
In Silico Identification of a BRCA1:miR-29:DNMT3 Axis Involved in the Control of Hormone Receptors in BRCA1-Associated Breast Cancers


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
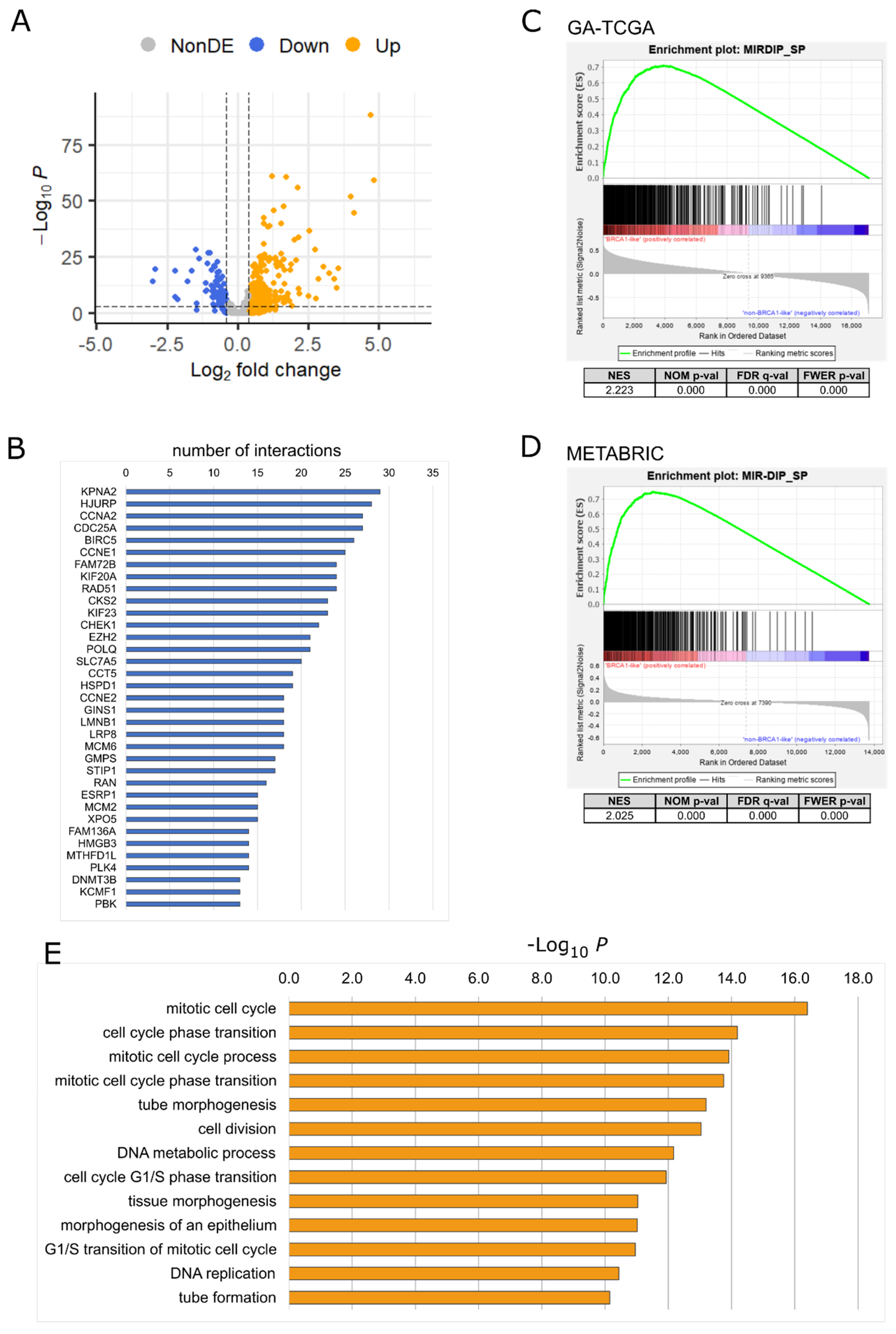
2.1. Downregulated miRNAs in BRCA1-like Tumors
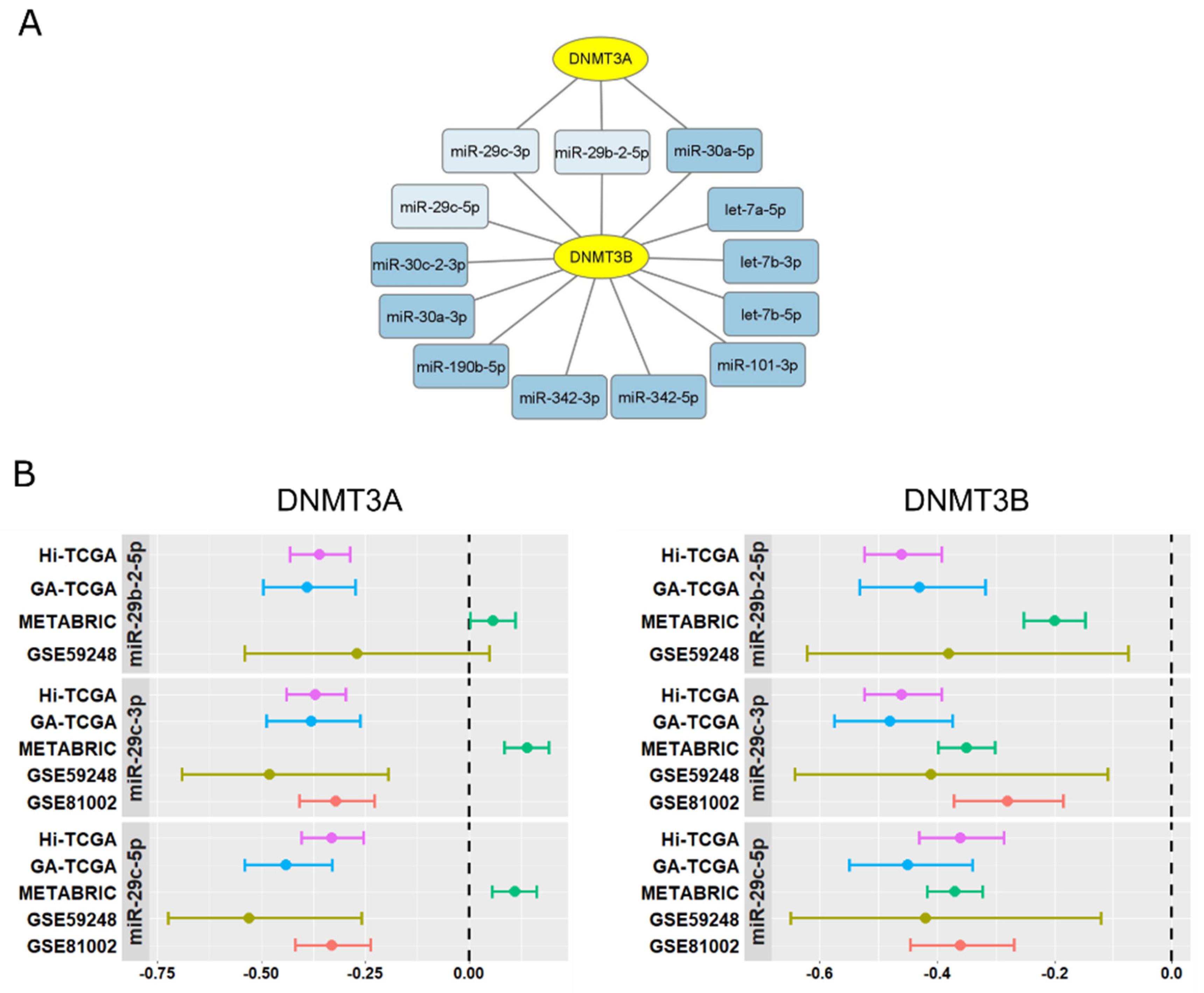
2.2. miR-29s:DNMT3A-DNMT3B Network Is Involved in BRCA1-like Tumors
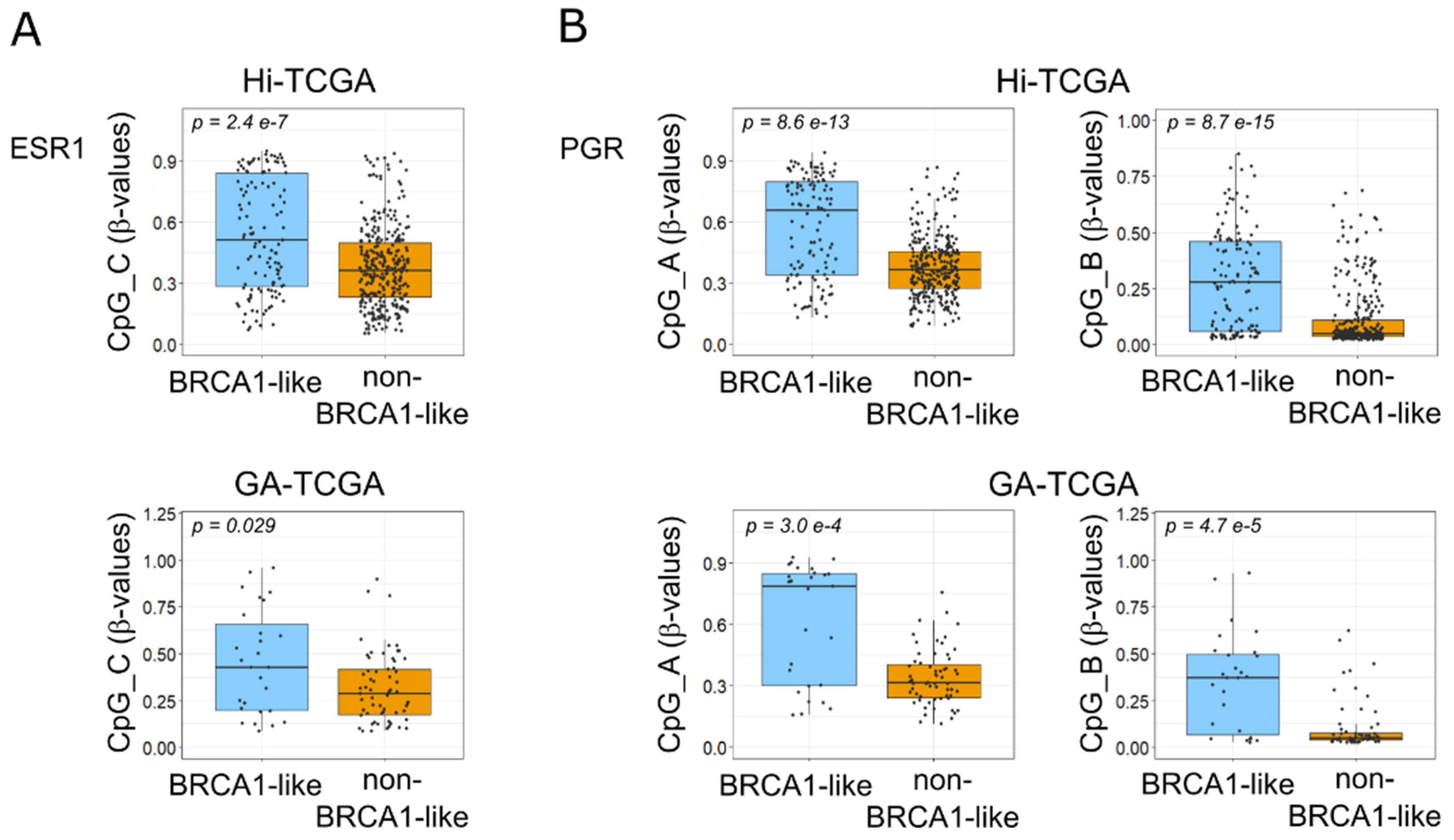
2.3. DNA Methyltransferases Affect ESR1 and PGR DNA Methylation in BRCA1-like Samples
3. Discussion
4. Materials and Methods
4.1. Data Sets
4.1.1. TCGA
4.1.2. METABRIC
4.1.3. Other Validation Data Sets
4.2. Differential Expression Analysis
4.3. GSEA Analyses and Gene Ontology (GO) Enrichment Analysis
4.4. Disease-Free Survival and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.-A.; Mooij, T.M.; Roos-Blom, M.-J.; Jervis, S.; van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. J. Am. Med. Assoc. 2017, 317, 2402–2416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santarosa, M.; Maestro, R. BRACking News on Triple-Negative/Basal-like Breast Cancers: How BRCA1 Deficiency May Result in the Development of a Selective Tumor Subtype. Cancer Metastasis Rev. 2012, 31, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Stevens, K.N.; Vachon, C.M.; Couch, F.J. Genetic Susceptibility to Triple-Negative Breast Cancer. Cancer Res. 2013, 73, 2025–2030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tung, N.M.; Garber, J.E. BRCA1/2 Testing: Therapeutic Implications for Breast Cancer Management. Br. J. Cancer 2018, 119, 141–152. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, Y.; Salas, L.A.; Miller, T.W.; Mark, K.; Marotti, J.D.; Kettenbach, A.N.; Cheng, C.; Christensen, B.C. Molecular and Epigenetic Profiles of BRCA1-like Hormone-Receptor-Positive Breast Tumors Identified with Development and Application of a Copy-Number-Based Classifier. Breast Cancer Res. 2019, 21, 14. [Google Scholar] [CrossRef] [Green Version]
- Glodzik, D.; Bosch, A.; Hartman, J.; Aine, M.; Vallon-Christersson, J.; Reuterswärd, C.; Karlsson, A.; Mitra, S.; Niméus, E.; Holm, K.; et al. Comprehensive Molecular Comparison of BRCA1 Hypermethylated and BRCA1 Mutated Triple Negative Breast Cancers. Nat. Commun. 2020, 11, 3747. [Google Scholar] [CrossRef]
- Catteau, A.; Harris, W.H.; Xu, C.F.; Solomon, E. Methylation of the BRCA1 Promoter Region in Sporadic Breast and Ovarian Cancer: Correlation with Disease Characteristics. Oncogene 1999, 18, 1957–1965. [Google Scholar] [CrossRef] [Green Version]
- Esteller, M.; Silva, J.M.; Dominguez, G.; Bonilla, F.; Matias-Guiu, X.; Lerma, E.; Bussaglia, E.; Prat, J.; Harkes, I.C.; Repasky, E.A.; et al. Promoter Hypermethylation and BRCA1 Inactivation in Sporadic Breast and Ovarian Tumors. J. Natl. Cancer Inst. 2000, 92, 564–569. [Google Scholar] [CrossRef] [Green Version]
- Rice, J.C.; Ozcelik, H.; Maxeiner, P.; Andrulis, I.; Futscher, B.W. Methylation of the BRCA1 Promoter Is Associated with Decreased BRCA1 MRNA Levels in Clinical Breast Cancer Specimens. Carcinogenesis 2000, 21, 1761–1765. [Google Scholar] [CrossRef] [Green Version]
- Grushko, T.A.; Dignam, J.J.; Das, S.; Blackwood, A.M.; Perou, C.M.; Ridderstråle, K.K.; Anderson, K.N.; Wei, M.-J.; Adams, A.J.; Hagos, F.G.; et al. MYC Is Amplified in BRCA1-Associated Breast Cancers. Clin. Cancer Res. 2004, 10, 499–507. [Google Scholar] [CrossRef] [Green Version]
- Turner, N.; Tutt, A.; Ashworth, A. Hallmarks of “BRCAness” in Sporadic Cancers. Nat. Rev. Cancer 2004, 4, 814–819. [Google Scholar] [CrossRef]
- Wessels, L.F.A.; van Welsem, T.; Hart, A.A.M.; van’t Veer, L.J.; Reinders, M.J.T.; Nederlof, P.M. Molecular Classification of Breast Carcinomas by Comparative Genomic Hybridization: A Specific Somatic Genetic Profile for BRCA1 Tumors. Cancer Res. 2002, 62, 7110–7117. [Google Scholar]
- Lord, C.J.; Ashworth, A. The DNA Damage Response and Cancer Therapy. Nature 2012, 481, 287–294. [Google Scholar] [CrossRef]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different Roles in a Common Pathway of Genome Protection. Nat. Rev. Cancer 2011, 12, 68–78. [Google Scholar] [CrossRef] [Green Version]
- Treszezamsky, A.D.; Kachnic, L.A.; Feng, Z.; Zhang, J.; Tokadjian, C.; Powell, S.N. BRCA1- and BRCA2-Deficient Cells Are Sensitive to Etoposide-Induced DNA Double-Strand Breaks via Topoisomerase II. Cancer Res. 2007, 67, 7078–7081. [Google Scholar] [CrossRef] [Green Version]
- Santarosa, M.; Del Col, L.; Tonin, E.; Caragnano, A.; Viel, A.; Maestro, R. Premature Senescence Is a Major Response to DNA Cross-Linking Agents in BRCA1-Defective Cells: Implication for Tailored Treatments of BRCA1 Mutation Carriers. Mol. Cancer Ther. 2009, 8, 844–854. [Google Scholar] [CrossRef] [Green Version]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA Repair Defect in BRCA Mutant Cells as a Therapeutic Strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Santarosa, M.; Del Col, L.; Viel, A.; Bivi, N.; D’Ambrosio, C.; Scaloni, A.; Tell, G.; Maestro, R. BRCA1 Modulates the Expression of HnRNPA2B1 and KHSRP. Cell Cycle 2010, 9, 4666–4673. [Google Scholar] [CrossRef] [Green Version]
- Savage, K.I.; Gorski, J.J.; Barros, E.M.; Irwin, G.W.; Manti, L.; Powell, A.J.; Pellagatti, A.; Lukashchuk, N.; McCance, D.J.; McCluggage, W.G.; et al. Identification of a BRCA1-MRNA Splicing Complex Required for Efficient DNA Repair and Maintenance of Genomic Stability. Mol. Cell 2014, 54, 445–459. [Google Scholar] [CrossRef] [Green Version]
- Kawai, S.; Amano, A. BRCA1 Regulates MicroRNA Biogenesis via the DROSHA Microprocessor Complex. J. Cell Biol. 2012, 197, 201–208. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.; Gregory, R.I. MicroRNA Biogenesis Pathways in Cancer. Nat. Rev. Cancer 2015, 15, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Chipman, L.B.; Pasquinelli, A.E. MiRNA Targeting: Growing beyond the Seed. Trends Genet. 2019, 35, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Wang, R.-H.; Akagi, K.; Kim, K.-A.; Martin, B.K.; Cavallone, L.; Kathleen Cuningham Foundation Consortium for Research into Familial Breast Cancer (kConFab); Haines, D.C.; Basik, M.; Mai, P.; et al. Tumor Suppressor BRCA1 Epigenetically Controls Oncogenic MicroRNA-155. Nat. Med. 2011, 17, 1275–1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, S.; Sharan, S.K. BRCA1 and MicroRNAs: Emerging Networks and Potential Therapeutic Targets. Mol. Cells 2012, 34, 425–432. [Google Scholar] [CrossRef] [Green Version]
- Kumaraswamy, E.; Wendt, K.L.; Augustine, L.A.; Stecklein, S.R.; Sibala, E.C.; Li, D.; Gunewardena, S.; Jensen, R.A. BRCA1 Regulation of Epidermal Growth Factor Receptor (EGFR) Expression in Human Breast Cancer Cells Involves MicroRNA-146a and Is Critical for Its Tumor Suppressor Function. Oncogene 2015, 34, 4333–4346. [Google Scholar] [CrossRef] [Green Version]
- Tanić, M.; Yanowski, K.; Andrés, E.; Gómez-López, G.; Socorro, M.R.-P.; Pisano, D.G.; Martinez-Delgado, B.; Benítez, J. MiRNA Expression Profiling of Formalin-Fixed Paraffin-Embedded (FFPE) Hereditary Breast Tumors. Genom. Data 2015, 3, 75–79. [Google Scholar] [CrossRef] [Green Version]
- Vos, S.; Vesuna, F.; Raman, V.; van Diest, P.J.; van der Groep, P. MiRNA Expression Patterns in Normal Breast Tissue and Invasive Breast Cancers of BRCA1 and BRCA2 Germ-Line Mutation Carriers. Oncotarget 2015, 6, 32115–32137. [Google Scholar] [CrossRef] [Green Version]
- Chu, A.; Robertson, G.; Brooks, D.; Mungall, A.J.; Birol, I.; Coope, R.; Ma, Y.; Jones, S.; Marra, M.A. Large-Scale Profiling of MicroRNAs for The Cancer Genome Atlas. Nucleic Acids Res. 2016, 44, e3. [Google Scholar] [CrossRef]
- Tokar, T.; Pastrello, C.; Rossos, A.E.M.; Abovsky, M.; Hauschild, A.-C.; Tsay, M.; Lu, R.; Jurisica, I. MirDIP 4.1-Integrative Database of Human MicroRNA Target Predictions. Nucleic Acids Res. 2018, 46, D360–D370. [Google Scholar] [CrossRef] [Green Version]
- Dvinge, H.; Git, A.; Gräf, S.; Salmon-Divon, M.; Curtis, C.; Sottoriva, A.; Zhao, Y.; Hirst, M.; Armisen, J.; Miska, E.A.; et al. The Shaping and Functional Consequences of the MicroRNA Landscape in Breast Cancer. Nature 2013, 497, 378–382. [Google Scholar] [CrossRef]
- Edwards, J.R.; Yarychkivska, O.; Boulard, M.; Bestor, T.H. DNA Methylation and DNA Methyltransferases. Epigenetics Chromatin 2017, 10, 23. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Yang, C.; Wu, C.; Cui, W.; Wang, L. DNA Methyltransferases in Cancer: Biology, Paradox, Aberrations, and Targeted Therapy. Cancers 2020, 12, 2123. [Google Scholar] [CrossRef]
- Man, X.; Li, Q.; Wang, B.; Zhang, H.; Zhang, S.; Li, Z. DNMT3A and DNMT3B in Breast Tumorigenesis and Potential Therapy. Front. Cell Dev. Biol. 2022, 10, 916725. [Google Scholar] [CrossRef]
- Lesurf, R.; Aure, M.R.; Mørk, H.H.; Vitelli, V.; Oslo Breast Cancer Research Consortium (OSBREAC); Lundgren, S.; Børresen-Dale, A.-L.; Kristensen, V.; Wärnberg, F.; Hallett, M.; et al. Molecular Features of Subtype-Specific Progression from Ductal Carcinoma In Situ to Invasive Breast Cancer. Cell Rep. 2016, 16, 1166–1179. [Google Scholar] [CrossRef] [Green Version]
- Aure, M.R.; Vitelli, V.; Jernström, S.; Kumar, S.; Krohn, M.; Due, E.U.; Haukaas, T.H.; Leivonen, S.-K.; Vollan, H.K.M.; Lüders, T.; et al. Integrative Clustering Reveals a Novel Split in the Luminal A Subtype of Breast Cancer with Impact on Outcome. Breast Cancer Res. 2017, 19, 44. [Google Scholar] [CrossRef] [Green Version]
- Lakhani, S.R. The Pathology of Familial Breast Cancer: Predictive Value of Immunohistochemical Markers Estrogen Receptor, Progesterone Receptor, HER-2, and P53 in Patients With Mutations in BRCA1 and BRCA2. J. Clin. Oncol. 2002, 20, 2310–2318. [Google Scholar] [CrossRef] [Green Version]
- Lapidus, R.G.; Ferguson, A.T.; Ottaviano, Y.L.; Parl, F.F.; Smith, H.S.; Weitzman, S.A.; Baylin, S.B.; Issa, J.P.; Davidson, N.E. Methylation of Estrogen and Progesterone Receptor Gene 5′ CpG Islands Correlates with Lack of Estrogen and Progesterone Receptor Gene Expression in Breast Tumors. Clin. Cancer Res. 1996, 2, 805–810. [Google Scholar]
- Gaudet, M.M.; Campan, M.; Figueroa, J.D.; Yang, X.R.; Lissowska, J.; Peplonska, B.; Brinton, L.A.; Rimm, D.L.; Laird, P.W.; Garcia-Closas, M.; et al. DNA Hypermethylation of ESR1 and PGR in Breast Cancer: Pathologic and Epidemiologic Associations. Cancer Epidemiol. Biomark. Prev. 2009, 18, 3036–3043. [Google Scholar] [CrossRef] [Green Version]
- Su, R.-W.; Strug, M.R.; Jeong, J.-W.; Miele, L.; Fazleabas, A.T. Aberrant Activation of Canonical Notch1 Signaling in the Mouse Uterus Decreases Progesterone Receptor by Hypermethylation and Leads to Infertility. Proc. Natl. Acad. Sci. USA 2016, 113, 2300–2305. [Google Scholar] [CrossRef] [Green Version]
- Kirn, V.; Strake, L.; Thangarajah, F.; Richters, L.; Eischeid, H.; Koitzsch, U.; Odenthal, M.; Fries, J. ESR1-Promoter-Methylation Status in Primary Breast Cancer and Its Corresponding Metastases. Clin. Exp. Metastasis 2018, 35, 707–712. [Google Scholar] [CrossRef]
- Kobayashi, M.; Ishii, H.; Sakuma, Y. Identification of Novel Splicing Events and Post-Transcriptional Regulation of Human Estrogen Receptor α F Isoforms. Mol. Cell. Endocrinol. 2011, 333, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Kastner, P.; Krust, A.; Turcotte, B.; Stropp, U.; Tora, L.; Gronemeyer, H.; Chambon, P. Two Distinct Estrogen-Regulated Promoters Generate Transcripts Encoding the Two Functionally Different Human Progesterone Receptor Forms A and B. EMBO J. 1990, 9, 1603–1614. [Google Scholar] [CrossRef] [PubMed]
- Stoppa-Lyonnet, D. The Biological Effects and Clinical Implications of BRCA Mutations: Where Do We Go from Here? Eur. J. Hum. Genet. 2016, 24 (Suppl. 1), S3–S9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, S.; Ma, Y.X.; Wang, C.; Yuan, R.Q.; Meng, Q.; Wang, J.A.; Erdos, M.; Goldberg, I.D.; Webb, P.; Kushner, P.J.; et al. Role of Direct Interaction in BRCA1 Inhibition of Estrogen Receptor Activity. Oncogene 2001, 20, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Rosen, E.M.; Fan, S.; Pestell, R.G.; Goldberg, I.D. BRCA1 in Hormone-Responsive Cancers. Trends Endocrinol. Metab. 2003, 14, 378–385. [Google Scholar] [CrossRef]
- Hosey, A.M.; Gorski, J.J.; Murray, M.M.; Quinn, J.E.; Chung, W.Y.; Stewart, G.E.; James, C.R.; Farragher, S.M.; Mulligan, J.M.; Scott, A.N.; et al. Molecular Basis for Estrogen Receptor Alpha Deficiency in BRCA1-Linked Breast Cancer. J. Natl. Cancer Inst. 2007, 99, 1683–1694. [Google Scholar] [CrossRef] [Green Version]
- Harte, M.T.; O’Brien, G.J.; Ryan, N.M.; Gorski, J.J.; Savage, K.I.; Crawford, N.T.; Mullan, P.B.; Harkin, D.P. BRD7, a Subunit of SWI/SNF Complexes, Binds Directly to BRCA1 and Regulates BRCA1-Dependent Transcription. Cancer Res. 2010, 70, 2538–2547. [Google Scholar] [CrossRef] [Green Version]
- Chiang, H.-C.; Zhang, X.; Li, J.; Zhao, X.; Chen, J.; Wang, H.T.-H.; Jatoi, I.; Brenner, A.; Hu, Y.; Li, R. BRCA1-Associated R-Loop Affects Transcription and Differentiation in Breast Luminal Epithelial Cells. Nucleic Acids Res. 2019, 47, 5086–5099. [Google Scholar] [CrossRef] [Green Version]
- Bonéy-Montoya, J.; Ziegler, Y.S.; Curtis, C.D.; Montoya, J.A.; Nardulli, A.M. Long-Range Transcriptional Control of Progesterone Receptor Gene Expression. Mol. Endocrinol. 2010, 24, 346–358. [Google Scholar] [CrossRef]
- Ma, Y.; Katiyar, P.; Jones, L.P.; Fan, S.; Zhang, Y.; Furth, P.A.; Rosen, E.M. The Breast Cancer Susceptibility Gene BRCA1 Regulates Progesterone Receptor Signaling in Mammary Epithelial Cells. Mol. Endocrinol. 2006, 20, 14–34. [Google Scholar] [CrossRef] [Green Version]
- Fabbri, M.; Garzon, R.; Cimmino, A.; Liu, Z.; Zanesi, N.; Callegari, E.; Liu, S.; Alder, H.; Costinean, S.; Fernandez-Cymering, C.; et al. MicroRNA-29 Family Reverts Aberrant Methylation in Lung Cancer by Targeting DNA Methyltransferases 3A and 3B. Proc. Natl. Acad. Sci. USA 2007, 104, 15805–15810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Yi, J.; Zheng, X.; Liu, S.; Fu, W.; Ren, L.; Li, L.; Hoon, D.S.B.; Wang, J.; Du, G. MiR-29c Plays a Suppressive Role in Breast Cancer by Targeting the TIMP3/STAT1/FOXO1 Pathway. Clin. Epigenetics 2018, 10, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milevskiy, M.J.G.; Sandhu, G.K.; Wronski, A.; Korbie, D.; Brewster, B.L.; Shewan, A.; Edwards, S.L.; French, J.D.; Brown, M.A. MiR-29b-1-5p Is Altered in BRCA1 Mutant Tumours and Is a Biomarker in Basal-like Breast Cancer. Oncotarget 2018, 9, 33577–33588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nygren, M.K.; Tekle, C.; Ingebrigtsen, V.A.; Mäkelä, R.; Krohn, M.; Aure, M.R.; Nunes-Xavier, C.E.; Perälä, M.; Tramm, T.; Alsner, J.; et al. Identifying MicroRNAs Regulating B7-H3 in Breast Cancer: The Clinical Impact of MicroRNA-29c. Br. J. Cancer 2014, 110, 2072–2080. [Google Scholar] [CrossRef] [Green Version]
- Tokumaru, Y.; Oshi, M.; Huyser, M.R.; Yan, L.; Fukada, M.; Matsuhashi, N.; Futamura, M.; Akao, Y.; Yoshida, K.; Takabe, K. Low Expression of MiR-29a Is Associated with Aggressive Biology and Worse Survival in Gastric Cancer. Sci. Rep. 2021, 11, 14134. [Google Scholar] [CrossRef]
- Pan, D.; Du, Y.; Li, R.; Shen, A.; Liu, X.; Li, C.; Hu, B. MiR-29b-3p Increases Radiosensitivity in Stemness Cancer Cells via Modulating Oncogenes Axis. Front. Cell Dev. Biol. 2021, 9, 741074. [Google Scholar] [CrossRef]
- Girault, I.; Tozlu, S.; Lidereau, R.; Bièche, I. Expression Analysis of DNA Methyltransferases 1, 3A, and 3B in Sporadic Breast Carcinomas. Clin. Cancer Res. 2003, 9, 4415–4422. [Google Scholar]
- Yu, Z.; Xiao, Q.; Zhao, L.; Ren, J.; Bai, X.; Sun, M.; Wu, H.; Liu, X.; Song, Z.; Yan, Y.; et al. DNA Methyltransferase 1/3a Overexpression in Sporadic Breast Cancer Is Associated with Reduced Expression of Estrogen Receptor-Alpha/Breast Cancer Susceptibility Gene 1 and Poor Prognosis. Mol. Carcinog. 2015, 54, 707–719. [Google Scholar] [CrossRef]
- Jahangiri, R.; Jamialahmadi, K.; Gharib, M.; Emami Razavi, A.; Mosaffa, F. Expression and Clinicopathological Significance of DNA Methyltransferase 1, 3A and 3B in Tamoxifen-Treated Breast Cancer Patients. Gene 2019, 685, 24–31. [Google Scholar] [CrossRef]
- Jung, Y.-D.; Park, S.-K.; Kang, D.; Hwang, S.; Kang, M.-H.; Hong, S.-W.; Moon, J.-H.; Shin, J.-S.; Jin, D.-H.; You, D.; et al. Epigenetic Regulation of MiR-29a/MiR-30c/DNMT3A Axis Controls SOD2 and Mitochondrial Oxidative Stress in Human Mesenchymal Stem Cells. Redox Biol. 2020, 37, 101716. [Google Scholar] [CrossRef]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA Methyltransferases Dnmt3a and Dnmt3b Are Essential for de Novo Methylation and Mammalian Development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Lyko, F. The DNA Methyltransferase Family: A Versatile Toolkit for Epigenetic Regulation. Nat. Rev. Genet. 2018, 19, 81–92. [Google Scholar] [CrossRef]
- Poli, E.; Zhang, J.; Nwachukwu, C.; Zheng, Y.; Adedokun, B.; Olopade, O.I.; Han, Y.-J. Molecular Subtype-Specific Expression of MicroRNA-29c in Breast Cancer Is Associated with CpG Dinucleotide Methylation of the Promoter. PLoS ONE 2015, 10, e0142224. [Google Scholar] [CrossRef] [Green Version]
- Chantalat, E.; Boudou, F.; Laurell, H.; Palierne, G.; Houtman, R.; Melchers, D.; Rochaix, P.; Filleron, T.; Stella, A.; Burlet-Schiltz, O.; et al. The AF-1-Deficient Estrogen Receptor ER\alpha46 Isoform Is Frequently Expressed in Human Breast Tumors. Breast Cancer Res. 2016, 18, 123. [Google Scholar] [CrossRef] [Green Version]
- Konan, H.-P.; Kassem, L.; Omarjee, S.; Surmieliova-Garnès, A.; Jacquemetton, J.; Cascales, E.; Rezza, A.; Trédan, O.; Treilleux, I.; Poulard, C.; et al. ER\alpha-36 Regulates Progesterone Receptor Activity in Breast Cancer. Breast Cancer Res. 2020, 22, 50. [Google Scholar] [CrossRef]
- Pagano, M.T.; Ortona, E.; Dupuis, M.L. A Role for Estrogen Receptor Alpha36 in Cancer Progression. Front. Endocrinol. 2020, 11, 506. [Google Scholar] [CrossRef]
- Clusan, L.; Ferrière, F.; Flouriot, G.; Pakdel, F. A Basic Review on Estrogen Receptor Signaling Pathways in Breast Cancer. Int. J. Mol. Sci. 2023, 24, 6834. [Google Scholar] [CrossRef]
- Anastasiadi, D.; Esteve-Codina, A.; Piferrer, F. Consistent Inverse Correlation between DNA Methylation of the First Intron and Gene Expression across Tissues and Species. Epigenetics Chromatin 2018, 11, 37. [Google Scholar] [CrossRef]
- Lea, A.J.; Vockley, C.M.; Johnston, R.A.; Del Carpio, C.A.; Barreiro, L.B.; Reddy, T.E.; Tung, J. Genome-Wide Quantification of the Effects of DNA Methylation on Human Gene Regulation. eLife 2018, 7, e37513. [Google Scholar] [CrossRef]
- Archey, W.B.; McEachern, K.A.; Robson, M.; Offit, K.; Vaziri, S.A.J.; Casey, G.; Borg, A.; Arrick, B.A. Increased CpG Methylation of the Estrogen Receptor Gene in BRCA1-Linked Estrogen Receptor-Negative Breast Cancers. Oncogene 2002, 21, 7034–7041. [Google Scholar] [CrossRef] [Green Version]
- Pathiraja, T.N.; Shetty, P.B.; Jelinek, J.; He, R.; Hartmaier, R.; Margossian, A.L.; Hilsenbeck, S.G.; Issa, J.-P.J.; Oesterreich, S. Progesterone Receptor Isoform-Specific Promoter Methylation: Association of PRA Promoter Methylation with Worse Outcome in Breast Cancer Patients. Clin. Cancer Res. 2011, 17, 4177–4186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curtis, C.; Shah, S.P.; Chin, S.-F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The Genomic and Transcriptomic Architecture of 2000 Breast Tumours Reveals Novel Subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.A.; Cai, C.; Langfelder, P.; Geschwind, D.H.; Kurian, S.M.; Salomon, D.R.; Horvath, S. Strategies for Aggregating Gene Expression Data: The CollapseRows R Function. BMC Bioinform. 2011, 12, 322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.-F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1alpha-Responsive Genes Involved in Oxidative Phosphorylation Are Coordinately Downregulated in Human Diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape Provides a Biologist-Oriented Resource for the Analysis of Systems-Level Datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef] [Green Version]
- Kassambara, A.; Kosinski, M.; Biecek, P.; Fabian, S. Package ‘Survminer’ v0.4.9. Available online: https://cran.r-project.org/web/packages/survminer/survminer.pdf (accessed on 10 May 2023).
- Therneau, T.M.; Lumley, T.; Elizabeth, A.; Cynthia, C. Package ‘Survival’ v3.5-5. Available online: https://cran.r-project.org/web/packages/survival/survival.pdf (accessed on 10 May 2023).








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santarosa, M.; Baldazzi, D.; Armellin, M.; Maestro, R. In Silico Identification of a BRCA1:miR-29:DNMT3 Axis Involved in the Control of Hormone Receptors in BRCA1-Associated Breast Cancers. Int. J. Mol. Sci. 2023, 24, 9916. https://doi.org/10.3390/ijms24129916
Santarosa M, Baldazzi D, Armellin M, Maestro R. In Silico Identification of a BRCA1:miR-29:DNMT3 Axis Involved in the Control of Hormone Receptors in BRCA1-Associated Breast Cancers. International Journal of Molecular Sciences. 2023; 24(12):9916. https://doi.org/10.3390/ijms24129916
Chicago/Turabian StyleSantarosa, Manuela, Davide Baldazzi, Michela Armellin, and Roberta Maestro. 2023. "In Silico Identification of a BRCA1:miR-29:DNMT3 Axis Involved in the Control of Hormone Receptors in BRCA1-Associated Breast Cancers" International Journal of Molecular Sciences 24, no. 12: 9916. https://doi.org/10.3390/ijms24129916
APA StyleSantarosa, M., Baldazzi, D., Armellin, M., & Maestro, R. (2023). In Silico Identification of a BRCA1:miR-29:DNMT3 Axis Involved in the Control of Hormone Receptors in BRCA1-Associated Breast Cancers. International Journal of Molecular Sciences, 24(12), 9916. https://doi.org/10.3390/ijms24129916