
RNA Overwriting of Cellular mRNA by Cas13b-Directed RNA-Dependent RNA Polymerase of Influenza A Virus
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
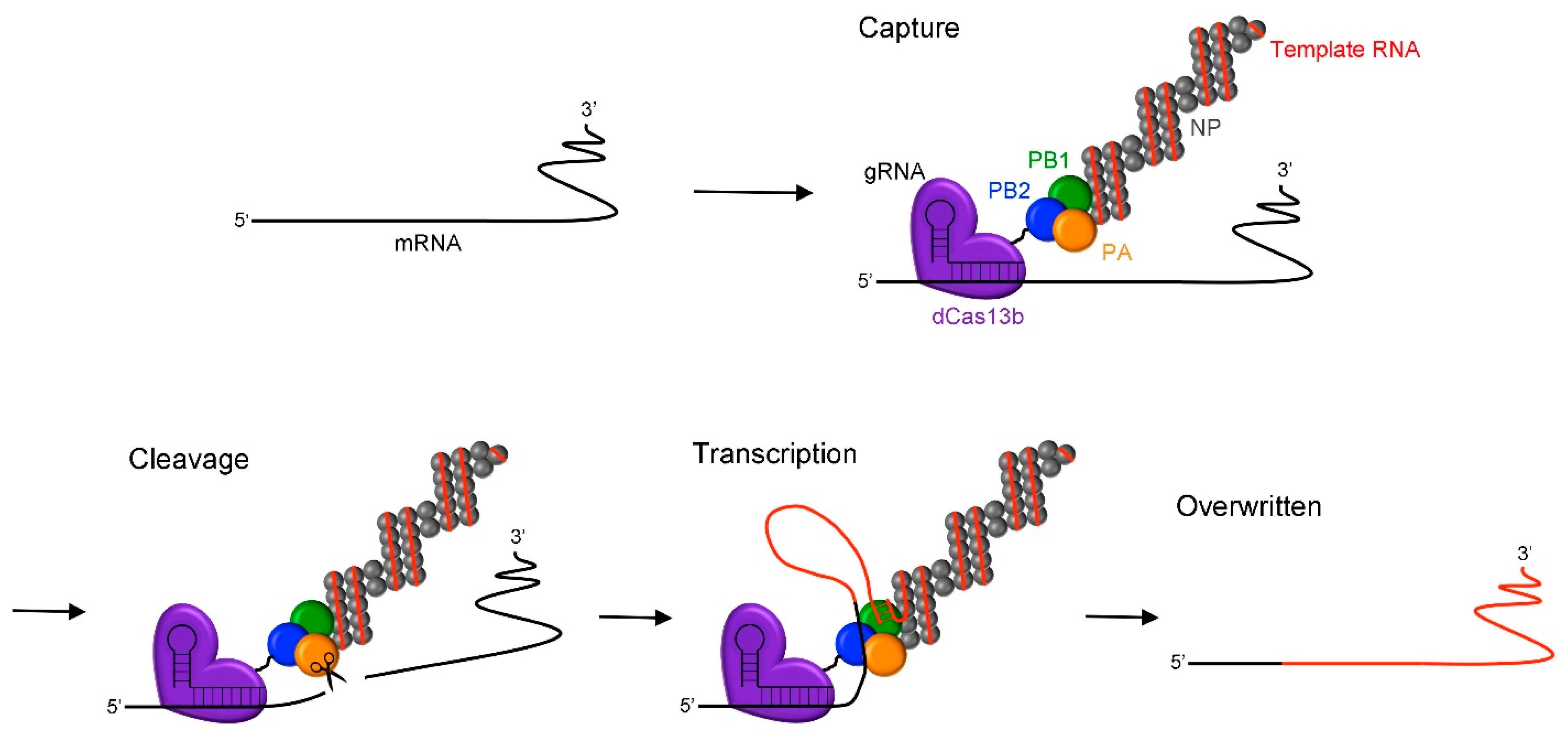
:1. Introduction
2. Results and Discussion
2.1. Investigation of PB2 Mutant Lacking the Cap-Binding Ability
2.2. Influence of Fusing dCas13b with RdRp Subunits on RNP Activity
2.3. RNA Overwriting in Cells
2.4. RNA Overwriting within an Open Reading Frame
3. Materials and Methods
3.1. Cells
3.2. Construction of Plasmids
3.3. RNP Activity Assay
3.4. RNP Formation Assay
3.5. Western Blot
3.6. Primer Extension Assay
3.7. RNA Overwriting
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cooper, T.A.; Wan, L.; Dreyfuss, G. RAN and Disease. Cell 2009, 136, 777–793. [Google Scholar] [CrossRef] [Green Version]
- Kataoka, N.; Mayeda, A.; Ohno, K. (Eds.) RNA Diseases in Humans-From Fundamental Research to Therapeutic Application; Frontiers: Lausanne, Switzerland, 2019. [Google Scholar]
- Kim, Y.G.; Cha, J.; Chandrasegaran, S. Hybrid restriction enzymes: Zinc finger fusions to Fok I cleavage domain. Proc. Natl. Acad. Sci. USA 1996, 93, 1156–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boch, J.; Scholze, H.; Schornack, S.; Landgraf, A.; Hahn, S.; Kay, S.; Lahaye, T.; Nickstadt, A.; Bonas, U. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 2009, 326, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Christian, M.; Cermak, T.; Doyle, E.L.; Schmidt, C.; Zhang, F.; Hummel, A.; Bogdanove, A.J.; Voytas, D.F. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics 2010, 186, 757–761. [Google Scholar] [CrossRef] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [Green Version]
- Booth, B.J.; Nourreddine, S.; Katrekar, D.; Savva, Y.; Bose, D.; Long, T.J.; Huss, D.J.; Mali, P. RNA editing: Expanding the potential of RNA therapeutics. Mol. Ther. 2023, 31, 1533–1549. [Google Scholar] [CrossRef] [PubMed]
- Kim, U.; Wang, Y.; Sanford, T.; Zeng, Y.; Nishikura, K. Molecular cloning of cDNA for double-stranded RNA adenosine deaminase, a candidate enzyme for nuclear RNA editing. Proc. Natl. Acad. Sci. USA 1994, 91, 11457–11461. [Google Scholar] [CrossRef] [Green Version]
- Stafforst, T.; Schneider, M.F. An RNA-deaminase conjugate selectively repairs point mutations. Angew. Chem. Int. Ed. 2012, 51, 11166–11169. [Google Scholar] [CrossRef] [PubMed]
- Montiel-Gonzalez, M.F.; Vallecillo-Viejo, I.; Yudowski, G.A.; Rosenthal, J.J.C. Correction of mutations within the cystic fibrosis transmembrane conductance regulator by site-directed RNA editing. Proc. Natl. Acad. Sci. USA 2013, 110, 18285–18290. [Google Scholar] [CrossRef] [Green Version]
- Montiel-Gonzalez, M.F.; Vallecillo-Viejo, I.C.; Rosenthal, J.J.C. An efficient system for selectively altering genetic information within mRNAs. Nucleic Acids Res. 2016, 44, e157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, J.; Rahman, R.; Gupta, V.; Rosbash, M.; Singer, R.H. MS2-TRIBE evaluates both protein-RNA interaction and nuclear organization of transcription by RNA editing. iScience 2020, 23, 101318. [Google Scholar] [CrossRef] [PubMed]
- Tohama, T.; Sakari, M.; Tsukahara, T. Development of a single construct system for site-directed RNA editing using MS2-ADAR. Int. J. Mol. Sci. 2020, 21, 4943. [Google Scholar] [CrossRef]
- Cox, D.B.; Gootenherg, J.S.; Abudayyeh, O.O.; Franklin, B.; Kellner, M.J.; Joung, J.; Zhang, F. RNA editing with CRISPR-Cas13. Science 2017, 358, 1019–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuda, M.; Umeno, H.; Nose, K.; Nishitarumizu, A.; Noguchi, R.; Nakagawa, H. Construction of a guide-RNA for site-directed RNA mutagenesis utilising intracellular A-to-I RNA editing. Sci. Rep. 2017, 7, 42478. [Google Scholar] [CrossRef] [Green Version]
- Wettengel, J.; Reautschnig, P.; Geisler, S.; Kahle, P.J.; Stafforst, T. Harnessing human ADAR2 for RNA repair—Recoding a PINK1 mutation rescues mitophagy. Nucleic Acids Res. 2017, 45, 2797–2808. [Google Scholar] [CrossRef] [Green Version]
- Qu, L.; Yi, Z.; Zhu, S.; Wang, C.; Cao, Z.; Zhou, Z.; Yuan, P.; Yu, Y.; Tian, F.; Liu, Z.; et al. Programmable RNA editing by recruiting endogenous ADAR using engineered RNAs. Nat. Biotechnol. 2019, 37, 1059–1069. [Google Scholar] [CrossRef] [PubMed]
- Merkle, T.; Merz, S.; Reautschnig, P.; Blaha, A.; Li, Q.; Vogel, P.; Wettengel, J.; Li, J.B.; Stafforst, T. Precise RNA editing by recruiting endogenous ADARs with antisense oligonucleotides. Nat. Biotechnol. 2019, 37, 133–138. [Google Scholar] [CrossRef]
- Vargeese, C.; Shivalila, C.; Lu, G.; Shimizu, M.; Boulay, D.; Bussow, K.; Byrne, M.; Bezigian, A.; Chatterjee, A.; Chew, D.; et al. Endogenous ADAR-mediated RNA editing in non-human primates using stereopure chemically modified oligonucleotides. Nat. Biotechnol. 2022, 40, 1093–1102. [Google Scholar]
- Katrekar, D.; Yen, J.; Xiang, Y.; Saha, A.; Meluzzi, D.; Savva, Y.; Mali, P. Efficient in vitro and in vivo RNA editing via recruitment of endogenous ADARs using circular guide RNAs. Nat. Biotechnol. 2022, 40, 938–945. [Google Scholar] [CrossRef]
- Du, M.; Jillette, N.; Zhu, J.J.; Li, S.; Cheng, A.W. CRISPR artificial splicing factors. Nat. Commun. 2020, 12, 2973. [Google Scholar] [CrossRef]
- Wilson, C.; Chen, P.J.; Miao, Z.; Liu, D.R. Programmable m6A modification of cellular RNAs with a Cas13-directed methyltransferase. Nat. Biotechnol. 2020, 38, 1431–1440. [Google Scholar] [CrossRef]
- Shi, H.; Xu, Y.; Tian, N.; Yang, M.; Liang, F.S. Inducible and reversible RNA N6-methyladenosine editing. Nat. Commun. 2022, 13, 1958. [Google Scholar] [CrossRef]
- Blijlevens, M.; Li, J.; van Beusechem, V.W. Biology of the mRNA splicing machinery and its dysregulation in cancer providing therapeutic opportunities. Int. J. Mol. Sci. 2021, 22, 5110. [Google Scholar] [CrossRef] [PubMed]
- Ogasawara, S.; Yamada, I. RNA editing with viral RNA-dependent RNA polymerase. ACS Synth. Biol. 2022, 11, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Vlugt, C.D.; Sikora, D.; Pelchat, M. Insight into influenza: A virus cap-snatching. Viruses 2018, 10, 641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turrell, L.; Lyall, J.W.; Tiley, L.S.; Fodor, E.; Vreede, F.T. The role and assembly mechanism of nucleoprotein in influenza A virus ribonucleoprotein complexes. Nat. Commun. 2013, 4, 1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fechter, P.; Mingay, L.; Sharps, J.; Chambers, A.; Fodor, E. Two aromatic residues in the PB2 subunit of influenza A RNA polymerase are crucial for cap binding. J. Biol. Chem. 2003, 278, 20381–20388. [Google Scholar] [CrossRef] [Green Version]
- Guilligay, D.; Tarendeau, F.; Resa-Infante, P.; Coloma, R.; Crepin, T.; Sehr, P.; Lewis, J.; Ruigrok, R.W.H.; Ortin, J.; Hart, D.J.; et al. The structural basis for cap binding by influenza virus polymerase subunit PB2. Nat. Struct. Mol. Biol. 2008, 15, 500–506. [Google Scholar] [CrossRef]
- Chattopadhyay, S.; Garcia-Mena, J.; DeVito, J.; Wolska, K.; Das, A. Bipartite function of a small RNA hairpin in transcription antitermination in bacteriophage lambda. Proc. Natl. Acad. Sci. USA 1995, 92, 4061–4065. [Google Scholar] [CrossRef] [Green Version]
- Smargon, A.A.; Cox, D.B.T.; Pyzocha, N.K.; Zheng, K.; Slaymaker, I.M.; Gootenberg, J.S.; Abudyyeh, O.A.; Essletzbichler, P.; Shmakov, S.; Makarova, K.S.; et al. Cas13b is a type VI-B CRISPR-associated RNA-guided RNase differentially regulated by accessory proteins Csx27 and Csx28. Mol. Cell 2017, 65, 618–630.e617. [Google Scholar] [CrossRef] [Green Version]
- Biswas, S.K.; Boutz, P.L.; Nayak, D.P. Influenza virus nucleoprotein interacts with influenza virus polymerase protein. J. Virol. 1998, 72, 5493–5501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poole, E.; Elton, D.; Medcalf, L.; Digard, P. Function domains of the influenza virus PB2 protein: Identification of NP- and PB1-binding sites. Virology 2004, 321, 120133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidic, J.; Noiray, M.; Bagchi, A.; Slama-Schwak, A. Identification of a novel complex between the nucleoprotein and PA(1-27) of influenza A virus polymerase. Biochemistry 2016, 55, 4259–4262. [Google Scholar] [CrossRef] [PubMed]
- Ju, H.; Zhang, J.; Huang, B.; Kang, D.; Huang, B.; Liu, X.; Zhan, P. Inhibitors of influenza virus polymerase acidic (PA) endonuclease: Contemporary developments and perspectives. J. Med. Chem. 2017, 60, 3533–3551. [Google Scholar] [CrossRef]
- Neumann, G.; Hobom, G. Mutational analysis of influenza virus promoter elements in vivo. J. Gen. Virol. 1995, 76, 1709–1717. [Google Scholar] [CrossRef]
- Rao, P.; Yuan, W.; Krug, R.M. Crucial role of CA cleavage sites in the cap-snatching mechanism for initiating viral mRNA synthesis. EMBO J. 2003, 22, 1188–1198. [Google Scholar] [CrossRef] [Green Version]
- Vreede, F.T.; Gifford, H.; Brownlee, G.G. Role of initiating nucleoside triphosphate concentrations in the regulation of influenza virus replication and transcription. J. Virol. 2008, 82, 6902–6910. [Google Scholar] [CrossRef] [Green Version]
- Belicha-Villanueva, A.; Rodriguez-Madoz, J.R.; Maamary, J.; Baum, A.; Bernal-Rubio, D.; de la Escalera, M.M.; Fernandez-Sesma, A.; García-Sastre, A. Recombinant influenza A viruses with enhanced levels of PB1 and PA viral protein expression. J. Virol. 2012, 86, 5926–5930. [Google Scholar] [CrossRef] [Green Version]
- Paterson, D.; Te Velthuis, A.J.W.; Vreede, F.T.; Fodor, E. Host restriction of influenza virus polymerase activity by PB2 6227E is diminished on viral templates in a nucleoprotein-independent manner. J. Virol. 2014, 88, 339–344. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhao, X.; Fang, Y.; Jiang, X.; Duong, T.; Fan, C.; Huang, C.C.; Kain, S.R. Generation of destabilized green fluorescent protein as a transcription reporter. J. Biol. Chem. 1998, 273, 34970–34975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Sanyal, S. p53 isoforms as cancer biomarkers and therapeutic targets. Cancers 2022, 14, 3145. [Google Scholar] [CrossRef] [PubMed]
- de Silva, S.; Bowers, W.J. Herpes virus amplicon vectors. Viruses 2009, 1, 594–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ogasawara, S.; Ebashi, S. RNA Overwriting of Cellular mRNA by Cas13b-Directed RNA-Dependent RNA Polymerase of Influenza A Virus. Int. J. Mol. Sci. 2023, 24, 10000. https://doi.org/10.3390/ijms241210000
Ogasawara S, Ebashi S. RNA Overwriting of Cellular mRNA by Cas13b-Directed RNA-Dependent RNA Polymerase of Influenza A Virus. International Journal of Molecular Sciences. 2023; 24(12):10000. https://doi.org/10.3390/ijms241210000
Chicago/Turabian StyleOgasawara, Shinzi, and Sae Ebashi. 2023. "RNA Overwriting of Cellular mRNA by Cas13b-Directed RNA-Dependent RNA Polymerase of Influenza A Virus" International Journal of Molecular Sciences 24, no. 12: 10000. https://doi.org/10.3390/ijms241210000
APA StyleOgasawara, S., & Ebashi, S. (2023). RNA Overwriting of Cellular mRNA by Cas13b-Directed RNA-Dependent RNA Polymerase of Influenza A Virus. International Journal of Molecular Sciences, 24(12), 10000. https://doi.org/10.3390/ijms241210000