612. [Google Scholar] [CrossRef]
- Sano, M. A new class of drugs for heart failure: SGLT2 inhibitors reduce sympathetic overactivity. J. Cardiol. 2018, 71, 471–476. [Google Scholar] [CrossRef]
- Verma, S.; Garg, A.; Yan, A.T.; Gupta, A.K.; Al-Omran, M.; Sabongui, A.; Teoh, H.; Mazer, C.D.; Connelly, K.A. Effect of Empagliflozin on Left Ventricular Mass and Diastolic Function in Individuals With Diabetes: An Important Clue to the EMPA-REG OUTCOME Trial? Diabetes Care 2016, 39, e212–e213. [Google Scholar] [CrossRef]
- Lim, V.G.; Bell, R.M.; Arjun, S.; Kolatsi-Joannou, M.; Long, D.A.; Yellon, D.M. SGLT2 Inhibitor, Canagliflozin, Attenuates Myocardial Infarction in the Diabetic and Nondiabetic Heart. JACC Basic Transl. Sci. 2019, 4, 15–26. [Google Scholar] [CrossRef]
- Iborra-Egea, O.; Santiago-Vacas, E.; Yurista, S.R.; Lupón, J.; Packer, M.; Heymans, S.; Zannad, F.; Butler, J.; Pascual-Figal, D.; Lax, A.; et al. Unraveling the Molecular Mechanism of Action of Empagliflozin in Heart Failure With Reduced Ejection Fraction With or Without Diabetes. JACC Basic Transl. Sci. 2019, 4, 831–840. [Google Scholar] [CrossRef]
- Chino, Y.; Samukawa, Y.; Sakai, S.; Nakai, Y.; Yamaguchi, J.; Nakanishi, T.; Tamai, I. SGLT2 inhibitor lowers serum uric acid through alteration of uric acid transport activity in renal tubule by increased glycosuria. Biopharm. Drug Dispos. 2014, 35, 391–404. [Google Scholar] [CrossRef]
- Esterline, R.L.; Vaag, A.; Oscarsson, J.; Vora, J. Mechanisms in endocrinology: SGLT2 inhibitors: Clinical benefits by restoration of normal diurnal metabolism? Eur. J. Endocrinol. 2018, 178, R113–R125. [Google Scholar] [CrossRef] [PubMed]
- Mazer, C.D.; Hare, G.M.; Connelly, P.W.; Gilbert, R.E.; Shehata, N.; Quan, A.; Teoh, H.; Leiter, L.A.; Zinman, B.; Jüni, P.; et al. Effect of Empagliflozin on Erythropoietin Levels, Iron Stores, and Red Blood Cell Morphology in Patients With Type 2 Diabetes Mellitus and Coronary Artery Disease. Circulation 2020, 141, 704–707. [Google Scholar] [CrossRef] [PubMed]
- Gaspari, T.; Spizzo, I.; Liu, H.; Hu, Y.; Simpson, R.W.; Widdop, R.E.; Dear, A.E. Dapagliflozin attenuates human vascular endothelial cell activation and induces vasorelaxation: A potential mechanism for inhibition of atherogenesis. Diabetes Vasc. Dis. Res. 2018, 15, 64–73, Erratum in: Diabetes Vasc. Dis. Res. 2018, 15, 364. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, F.; Scheen, A.J. Effects of SGLT2 inhibitors on systemic and tissue low-grade inflammation: The potential contribution to diabetes complications and cardiovascular disease. Diabetes Metab. 2018, 44, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Ota, T. Emerging roles of SGLT2 inhibitors in obesity and insulin resistance: Focus on fat browning and macrophage polarization. Adipocyte 2017, 7, 1–8. [Google Scholar] [CrossRef]
- Packer, M. Do sodium-glucose co-transporter-2 inhibitors prevent heart failure with a preserved ejection fraction by counterbalancing the effects of leptin? A novel hypothesis. Diabetes Obes. Metab. 2018, 20, 1361–1366. [Google Scholar] [CrossRef]
- Pedicino, D.; Severino, A.; Di Sante, G.; De Rosa, M.C.; Pirolli, D.; Vinci, R.; Pazzano, V.; Giglio, A.F.; Trotta, F.; Russo, G.; et al. Restricted T-Cell Repertoire in the Epicardial Adipose Tissue of Non-ST Segment Elevation Myocardial Infarction Patients. Front. Immunol. 2022, 13, 845526. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Sato, T.; Aizawa, Y.; Yuasa, S.; Kishi, S.; Fuse, K.; Fujita, S.; Ikeda, Y.; Kitazawa, H.; Takahashi, M.; Sato, M.; et al. The effect of dapagliflozin treatment on epicardial adipose tissue volume. Cardiovasc. Diabetol. 2018, 17, 6. [Google Scholar] [CrossRef]
- Ojima, A.; Matsui, T.; Nishino, Y.; Nakamura, N.; Yamagishi, S. Empagliflozin, an Inhibitor of Sodium-Glucose Cotransporter 2 Exerts Anti-Inflammatory and Antifibrotic Effects on Experimental Diabetic Nephropathy Partly by Suppressing AGEs-Receptor Axis. Horm. Metab. Res. 2015, 47, 686–692. [Google Scholar] [CrossRef]
- Pedicino, D.; Giglio, A.F.; Ruggio, A.; Massaro, G.; D’Aiello, A.; Trotta, F.; Lucci, C.; Graziani, F.; Biasucci, L.M.; Crea, F.; et al. Inflammasome, T Lymphocytes and Innate-Adaptive Immunity Crosstalk: Role in Cardiovascular Disease and Therapeutic Perspectives. Thromb. Haemost. 2018, 118, 1352–1369. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Bajaj, M.; Yang, H.-C.; Perez-Polo, J.R.; Birnbaum, Y. SGLT-2 Inhibition with Dapagliflozin Reduces the Activation of the Nlrp3/ASC Inflammasome and Attenuates the Development of Diabetic Cardiomyopathy in Mice with Type 2 Diabetes. Further Augmentation of the Effects with Saxagliptin, a DPP4 Inhibitor. Cardiovasc. Drugs Ther. 2017, 31, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Verma, S. Mechanisms of Cardiovascular Benefits of Sodium Glucose Co-Transporter 2 (SGLT2) Inhibitors: A State-of-the-Art Review. JACC Basic Transl. Sci. 2020, 5, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Nasiri-Ansari, N.; Dimitriadis,, G.K.; Agrogiannis, G.; Perrea, D.; Kostakis, I.D.; Kaltsas, G.; Papavassiliou, A.G.; Randeva, H.S.; Kassi, E. Canagliflozin attenuates the progression of atherosclerosis and inflammation process in APOE knockout mice. Cardiovasc. Diabetol. 2018, 17, 106. [Google Scholar] [CrossRef]
- Leng, W.; Ouyang, X.; Lei, X.; Wu, M.; Chen, L.; Wu, Q.; Deng, W.; Liang, Z. The SGLT-2 Inhibitor Dapagliflozin Has a Therapeutic Effect on Atherosclerosis in Diabetic ApoE−/−Mice. Mediat. Inflamm. 2016, 2016, 6305735. [Google Scholar] [CrossRef]
- Lee, S.-G.; Lee, S.-J.; Lee, J.-J.; Kim, J.-S.; Lee, O.-H.; Kim, C.-K.; Kim, D.; Lee, Y.-H.; Oh, J.; Park, S.; et al. Anti-Inflammatory Effect for Atherosclerosis Progression by Sodium-Glucose Cotransporter 2 (SGLT-2) Inhibitor in a Normoglycemic Rabbit Model. Korean Circ. J. 2020, 50, 443–457. [Google Scholar] [CrossRef]
- White, J.R. A Brief History of the Development of Diabetes Medications. Diabetes Spectr. 2014, 27, 82–86. [Google Scholar] [CrossRef]
- Holman, R.R.; Bethel, M.A.; Mentz, R.J.; Thompson, V.P.; Lokhnygina, Y.; Buse, J.B.; Chan, J.C.; Choi, J.; Gustavson, S.M.; Iqbal, N.; et al. Effects of Once-Weekly Exenatide on Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 1228–1239. [Google Scholar] [CrossRef]
- Marso, S.P.; Daniels, G.H.; Brown-Frandsen, K.; Kristensen, P.; Mann, J.F.E.; Nauck, M.A.; Nissen, S.E.; Pocock, S.; Poulter, N.R.; Ravn, L.S.; et al. Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 311–322. [Google Scholar] [CrossRef]
- Marso, S.P.; Bain, S.C.; Consoli, A.; Eliaschewitz, F.G.; Jódar, E.; Leiter, L.A.; Lingvay, I.; Rosenstock, J.; Seufert, J.; Warren, M.L.; et al. Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 1834–1844. [Google Scholar] [CrossRef]
- Pfeffer, M.A.; Claggett, B.; Diaz, R.; Dickstein, K.; Gerstein, H.C.; Køber, L.V.; Lawson, F.C.; Ping, L.; Wei, X.; Lewis, E.F.; et al. Lixisenatide in Patients with Type 2 Diabetes and Acute Coronary Syndrome. N. Engl. J. Med. 2015, 373, 2247–2257. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, A.F.; Green, J.B.; Janmohamed, S.; D’Agostino, R.B.; Granger, C.B.; Jones, N.P.; Leiter, L.A.; Rosenberg, A.E.; Sigmon, K.N.; Somerville, M.C.; et al. Albiglutide and cardiovascular outcomes in patients with type 2 diabetes and cardiovascular disease (Harmony Outcomes): A double-blind, randomised placebo-controlled trial. Lancet 2018, 392, 1519–1529. [Google Scholar] [CrossRef] [PubMed]
- Gerstein, H.C.; Colhoun, H.M.; Dagenais, G.R.; Diaz, R.; Lakshmanan, M.; Pais, P.; Probstfield, J.; Riesmeyer, J.S.; Riddle, M.C.; Rydén, L.; et al. Dulaglutide and cardiovascular outcomes in type 2 diabetes (REWIND): A double-blind, randomised placebo-controlled trial. Lancet 2019, 394, 121–130. [Google Scholar] [CrossRef]
- Husain, M.; Birkenfeld, A.L.; Donsmark, M.; Dungan, K.; Eliaschewitz, F.G.; Franco, D.R.; Jeppesen, O.K.; Lingvay, I.; Mosenzon, O.; Pedersen, S.D.; et al. Oral Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N. Engl. J. Med. 2019, 381, 841–851. [Google Scholar] [CrossRef]
- Newman, J.D.; Vani, A.K.; Aleman, J.O.; Weintraub, H.S.; Berger, J.S.; Schwartzbard, A.Z. The Changing Landscape of Diabetes Therapy for Cardiovascular Risk Reduction: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2018, 72, 1856–1869. [Google Scholar] [CrossRef]
- Liuzzo, G.; Patrono, C. Dual glucose-dependent insulinotropic polypeptide/glucagon-like peptide-1 receptor agonism for substantial body weight reduction in obese subjects. Eur. Heart J. 2022, 43, 3288–3289. [Google Scholar] [CrossRef]
- Mann, J.F.E.; Fonseca, V.; Mosenzon, O.; Raz, I.; Goldman, B.; Idorn, T.; von Scholten, B.J.; Poulter, N.R.; The LEADER Publication Committee on behalf of the LEADER Trial Investigators. Effects of Liraglutide Versus Placebo on Cardiovascular Events in Patients With Type 2 Diabetes Mellitus and Chronic Kidney Disease. Circulation 2018, 138, 2908–2918. [Google Scholar] [CrossRef]
- Heuvelman, V.D.; Van Raalte, D.H.; Smits, M.M. Cardiovascular effects of glucagon-like peptide 1 receptor agonists: From mechanistic studies in humans to clinical outcomes. Cardiovasc. Res. 2020, 116, 916–930. [Google Scholar] [CrossRef] [PubMed]
- Hermansen, K.; Baekdal, T.A.; Düring, M.; Pietraszek, A.; Mortensen, L.S.; Jørgensen, H.; Flint, A. Liraglutide suppresses postprandial triglyceride and apolipoprotein B48 elevations after a fat-rich meal in patients with type 2 diabetes: A randomized, double-blind, placebo-controlled, cross-over trial. Diabetes Obes. Metab. 2013, 15, 1040–1048. [Google Scholar] [CrossRef]
- Drucker, D.J.; Nauck, M.A. The incretin system: Glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet 2006, 368, 1696–1705. [Google Scholar] [CrossRef]
- Yusta, B.; Baggio, L.L.; Koehler, J.; Holland, D.; Cao, X.; Pinnell, L.J.; Johnson-Henry, K.C.; Yeung, W.; Surette, M.G.; Bang, K.A.; et al. GLP-1R Agonists Modulate Enteric Immune Responses Through the Intestinal Intraepithelial Lymphocyte GLP-1R. Diabetes 2015, 64, 2537–2549. [Google Scholar] [CrossRef] [PubMed]
- Burgmaier, M.; Liberman, A.; Möllmann, J.; Kahles, F.; Reith, S.; Lebherz, C.; Marx, N.; Lehrke, M. Glucagon-like peptide-1 (GLP-1) and its split products GLP-1(9-37) and GLP-1(28-37) stabilize atherosclerotic lesions in apoe−/− mice. Atherosclerosis 2013, 231, 427–435. [Google Scholar] [CrossRef]
- Chaudhuri, A.; Ghanim, H.; Vora, M.; Sia, C.L.; Korzeniewski, K.; Dhindsa, S.; Makdissi, A.; Dandona, P. Exenatide Exerts a Potent Antiinflammatory Effect. J. Clin. Endocrinol. Metab. 2012, 97, 198–207. [Google Scholar] [CrossRef]
- Bajaj, H.S.; Al-Jabri, B.; Verma, S. Glucagon-like peptide-1 receptor agonists and cardiovascular protection in type 2 diabetes: A pathophysiology-based review of clinical implications. Curr. Opin. Cardiol. 2018, 33, 665–675. [Google Scholar] [CrossRef]
- Ceriello, A.; Novials, A.; Ortega, E.; Canivell, S.; La Sala, L.; Pujadas, G.; Esposito, K.; Giugliano, D.; Genovese, S. Glucagon-Like Peptide 1 Reduces Endothelial Dysfunction, Inflammation, and Oxidative Stress Induced by Both Hyperglycemia and Hypoglycemia in Type 1 Diabetes. Diabetes Care 2013, 36, 2346–2350. [Google Scholar] [CrossRef]
- Koska, J.; Sands, M.; Burciu, C.; D’souza, K.M.; Raravikar, K.; Liu, J.; Truran, S.; Franco, D.A.; Schwartz, E.A.; Schwenke, D.C.; et al. Exenatide Protects Against Glucose- and Lipid-Induced Endothelial Dysfunction: Evidence for Direct Vasodilation Effect of GLP-1 Receptor Agonists in Humans. Diabetes 2015, 64, 2624–2635. [Google Scholar] [CrossRef]
- Cameron-Vendrig, A.; Reheman, A.; Siraj, M.A.; Xu, X.R.; Wang, Y.; Lei, X.; Afroze, T.; Shikatani, E.; El-Mounayri, O.; Noyan, H.; et al. Glucagon-Like Peptide 1 Receptor Activation Attenuates Platelet Aggregation and Thrombosis. Diabetes 2016, 65, 1714–1723. [Google Scholar] [CrossRef]
- Ussher, J.R.; Baggio, L.L.; Campbell, J.E.; Mulvihill, E.E.; Kim, M.; Kabir, M.G.; Cao, X.; Baranek, B.M.; Stoffers, D.A.; Seeley, R.J.; et al. Inactivation of the cardiomyocyte glucagon-like peptide-1 receptor (GLP-1R) unmasks cardiomyocyte-independent GLP-1R-mediated cardioprotection. Mol. Metab. 2014, 3, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.S.; Kim, W.; Ha, S.J.; Kim, J.B.; Kim, S.-J.; Kim, W.-S.; Seon, H.J.; Kim, K.S. Cardioprotective effects of exenatide in patients with ST-segment-elevation myocardial infarction undergoing primary percutaneous coronary intervention: Results of exenatide myocardial protection in revascularization study. Arter. Thromb. Vasc. Biol. 2013, 33, 2252–2260. [Google Scholar] [CrossRef] [PubMed]
- Iacobellis, G.; Leonetti, F. Epicardial Adipose Tissue and Insulin Resistance in Obese Subjects. J. Clin. Endocrinol. Metab. 2005, 90, 6300–6302. [Google Scholar] [CrossRef]
- Iacobellis, G.; Fricke, A.C.V. Effects of Semaglutide Versus Dulaglutide on Epicardial Fat Thickness in Subjects with Type 2 Diabetes and Obesity. J. Endocr. Soc. 2020, 4, bvz042. [Google Scholar] [CrossRef] [PubMed]
- Iacobellis, G.; Baroni, M.G. Cardiovascular risk reduction throughout GLP-1 receptor agonist and SGLT2 inhibitor modulation of epicardial fat. J. Endocrinol. Investig. 2022, 45, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Liuzzo, G.; Biasucci, L.M.; Gallimore, J.R.; Grillo, R.L.; Rebuzzi, A.G.; Pepys, M.B.; Maseri, A. The Prognostic Value of C-Reactive Protein and Serum Amyloid A Protein in Severe Unstable Angina. N. Engl. J. Med. 1994, 331, 417–424. [Google Scholar] [CrossRef] [PubMed]




| SGLT-2i Name | Clinical Trial Name (CVOT) | Brief Reference | Number of Patients | Definition of CV Outcomes | Median Follow Up (Months) | Principal Findings |
|---|---|---|---|---|---|---|
| Empagliflozin | EMPA-REG OUTCOME | Zinman B et al., New England Journal of Medicine, 2015 [44] | 7020 | CV, non-fatal myocardial infarction, or non-fatal stroke | 37.2 | The empagliflozin group presented significantly lower rates of death from CV causes (3.7% vs. 5.9% in the placebo group; 38% relative risk reduction), and hospitalization for HF (2.7% and 4.1%, respectively; 35% relative risk reduction). |
| Canagliflozin | CANVAS | Neal B et al., New England Journal of Medicine, 2017 [45] | 10,142 | CV death or hospitalization for HF | 29 | The rate of the primary outcome (composite of death from cardiovascular causes, non-fatal myocardial infarction, or non-fatal stroke) was lower with canagliflozin than with placebo (HR, 0.86; 95% CI, 0.75 to 0.97; p < 0.001 for non-inferiority; p = 0.02 for superiority). |
| Dapagliflozin | DECLARE-TIMI | Wiviott SD et al., New England Journal of Medicine, 2019 [47] | 17,160 | CV death or hospitalization for HF | 50.4 | Dapagliflozin did result in a lower rate of CV death or hospitalization for heart failure (4.9% vs. 5.8%; HR, 0.83; 95% CI, 0.73 to 0.95; p = 0.005), which reflected a lower rate of hospitalization for HF (hazard ratio, 0.73; 95% CI, 0.61 to 0.88) |
| Dapagliflozin | DAPA-HF | McMurray JJV et al., New England Journal of Medicine, 2019 [48] | 4744 | CV death or HF hospitalization/urgent HF visit | 18.2 | The primary outcome (composite of worsening heart failure or death from cardiovascular causes) was lower in the dapagliflozin group than in the placebo group (HR, 0.74; 95% CI, 0.65 to 0.85; p < 0.001). The first worsening HF event occurred in 10.0% of the dapagliflozin group and in 13.7% in the placebo group (HR, 0.70; 95% CI, 0.59 to 0.83). Death from cardiovascular causes occurred in 9.6% in the dapagliflozin group and in 11.5% in the placebo group (HR, 0.82; 95% CI, 0.69 to 0.98) |
| Ertugliflozin | VERTIS-CV | Cannon et al., New England Journal of Medicine, 2020 [49] | 8246 | CV death or hospitalization for HF | 42 | MACE occurred in 11.9% of the ertugliflozin group and in 11.9% of the placebo group (HR, 0.97; 95.6% CI, 0.85 to 1.11; p < 0.001 for non-inferiority). Death from CV causes or hospitalization for HF occurred in 8.1% in the ertugliflozin group and in 9.1% in the placebo group (HR, 0.88; 95.8% CI, 0.75 to 1.03; p = 0.11 for superiority). |
| Sotagliflozin | SCORED | Bahtt DL et al., New England Journal of Medicine, 2021 [50] | 10,584 | CV death or HF hospitalization/ urgent HF visit | 16 | The rate of primary end-point events was 5.6 events in the sotagliflozin group and 7.5 events per 100 patient-years in the placebo group (HR, 0.74; 95% [CI], 0.63 to 0.88; p < 0.001). The rate of deaths from cardiovascular causes per 100 patient-years was 2.2 with sotagliflozin and 2.4 with placebo (HR, 0.90; 95% CI, 0.73 to 1.12; p = 0.35). |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
d’Aiello, A.; Bonanni, A.; Vinci, R.; Pedicino, D.; Severino, A.; De Vita, A.; Filomia, S.; Brecciaroli, M.; Liuzzo, G. Meta-Inflammation and New Anti-Diabetic Drugs: A New Chance to Knock Down Residual Cardiovascular Risk. Int. J. Mol. Sci. 2023, 24, 8643. https://doi.org/10.3390/ijms24108643
d’Aiello A, Bonanni A, Vinci R, Pedicino D, Severino A, De Vita A, Filomia S, Brecciaroli M, Liuzzo G. Meta-Inflammation and New Anti-Diabetic Drugs: A New Chance to Knock Down Residual Cardiovascular Risk. International Journal of Molecular Sciences. 2023; 24(10):8643. https://doi.org/10.3390/ijms24108643
Chicago/Turabian Styled’Aiello, Alessia, Alice Bonanni, Ramona Vinci, Daniela Pedicino, Anna Severino, Antonio De Vita, Simone Filomia, Mattia Brecciaroli, and Giovanna Liuzzo. 2023. "Meta-Inflammation and New Anti-Diabetic Drugs: A New Chance to Knock Down Residual Cardiovascular Risk" International Journal of Molecular Sciences 24, no. 10: 8643. https://doi.org/10.3390/ijms24108643
APA Styled’Aiello, A., Bonanni, A., Vinci, R., Pedicino, D., Severino, A., De Vita, A., Filomia, S., Brecciaroli, M., & Liuzzo, G. (2023). Meta-Inflammation and New Anti-Diabetic Drugs: A New Chance to Knock Down Residual Cardiovascular Risk. International Journal of Molecular Sciences, 24(10), 8643. https://doi.org/10.3390/ijms24108643