

Genome-Wide Association Study of Lint Percentage in Gossypium hirsutum L. Races
 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Large Variation in Lint Percentage in G. hirsutum Races
2.2. Genome-Wide Association Study for Lint Percentage
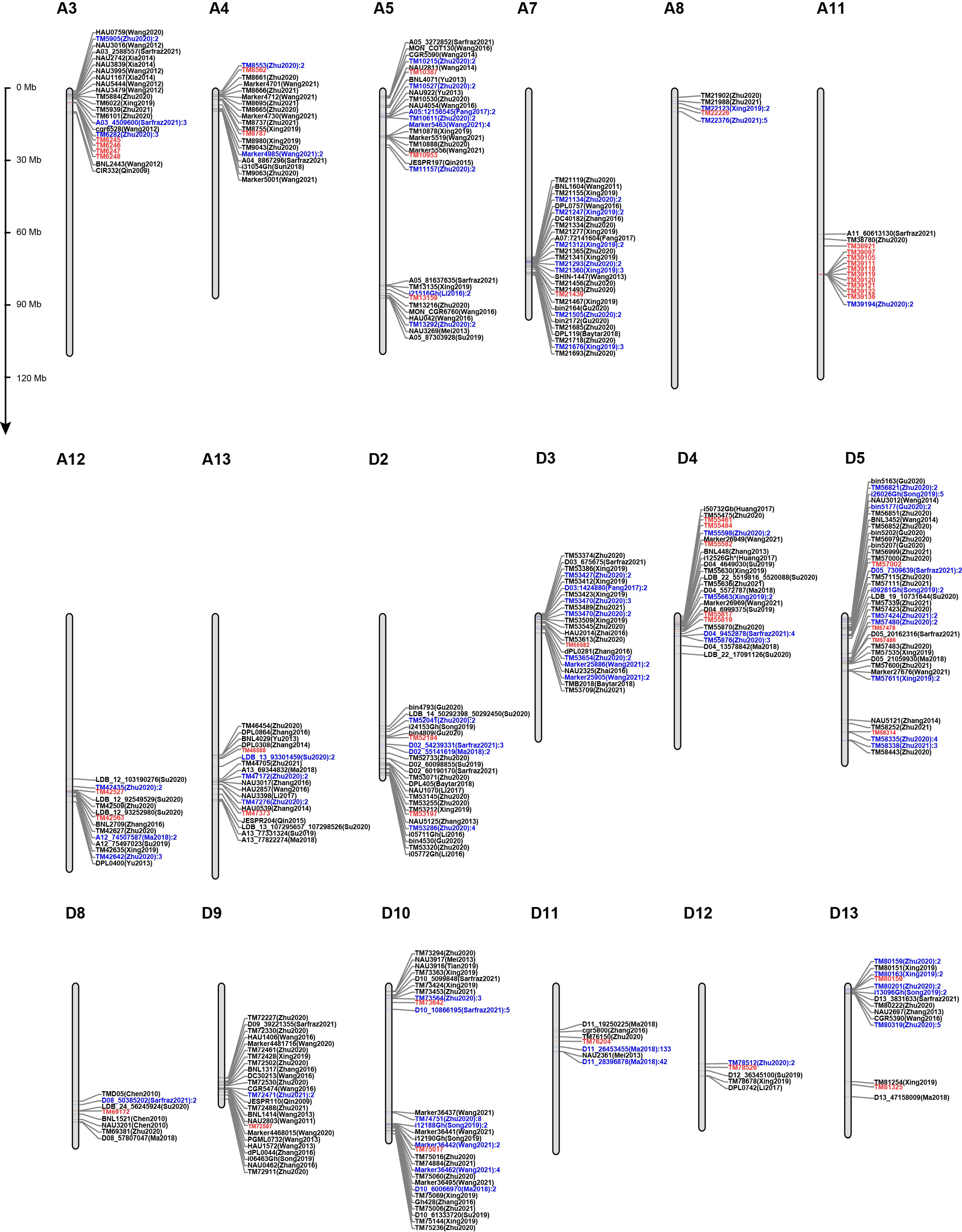
2.3. Validation of the Stability of 45 SNPs Associated with Lint Percentage
2.4. Discovery of Candidate Genes for Lint Percentage
2.5. Gh_A08G0526 and Gh_D12G0934 Were Candidate Genes for Lint Percentage
3. Materials and Methods
3.1. Natural Population Germplasm and Lint Percentage Evaluation
3.2. SNP Genotyping and Population Structure Assessment
3.3. Genome-Wide Association Study of Lint Percentage
3.4. Prediction and Identification of Related Candidate Gene
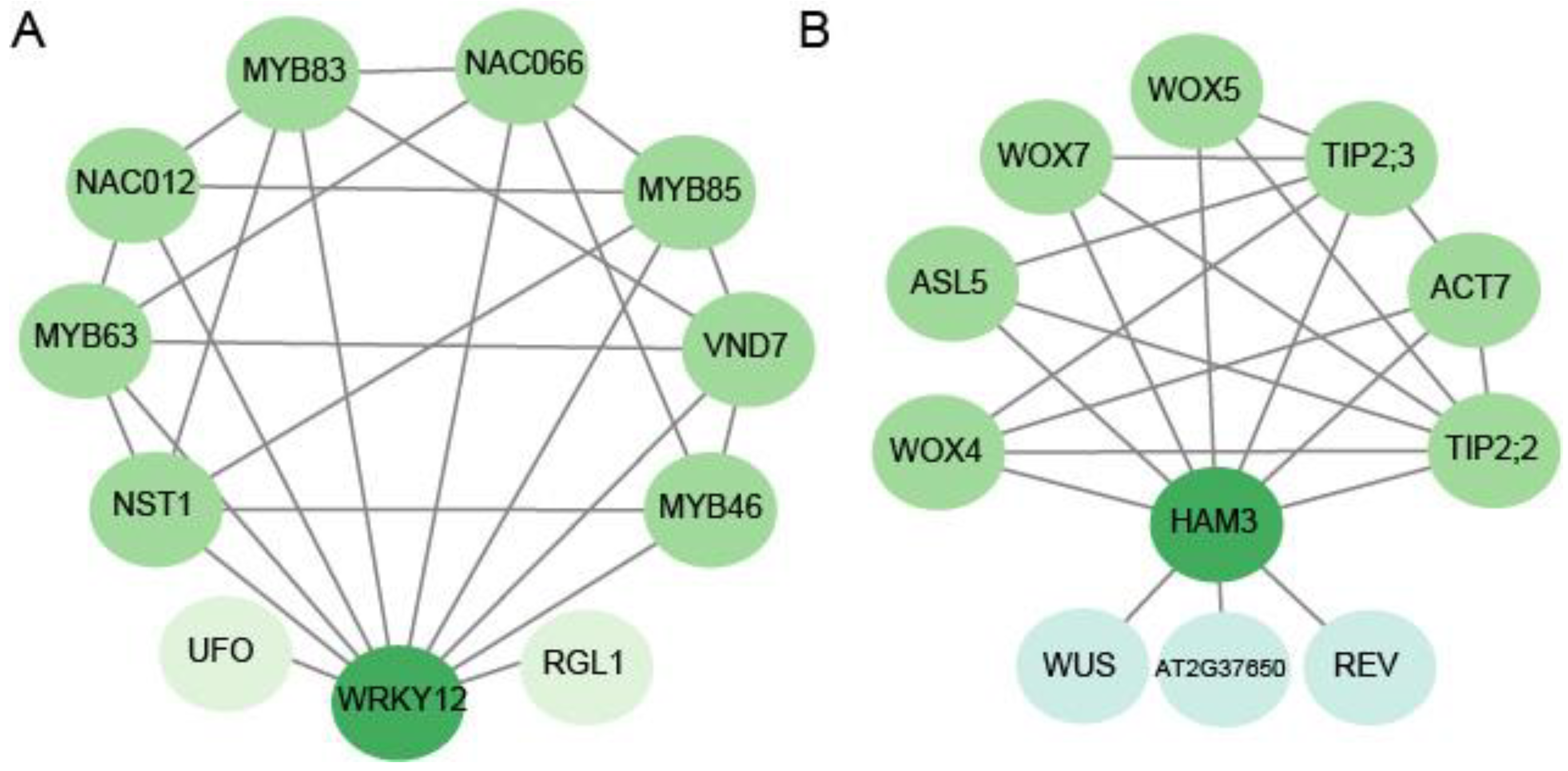
3.5. The Protein–Protein Interaction Analysis, the Cis-Elements, and the Predicted miRNA Targeting the Candidate Genes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Baytar, A.A.; Peynircioğlu, C.; Sezener, V.; Frary, A.; Doğanlar, S. Molecular mapping of QTLs for fiber quality traits in Gossypium hirsutum multi-parent recombinant inbred lines. Euphytica 2021, 217, 181. [Google Scholar] [CrossRef]
- Mehboob-ur-Rahman; Shaheen, T.; Tabbasam, N.; Iqbal, M.A.; Ashraf, M.; Zafar, Y.; Paterson, A.H. Cotton genetic resources. A review. Agron. Sustain. Dev. 2012, 32, 419–432. [Google Scholar]
- Imran, M.; Shakeel, A.; Azhar, F.M.; Farooq, J.; Saleem, M.F.; Saeed, A.; Nazeer, W.; Riaz, M.; Naeem, M.; Javaid, A. Combining ability analysis for within-boll yield components in upland cotton (Gossypium hirsutum L.). Genet. Mol. Res. 2012, 11, 2790–2800. [Google Scholar] [CrossRef]
- Su, J.; Wang, C.; Ma, Q.; Zhang, A.; Shi, C.; Liu, J.; Zhang, X.; Yang, D.; Ma, X. An RTM-GWAS procedure reveals the QTL alleles and candidate genes for three yield-related traits in upland cotton. BMC Plant Biol. 2020, 20, 416. [Google Scholar] [CrossRef]
- Song, C.; Li, W.; Pei, X.; Liu, Y.; Ren, Z.; He, K.; Zhang, F.; Sun, K.; Zhou, X.; Ma, X.; et al. Dissection of the genetic variation and candidate genes of lint percentage by a genome-wide association study in upland cotton. Theor. Appl. Genet. 2019, 132, 1991–2002. [Google Scholar] [CrossRef]
- Allen, E.; Xie, Z.; Gustafson, A.M.; Carrington, J.C. microRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell 2005, 121, 207–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdurakhmonov, I.Y.; Buriev, Z.T.; Saha, S.; Pepper, A.E.; Musaev, J.A.; Almatov, A.; Shermatov, S.E.; Kushanov, F.N.; Mavlonov, G.T.; Reddy, U.K.; et al. Microsatellite markers associated with lint percentage trait in cotton, Gossypium hirsutum. Euphytica 2007, 156, 141–156. [Google Scholar] [CrossRef]
- Sun, Z.; Wang, X.; Liu, Z.; Gu, Q.; Zhang, Y.; Li, Z.; Ke, H.; Yang, J.; Wu, J.; Wu, L.; et al. A genome-wide association study uncovers novel genomic regions and candidate genes of yield-related traits in upland cotton. Theor. Appl. Genet. 2018, 131, 2413–2425. [Google Scholar] [CrossRef]
- Endrizzi, J.E.; Turcotte, E.L.; Kohel, R.J. Genetics, Cytology, and evolution of Gossypium. In Advances in Genetics; Caspari, E.W., Scandalios, J.G., Eds.; Academic Press: Cambridge, MA, USA, 1985; Volume 23, pp. 271–375. [Google Scholar]
- Said, J.I.; Song, M.; Wang, H.; Lin, Z.; Zhang, X.; Fang, D.D.; Zhang, J. A comparative meta-analysis of QTL between intraspecific Gossypium hirsutum and interspecific G. hirsutum × G. barbadense populations. Mol. Genet. Genom. 2015, 290, 1003–1025. [Google Scholar] [CrossRef]
- Diouf, L.; Magwanga, R.O.; Gong, W.; He, S.; Pan, Z.; Jia, Y.H.; Kirungu, J.N.; Du, X. QTL Mapping of fiber quality and yield-related traits in an intra-specific Upland cotton using genotype by sequencing (GBS). Int. J. Mol. Sci. 2018, 19, 441. [Google Scholar] [CrossRef] [Green Version]
- Abdelraheem, A.; Fang, D.D.; Zhang, J.F. Quantitative trait locus mapping of drought and salt tolerance in an introgressed recombinant inbred line population of Upland cotton under the greenhouse and field conditions. Euphytica 2018, 214, 8. [Google Scholar] [CrossRef]
- Zhu, G.; Gao, W.; Song, X.; Sun, F.; Hou, S.; Liu, N.; Huang, Y.; Zhang, D.; Ni, Z.; Chen, Q.; et al. Genome-wide association reveals genetic variation of lint yield components under salty field conditions in cotton (Gossypium hirsutum L.). BMC Plant Biol. 2020, 20, 23. [Google Scholar] [CrossRef] [Green Version]
- Cavanagh, C.; Morell, M.; Mackay, I.; Powell, W. From mutations to MAGIC: Resources for gene discovery, validation and delivery in crop plants. Curr. Opin. Plant Biol. 2008, 11, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Nie, X.; Huang, C.; You, C.; Li, W.; Zhao, W.; Shen, C.; Zhang, B.; Wang, H.; Yan, Z.; Dai, B.; et al. Genome-wide SSR-based association mapping for fiber quality in nation-wide upland cotton inbreed cultivars in China. BMC Genom. 2016, 17, 352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, L.; Wang, Q.; Hu, Y.; Jia, Y.; Chen, J.; Liu, B.; Zhang, Z.; Guan, X.; Chen, S.; Zhou, B.; et al. Genomic analyses in cotton identify signatures of selection and loci associated with fiber quality and yield traits. Nat. Genet. 2017, 49, 1089–1098. [Google Scholar] [CrossRef]
- Huang, C.; Nie, X.; Shen, C.; You, C.; Li, W.; Zhao, W.; Zhang, X.; Lin, Z. Population structure and genetic basis of the agronomic traits of upland cotton in China revealed by a genome-wide association study using high-density SNPs. Plant Biotechnol. J. 2017, 15, 1374–1386. [Google Scholar] [CrossRef]
- Sun, Z.; Wang, X.; Liu, Z.; Gu, Q.; Zhang, Y.; Li, Z.; Ke, H.; Yang, J.; Wu, J.; Wu, L.; et al. Genome-wide association study discovered genetic variation and candidate genes of fibre quality traits in Gossypium hirsutum L. Plant Biotechnol. J. 2017, 15, 982–996. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; He, S.; Wang, X.; Sun, J.; Zhang, Y.; Zhang, G.; Wu, L.; Li, Z.; Liu, Z.; Sun, G.; et al. Resequencing a core collection of upland cotton identifies genomic variation and loci influencing fiber quality and yield. Nat. Genet. 2018, 50, 803–813. [Google Scholar] [CrossRef]
- Sarfraz, Z.; Iqbal, M.S.; Geng, X.; Iqbal, M.S.; Nazir, M.F.; Ahmed, H.; He, S.; Jia, Y.; Pan, Z.; Sun, G.; et al. GWAS mediated elucidation of heterosis for metric traits in cotton (Gossypium hirsutum L.) across multiple environments. Front. Plant Sci. 2021, 12, 565552. [Google Scholar] [CrossRef]
- Tyagi, P.; Gore, M.A.; Bowman, D.T.; Campbell, B.T.; Udall, J.A.; Kuraparthy, V. Genetic diversity and population structure in the US Upland cotton (Gossypium hirsutum L.). Theor. Appl. Genet. 2014, 127, 283–295. [Google Scholar] [CrossRef]
- Feng, L.; Zhang, S.; Xing, L.; Yang, B.; Gao, X.; Xie, X.; Zhou, B. QTL analysis for yield and fibre quality traits using three sets of introgression lines developed from three Gossypium hirsutum race stocks. Mol. Genet. Genom. 2019, 294, 789–810. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, T.; Liu, Q.; Gao, X.; Zhu, X.; Zhang, T.; Zhou, B. Quantitative trait locus analysis of boll-related traits in an intraspecific population of Gossypium hirsutum. Euphytica 2015, 203, 121–144. [Google Scholar] [CrossRef]
- Liu, D.; Liu, F.; Shan, X.; Zhang, J.; Tang, S.; Fang, X.; Liu, X.; Wang, W.; Tan, Z.; Teng, Z.; et al. Construction of a high-density genetic map and lint percentage and cottonseed nutrient trait QTL identification in upland cotton (Gossypium hirsutum L.). Mol. Genet. Genom. 2015, 290, 1683–1700. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Liu, X.; Su, Y.; Zhang, X.; Guo, K.; Teng, Z.; Zhang, J.; Liu, D.; Zhang, Z. Genetic mapping and identification of Lgf loci controlling green fuzz in Upland cotton (Gossypium hirsutum L.). Crop J. 2021, 9, 777–784. [Google Scholar] [CrossRef]
- Zhang, S.; Feng, L.; Xing, L.; Yang, B.; Gao, X.; Zhu, X.; Zhang, T.; Zhou, B. New QTLs for lint percentage and boll weight mined in introgression lines from two feral landraces into Gossypium hirsutum acc TM-1. Plant Breed. 2016, 135, 90–101. [Google Scholar] [CrossRef]
- Feng, L.; Zhou, C.; Su, Q.; Xu, M.; Yue, H.; Zhang, S.; Zhou, B. Fine-mapping and candidate gene analysis of qFS-Chr. D02, a QTL for fibre strength introgressed from a semi-wild cotton into Gossypium hirsutum. Plant Sci. 2020, 297, 110524. [Google Scholar] [CrossRef]
- Liu, X.; Yang, L.; Wang, J.; Wang, Y.; Guo, Z.; Li, Q.; Yang, J.; Wu, Y.; Chen, L.; Teng, Z.; et al. Analyzing quantitative trait Loci for fiber quality and yield-related traits from a recombinant inbred line population with Gossypium hirsutum race palmeri as one parent. Front. Plant Sci. 2021, 12, 817748. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Zhou, Z.L.; Wang, X.X.; Cai, X.Y.; Li, X.N.; Wang, C.Y.; Wang, Y.H.; Fang, L.; Wang, K.B. Genome-wide association mapping of glyphosate-resistance in Gossypium hirsutum races. Euphytica 2016, 209, 209–221. [Google Scholar] [CrossRef]
- Guo, X.; Wang, Y.; Hou, Y.; Zhou, Z.; Sun, R.; Qin, T.; Wang, K.; Liu, F.; Wang, Y.; Huang, Z.; et al. Genome-wide dissection of the genetic basis for drought tolerance in Gossypium hirsutum L. races. Front. Plant Sci. 2022, 13, 876095. [Google Scholar] [CrossRef]
- Xu, Y.; Magwanga, R.O.; Yang, X.; Jin, D.; Cai, X.; Hou, Y.; Wei, Y.; Zhou, Z.; Wang, K.; Liu, F. Genetic regulatory networks for salt-alkali stress in Gossypium hirsutum with differing morphological characteristics. BMC Genom. 2020, 21, 15. [Google Scholar] [CrossRef] [Green Version]
- Xing, H.; Yuan, Y.; Zhang, H.; Wang, L.; Mao, L.; Tao, J.; Wang, X.; Feng, W.; Wang, H.; Wang, Q.; et al. Multi-environments and multi-models association mapping identified candidate genes of lint percentage and seed index in Gossypium hirsutum L. Mol. Breed. 2019, 39, 149. [Google Scholar] [CrossRef]
- Qin, H.; Chen, M.; Yi, X.; Bie, S.; Zhang, C.; Zhang, Y.; Lan, J.; Meng, Y.; Yuan, Y.; Jiao, C. Identification of associated SSR markers for yield component and fiber quality traits based on frame map and Upland cotton collections. PLoS ONE 2015, 10, e0118073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, H.; Ge, Q.; Shang, H.; Yuan, Y. Inheritance, QTLs, and candidate genes of lint percentage in Upland cotton. Front. Genet. 2022, 13, 855574. [Google Scholar] [CrossRef] [PubMed]
- Said, J.I.; Lin, Z.; Zhang, X.; Song, M.; Zhang, J. A comprehensive meta QTL analysis for fiber quality, yield, yield related and morphological traits, drought tolerance, and disease resistance in tetraploid cotton. BMC Genom. 2013, 14, 776. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Hu, Y.; Jiang, W.; Fang, L.; Guan, X.; Chen, J.; Zhang, J.; Saski, C.A.; Scheffler, B.E.; Stelly, D.M.; et al. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 2015, 33, 531–537. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, M.D.; Chen-Plotkin, A.S. The post-GWAS era: From association to function. Am. J. Hum. Genet. 2018, 102, 717–730. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, T.; Guo, W. The im mutant gene negatively affects many aspects of fiber quality traits and lint percentage in cotton. Crop Sci. 2013, 53, 27–37. [Google Scholar] [CrossRef]
- Yang, P.; Sun, X.; Liu, X.; Wang, W.; Hao, Y.; Chen, L.; Liu, J.; He, H.; Zhang, T.; Bao, W.; et al. Identification of candidate genes for lint percentage and fiber quality through QTL mapping and transcriptome analysis in an allotetraploid interspecific cotton CSSLs population. Front. Plant Sci. 2022, 13, 882051. [Google Scholar] [CrossRef]
- Wang, H.; Jia, X.; Kang, M.; Li, W.; Fu, X.; Ma, L.; Lu, J.; Wei, H.; Yu, S. QTL mapping and candidate gene identification of lint percentage based on a recombinant inbred line population of upland cotton. Euphytica 2021, 217, 102. [Google Scholar] [CrossRef]
- Su, J.; Fan, S.; Li, L.; Wei, H.; Wang, C.; Wang, H.; Song, M.; Zhang, C.; Gu, L.; Zhao, S.; et al. Detection of favorable QTL alleles and candidate genes for lint percentage by GWAS in Chinese Upland cotton. Front. Plant Sci. 2016, 7, 1576. [Google Scholar] [CrossRef] [Green Version]
- Su, J.; Wang, C.; Hao, F.; Ma, Q.; Wang, J.; Li, J.; Ning, X. Genetic detection of lint percentage applying single-locus and multi-locus genome-wide association studies in Chinese early-maturity Upland cotton. Front. Plant Sci. 2019, 10, 964. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Andres, R.J.; Zhang, K.; Kuraparthy, V. High-density linkage map construction and QTL analysis of fiber quality and lint percentage in tetraploid cotton. Crop Sci. 2021, 61, 3340–3360. [Google Scholar] [CrossRef]
- Wang, H.; Zhao, Q.; Chen, F.; Wang, M.; Dixon, R.A. NAC domain function and transcriptional control of a secondary cell wall master switch. Plant J. 2011, 68, 1104–1114. [Google Scholar] [CrossRef]
- Sun, W.; Gao, Z.; Wang, J.; Huang, Y.; Chen, Y.; Li, J.; Lv, M.; Wang, J.; Luo, M.; Zuo, K. Cotton fiber elongation requires the transcription factor GhMYB212 to regulate sucrose transportation into expanding fibers. New Phytol. 2019, 222, 864–881. [Google Scholar] [CrossRef]
- Walford, S.A.; Wu, Y.; Llewellyn, D.J.; Dennis, E.S. GhMYB25-like: A key factor in early cotton fibre development. Plant J. 2011, 65, 785–797. [Google Scholar] [CrossRef]
- Machado, A.; Wu, Y.; Yang, Y.; Llewellyn, D.J.; Dennis, E.S. The MYB transcription factor GhMYB25 regulates early fibre and trichome development. Plant J. 2009, 59, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Chen, F.; Guo, Y.; Gan, X.; Yang, M.; Zeng, W.; Persson, S.; Li, J.; Xu, W. GhMYB7 promotes secondary wall cellulose deposition in cotton fibres by regulating GhCesA gene expression through three distinct cis-elements. New Phytol. 2021, 232, 1718–1737. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Luo, F.; Zhong, Y.; He, J.; Li, L. Modulation of NAC transcription factor NST1 activity by XYLEM NAC DOMAIN1 regulates secondary cell wall formation in Arabidopsis. J. Exp. Bot. 2020, 71, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Huang, G.Q.; Zou, D.; Yan, J.Q.; Li, Y.; Hu, S.; Li, X.B. The cotton (Gossypium hirsutum) NAC transcription factor (FSN1) as a positive regulator participates in controlling secondary cell wall biosynthesis and modification of fibers. New Phytol. 2018, 217, 625–640. [Google Scholar] [CrossRef] [Green Version]
- Zhong, R.; Lee, C.; Ye, Z.H. Global analysis of direct targets of secondary wall NAC master switches in Arabidopsis. Mol. Plant 2010, 3, 1087–1103. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, M.; Mitsuda, N.; Ohtani, M.; Ohme-Takagi, M.; Kato, K.; Demura, T. VASCULAR-RELATED NAC-DOMAIN7 directly regulates the expression of a broad range of genes for xylem vessel formation. Plant J. 2011, 66, 579–590. [Google Scholar] [CrossRef]
- Ohashi-Ito, K.; Iwamoto, K.; Fukuda, H. LOB DOMAIN-CONTAINING PROTEIN 15 positively regulates expression of VND7, a master regulator of tracheary elements. Plant Cell Physiol. 2018, 59, 989–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarthy, R.L.; Zhong, R.; Ye, Z.H. MYB83 is a direct target of SND1 and acts redundantly with MYB46 in the regulation of secondary cell wall biosynthesis in Arabidopsis. Plant Cell Physiol. 2009, 50, 1950–1964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, N.N.; Li, Y.; Chen, Y.H.; Lu, R.; Zhou, L.; Wang, Y.; Zheng, Y.; Li, X.B. Phosphorylation of WRKY16 by MPK3-1 is essential for its transcriptional activity during fiber initiation and elongation in cotton (Gossypium hirsutum). Plant Cell 2021, 33, 2736–2752. [Google Scholar] [CrossRef] [PubMed]
- Kandasamy, M.K.; McKinney, E.C.; Meagher, R.B. A single vegetative actin isovariant overexpressed under the control of multiple regulatory sequences is sufficient for normal Arabidopsis development. Plant Cell 2009, 21, 701–718. [Google Scholar] [CrossRef] [Green Version]
- Gilliland, L.U.; Pawloski, L.C.; Kandasamy, M.K.; Meagher, R.B. Arabidopsis actin gene ACT7 plays an essential role in germination and root growth. Plant J. 2003, 33, 319–328. [Google Scholar] [CrossRef]
- Ji, J.; Strable, J.; Shimizu, R.; Koenig, D.; Sinha, N.; Scanlon, M.J. WOX4 promotes procambial development. Plant Physiol. 2010, 152, 1346–1356. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Xu, L. Transcription factors WOX11/12 directly activate WOX5/7 to promote root primordia initiation and organogenesis. Plant Physiol. 2016, 172, 2363–2373. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Feng, Z.; Meng, L. Ectopic expression of the Arabidopsis ASYMMETRIC LEAVES2-LIKE5 (ASL5) gene in cockscomb (Celosia cristata) generates vascular-pattern modifications in lateral organs. Plant Cell Tissue Organ Cult. 2012, 110, 163–169. [Google Scholar] [CrossRef]
- Engstrom, E.M.; Andersen, C.M.; Gumulak-Smith, J.; Hu, J.; Orlova, E.; Sozzani, R.; Bowman, J.L. Arabidopsis homologs of the petunia hairy meristem gene are required for maintenance of shoot and root indeterminacy. Plant Physiol. 2011, 155, 735–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, R.; Li, C.; Zhang, J.; Li, F.; Ma, L.; Tan, Y.; Wang, Q.; Zhang, B. Differential expression of microRNAs during fiber development between fuzzless-lintless mutant and its wild-type allotetraploid cotton. Sci. Rep. 2017, 7, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, N.; Tu, L.; Tang, W.; Gao, W.; Lindsey, K.; Zhang, X. Small RNA and degradome profiling reveals a role for miRNAs and their targets in the developing fibers of Gossypium barbadense. Plant J. 2014, 80, 331–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bates, D.; Mächler, M.; Bolker, B.; Walker, S. Fitting linear mixed-effects models using lme4. J. Stat. Softw. 2015, 67, 1–48. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis, 2nd ed.; Spring: New York, NY, USA, 2018. [Google Scholar]
- Lipka, A.E.; Tian, F.; Wang, Q.; Peiffer, J.; Li, M.; Bradbury, P.J.; Gore, M.A.; Buckler, E.S.; Zhang, Z. GAPIT: Genome association and prediction integrated tool. Bioinformatics 2012, 28, 2397–2399. [Google Scholar] [CrossRef] [Green Version]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [Green Version]
- Artico, S.; Nardeli, S.M.; Brilhante, O.; Grossi-de-Sa, M.F.; Alves-Ferreira, M. Identification and evaluation of new reference genes in Gossypium hirsutum for accurate normalization of real-time quantitative RT-PCR data. BMC Plant Biol. 2010, 10, 49. [Google Scholar] [CrossRef] [Green Version]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Saeed, A.I.; Bhagabati, N.K.; Braisted, J.C.; Liang, W.; Sharov, V.; Howe, E.A.; Li, J.; Thiagarajan, M.; White, J.A.; Quackenbush, J. TM4 microarray software suite. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2006; Volume 411, pp. 134–193. [Google Scholar]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Lescot, M.; Dehais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouze, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yu, J.; Li, D.; Zhang, Z.; Liu, F.; Zhou, X.; Wang, T.; Ling, Y.; Su, Z. PMRD: Plant microRNA database. Nucleic Acids Res. 2010, 38, D806–D813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, X.; Zhuang, Z.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server (2017 release). Nucleic Acids Res. 2018, 46, W49–W54. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Env. | Max | Min | Mean | Std. | CV | Skewness | Kurtosis |
|---|---|---|---|---|---|---|---|
| E1 | 47.55 | 12.03 | 30.31 | 5.86 | 19.34 | 0.14 | −0.15 |
| E2 | 54.77 | 8.29 | 28 | 6.55 | 23.39 | 0.43 | 0.85 |
| E3 | 43.8 | 14.47 | 28.04 | 6.21 | 22.14 | 0.48 | −0.24 |
| E4 | 43.86 | 12.35 | 26.61 | 6.21 | 23.34 | 0.39 | −0.39 |
| E5 | 42.43 | 14.87 | 26.37 | 5.76 | 21.83 | 0.51 | −0.03 |
| SNP | Chr | Pos. (bp) | GAPIT | Tassel | ||||
|---|---|---|---|---|---|---|---|---|
| FaST-LMM | FarmCPU | BLINK | MLMM | MLM | TMLM | |||
| TM6245 | A3 | 6,000,457 | BLUP (4.90) | BLUP (4.90) | ||||
| TM6246 | A3 | 6,008,064 | BLUP (5.10) | BLUP (6.99) | BLUP (5.10) | |||
| TM6247 | A3 | 6,015,363 | BLUP (5.10) | BLUP (5.10) | ||||
| TM6248 | A3 | 6,020,227 | BLUP (4.90) | BLUP (4.90) | ||||
| TM8562 | A4 | 1,704,160 | E5 (6.37) | E5 (6.23) | ||||
| TM8787 | A4 | 5,125,340 | E1 (7.94) | E1 (10.43) | ||||
| TM10387 | A5 | 7,465,603 | E5 (4.71) | E5 (4.71) | ||||
| TM10953 | A5 | 21,122,062 | E3 (4.97) | E3 (9.45) | ||||
| TM13159 | A5 | 83,646,730 | E3 (6.75); E4 (6.30); E5 (6.66) | E3 (17.39); E4 (6.90); E5 (9.84) | ||||
| TM21439 | A7 | 73,820,001 | E2 (5.42) | E2 (5.42) | ||||
| TM22226 | A8 | 8,180,911 | E4 (15.51) | E4 (11.19); E5 (6.70) | ||||
| TM38921 | A11 | 65,498,934 | E2 (6.45) | E2 (7.92) | ||||
| TM39097 | A11 | 77,128,215 | E2 (5.41) | E2 (5.01) | ||||
| TM39105 | A11 | 77,162,385 | E2 (4.94) | E2 (5.23) | ||||
| TM39111 | A11 | 77,201,721 | E2 (4.85) | E2 (5.06) | ||||
| TM39118 | A11 | 77,245,152 | E2 (5.41) | E2 (4.73) | ||||
| TM39119 | A11 | 77,250,629 | E2 (6.57) | E2 (7.05) | E2 (4.99) | E2 (6.57) | E2 (5.48) | E2 (4.80) |
| TM39120 | A11 | 77,254,990 | E2 (5.35) | E2 (5.42) | ||||
| TM39121 | A11 | 77,259,287 | E2 (5.39) | E2 (4.72) | ||||
| TM39122 | A11 | 77,267,198 | E2 (5.33) | E2 (5.42) | ||||
| TM39136 | A11 | 77,356,582 | E3 (9.26); E5 (12.70) | E1 (5.59); E2 (4.84); E3 (14.49); E4 (5.33); E5 (13.96) | ||||
| TM42527 | A12 | 73,222,779 | E1 (5.91) | E1 (13.91) | ||||
| TM42563 | A12 | 73,856,986 | E2 (5.34) | E2 (7.14) | ||||
| TM46588 | A13 | 60,134,206 | E1 (6.86) | E1 (14.22) | ||||
| TM47373 | A13 | 72,835,782 | E3 (8.73) | E3 (7.06) | ||||
| TM52184 | D2 | 51,543,776 | E1 (4.89); E4 (10.43) | E4 (6.60) | ||||
| TM53197 | D2 | 64,206,361 | E1 (4.77) | E3 (6.30) | ||||
| TM53588 | D3 | 4,016,585 | E4 (15.33) | E1 (13.39); E4 (14.39); E5 (10.63) | ||||
| TM55461 | D4 | 2,087,134 | E2 (6.19) | E2 (9.29) | E2 (6.19) | E2 (5.23) | ||
| TM55484 | D4 | 2,902,882 | E5 (9.59) | E5 (13.00) | ||||
| TM55582 | D4 | 3,824,554 | E2 (6.80) | E2 (6.86) | ||||
| TM55817 | D4 | 7,622,861 | E2 (5.35) | E2 (5.35) | ||||
| TM55819 | D4 | 7,631,258 | E2 (5.35) | E2 (5.35) | ||||
| TM57002 | D5 | 6,810,289 | E1 (4.85); E3 (5.82) | |||||
| TM57478 | D5 | 19,820,229 | E4 (4.95) | E4 (22.33); E5 (4.79) | E4 (12.47) | E4 (4.95) | E4 (4.76) | |
| TM57486 | D5 | 20,068,395 | E2 (5.45) | E3 (4.87) | ||||
| TM58314 | D5 | 49,232,321 | E1 (4.90); E3 (4.77) | |||||
| TM69172 | D8 | 53,049,470 | E3 (13.74) | E3 (8.17) | ||||
| TM72587 | D9 | 43,752,600 | E3 (7.45) | E3 (7.83) | ||||
| TM73642 | D10 | 8,030,303 | E3 (5.42) | E3 (6.93) | ||||
| TM75017 | D10 | 58,300,092 | E4 (4.72) | E4 (13.12) | ||||
| TM76204 | D11 | 24,111,532 | E3 (8.00) | E3 (14.52) | ||||
| TM78526 | D12 | 34,893,826 | E2 (4.84) | E3 (5.16) | E2 (4.84) | |||
| TM80159 | D13 | 2,590,468 | E5 (5.39) | E5 (9.39) | ||||
| TM81325 | D13 | 42,144,468 | E1 (7.99); E2 (9.96) | E1 (6.54) | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Guo, X.; Cai, X.; Xu, Y.; Sun, R.; Umer, M.J.; Wang, K.; Qin, T.; Hou, Y.; Wang, Y.; et al. Genome-Wide Association Study of Lint Percentage in Gossypium hirsutum L. Races. Int. J. Mol. Sci. 2023, 24, 10404. https://doi.org/10.3390/ijms241210404
Wang Y, Guo X, Cai X, Xu Y, Sun R, Umer MJ, Wang K, Qin T, Hou Y, Wang Y, et al. Genome-Wide Association Study of Lint Percentage in Gossypium hirsutum L. Races. International Journal of Molecular Sciences. 2023; 24(12):10404. https://doi.org/10.3390/ijms241210404
Chicago/Turabian StyleWang, Yuanyuan, Xinlei Guo, Xiaoyan Cai, Yanchao Xu, Runrun Sun, Muhammad Jawad Umer, Kunbo Wang, Tengfei Qin, Yuqing Hou, Yuhong Wang, and et al. 2023. "Genome-Wide Association Study of Lint Percentage in Gossypium hirsutum L. Races" International Journal of Molecular Sciences 24, no. 12: 10404. https://doi.org/10.3390/ijms241210404
APA StyleWang, Y., Guo, X., Cai, X., Xu, Y., Sun, R., Umer, M. J., Wang, K., Qin, T., Hou, Y., Wang, Y., Zhang, P., Wang, Z., Liu, F., Wang, Q., & Zhou, Z. (2023). Genome-Wide Association Study of Lint Percentage in Gossypium hirsutum L. Races. International Journal of Molecular Sciences, 24(12), 10404. https://doi.org/10.3390/ijms241210404