
Role of the Sodium-Dependent Organic Anion Transporter (SOAT/SLC10A6) in Physiology and Pathophysiology



Abstract
:1. Introduction
2. The SLC10 Carrier Family
3. Genomic Organization of the SLC10A6/Slc10a6 Genes
4. SOAT Protein Structure, Sorting, and Dimerization
5. SOAT Substrate Docking and Proposed Transport Mechanism
6. SOAT Substrate Recognition

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrates | Substrate Km |
|---|---|
| Steroid 3’-monosulfates | |
| Pregnenolone sulfate (PREGS) | 11.3 µM [1,3] |
| Estrone sulfate (E1S) | 12.0 µM [1,13] |
| Dehydroepiandrosterone sulfate (DHEAS) | 28.7 µM [1] |
| 16α-OH-DHEAS | 319.0 µM [49] |
| 17α-OH-PREGS | n.d. [3] |
| Androstenediol-3-sulfate | n.d. [13] |
| Androsterone sulfate | n.d. [3] |
| Epiandrosterone sulfate | n.d. [3] |
| 17β-estradiol-3-sulfate | n.d. [13] |
| Steroid 17’-monosulfates | |
| 17β-estradiol-17-sulfate | n.d. [3] |
| 5α-dihydrotestosterone sulfate | n.d. [3] |
| Epitestosterone sulfate | n.d. [3] |
| Testosterone sulfate | n.d. [3] |
| Bile acids | |
| Taurolithocholic acid | 19.3 µM [14] |
| Taurolithocholic acid-3-sulfate | n.d. [1] |
| Non-steroidal organosulfates | |
| 2-Sulfooxymethylpyrene (2-SMP) | n.d. [1] |
| 4-Sulfooxymethylpyrene (4-SMP) | n.d. [1] |
7. Sodium-Dependent Transport Mode of SOAT
8. SOAT Genetic Variants
9. SOAT Inhibitors
| Inhibitor | Inhibitor IC50 |
|---|---|
| Monomeric and dimeric bile acids | |
| S 0960 (dimeric) | 0.15 µM [48] |
| Taurolithocholic acid-3-sulfate (TLCS) * | 0.5 µM [48] |
| S 1690 (dimeric) | 0.7 µM [48] |
| S 3068 (dimeric) | 1.0 µM [48] |
| Taurolithocholic acid (TLC) * | 3.0–3.9 µM [14,48] |
| Lithocholic acid-3-sulfate | 4.2 µM [48] |
| Lithocholic acid | 10.4 µM [48] |
| Chenodeoxycholic acid | 11.2 µM [48] |
| Glycochenodeoxycholic acid | 26.6 µM [48] |
| Taurochenodeoxycholic acid | 38.1 µM [48] |
| Glycodeoxycholic acid | 46.7 µM [48] |
| Glycolithocholic acid-3-sulfate | 46.7 µM [48] |
| Tauroursodeoxycholic acid | 49.6 µM [48] |
| Taurodeoxycholic acid | 76.3 µM [48] |
| Taurocholic acid | 65.3–99.7 µM [14,48] |
| Deoxycholic acid | 100.1 µM [48] |
| Glycoursodeoxycholic acid | 100.8 µM [48] |
| 7-Ketolithocholic acid | 164.2 µM [48] |
| Hyodeoxycholic acid | 172.6 µM [48] |
| Cholic acid | 177.6 µM [48] |
| Glycocholic acid | 284.8 µM [48] |
| Ursododeoxycholic acid | 384.8 µM [48] |
| Hyocholic acid | 971.8 µM [48] |
| Glycolithocholic acid | n.d. [1] |
| Steroids and steroid sulfates | |
| Digitonin | 4.1 µM [48] |
| Pregnenolone-3-sulfate (PREGS) * | 9.1 µM [48] |
| Estrone-3-sulfate (E1S) * | 22.1 µM [48] |
| RR Scymnol sulfate | 23.3 µM [48] |
| Cortisone | 29.6 µM [48] |
| Estriol | ~100 µM [48] |
| 17β-estradiol-3,17-disulfate | 133.2 µM [48] |
| 17β-estradiol-3-sulfate * | 145.9 µM [48] |
| Corticosterone-21-sulfate | 323.8 µM [48] |
| Betulin derivatives | |
| Betulinic acid | 1.2 µM [14] |
| SAL-II-68 | 3.6 µM [48] |
| EMe I 4 | 5.4 µM [48] |
| SAL-II-156 | 66.9 µM [48] |
| 3-O-Caffeoyl betulin | 301.1 µM [14] |
| Lupenone | 664.5 µM [14] |
| Betulin | 912.2 µM [14] |
| Non-steroidal organosulfates | |
| Bromosulfophthalein (BSP) | 3.6 µM [14,48] |
| 4-Methylumbelliferyl sulfate | 255.7 µM [48] |
| 1-(ω-sulfooxyethyl)pyrene | n.d. [1] |
| 2-Sulfooxymethylpyrene (2-SMP) * | n.d. [1] |
| 4-Sulfooxymethylpyrene (4-SMP) * | n.d. [1] |
| α-Naphthyl sulfate | n.d. [1] |
| Others | |
| S 1647 | 1.1 µM [48] |
| S 3740 | 1.1 µM [48] |
| T 5854015 | 9.0 µM [48] |
| T 0511-1698 | 15.0 µM [48] |
| L-Thyroxine | 49.5 µM [48] |
| T 5573915 | 57.0 µM [48] |
| S 0382 | 75.2 µM [48] |
| T 5239532 | 137.0 µM [48] |
| Cyclosporine A | n.d. [14] |
| Erythrosine B | n.d. [14] |
| Ginkgolic acid 17:1 | n.d. [14] |
| Troglitazone | n.d. [14] |
| Irbesartan | n.d. [14] |
| Losartan | n.d. [14] |
| Propanolamine derivatives | |
| S 8214 | 14.9 µM [48] |
| S 9202 | 18.7 µM [48] |
| S 9086 | 23.1 µM [48] |
| S 9203 | 37.7 µM [48] |
| S 9087 | 50.0 µM [48] |
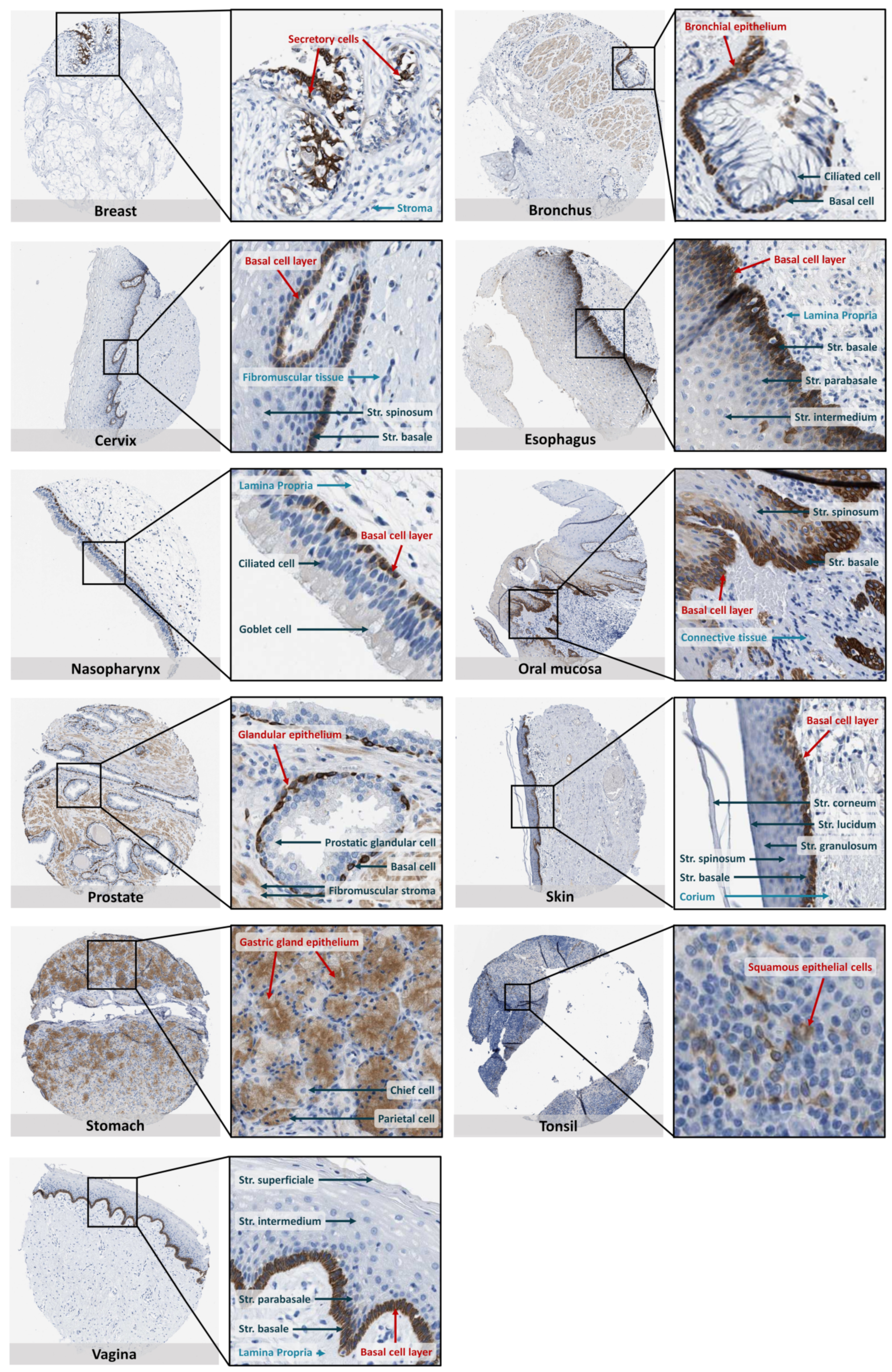
10. SOAT Tissue Expression Pattern


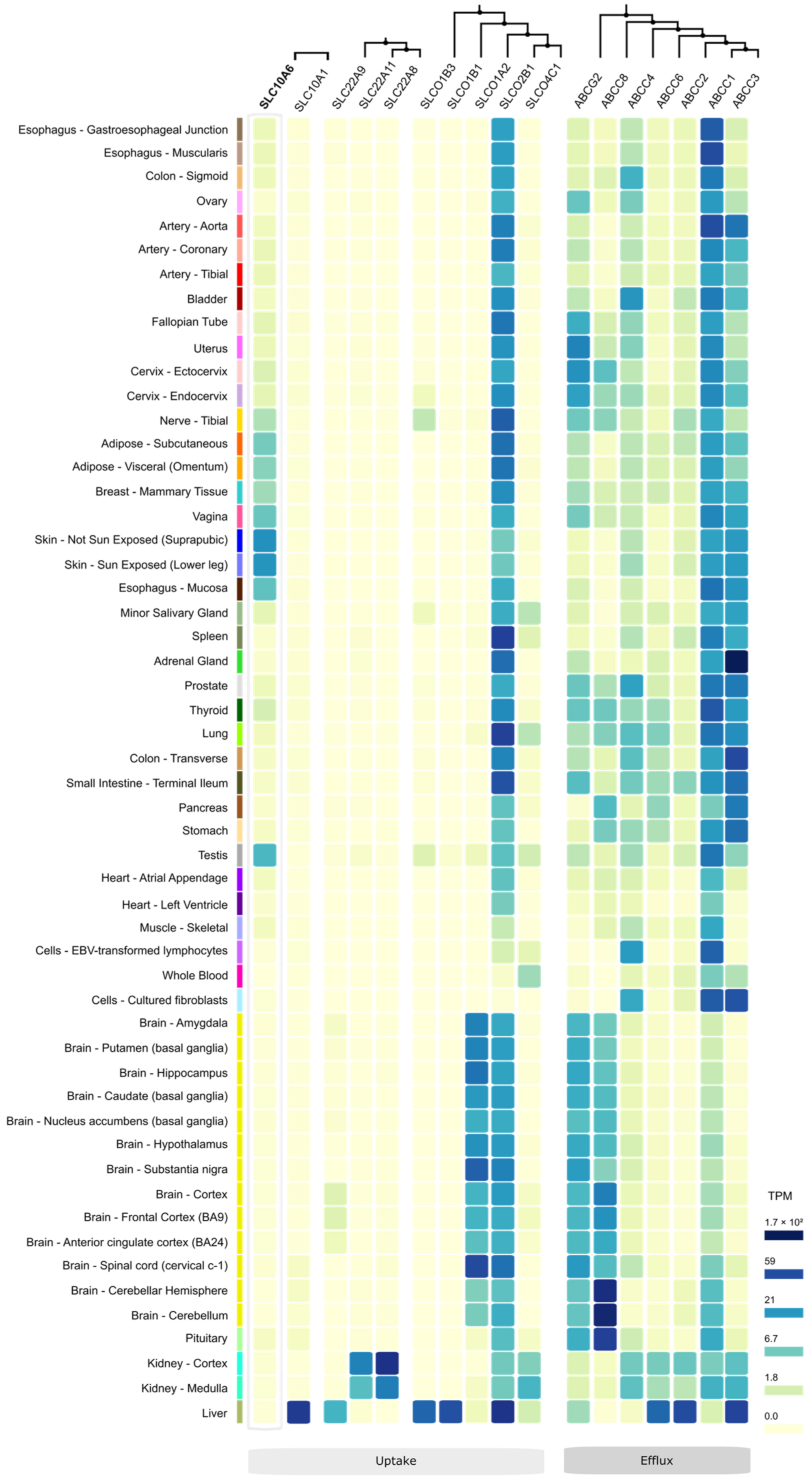
11. Other Steroid Sulfate Transporters

| Gene | Protein (Syn.) | DHEAS Transport Km | E1S Transport Km | Other Selected Endogenous Substrates | Substrate Pattern | Transport Mode |
|---|---|---|---|---|---|---|
| SLC10A1 | NTCP (LBAT) | 56.1 µM [14] | 57.6 µM [14] | Bile acids, taurolithocholic acid [14] | Multi-specific | Active, Na+-dependent |
| SLC10A6 | SOAT | 28.7 µM [1] | 12.0 µM [1] | Testosterone sulfate, taurolithocholic acid [3,14] | Specific | Active, Na+-dependent |
| SLCO1A2 | OATP1A2 (OATP-A) | 7 µM [59,60] | 16–59 µM [59,61,62,63] | Bile acids, bilirubin, thyroid hormones, prostaglandin E2 [64] | Multi-specific | Na+-independent |
| SLCO1B1 | OATP1B1 (OATP-C) | 22 µM [65,66] | 0.09–45 µM [59,65,67,68] | Bile acids, bilirubin, thyroid hormones, glucuronide conjugates, prostaglandin E2 [66,69] | Multi-specific | Na+-independent |
| SLCO1B3 | OATP1B3 (OATP8) | >30 µM [65] | 3–58 µM [70,71,72] | Bile acids, bilirubin, thyroid hormones, leukotriene C4 [73] | Multi-specific | Na+-independent |
| SLCO2B1 | OATP2B1 (OATP-B) | 9 µM [74] | 1.56–21 µM [67,74,75,76] | Taurocholic acid, prostaglandins, leukotriene C4, thromboxane B2 [77] | Multi-specific | Na+-independent |
| SLCO4C1 | OATP4C1 (OATP-H) | - | 26.6 µM [78] | Bile acids, conjugated steroids, thyroid hormones, eicosanoids [77,79] | Multi-specific | Na+-independent |
| SLC22A8 | OAT3 | 13 µM [80] | 2.2–21.2 µM [81] | Creatinine, cAMP, glutarate, oxalate, prostaglandins E2 and F2α [77] | Multi-specific | Na+-independent |
| SLC22A11 | OAT4 | 0.63–29.2 µM [81] | 1.01–21.7 µM [81] | Creatinine, glutarate, prostaglandins E2 and F2α [77] | Multi-specific | Na+-independent |
| SLC22A9 | OAT7 | 2.2 µM [81] | 8.7 µM [81] | Creatinine, cGMP [82] | Multi-specific | Na+-independent |
12. Role of SOAT in Health and Disease
12.1. Male Fertility/Infertility
12.2. Placenta Function during Pregnancy
12.3. Breast Cancer
12.4. Adiposity and Inflammation
12.5. SOAT Expression in Cancer Tissues


13. Slc10a6 Knockout Mouse Model
14. Cell Models

15. Summary and Further Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Geyer, J.; Döring, B.; Meerkamp, K.; Ugele, B.; Bakhiya, N.; Fernandes, C.F.; Godoy, J.R.; Glatt, H.; Petzinger, E. Cloning and functional characterization of human sodium-dependent organic anion transporter (SLC10A6). J. Biol. Chem. 2007, 282, 19728–19741. [Google Scholar] [CrossRef] [Green Version]
- Geyer, J.; Godoy, J.R.; Petzinger, E. Identification of a sodium-dependent organic anion transporter from rat adrenal gland. Biochem. Biophys. Res. Commun. 2004, 316, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Grosser, G.; Bennien, J.; Sanchez-Guijo, A.; Bakhaus, K.; Döring, B.; Hartmann, M.; Wudy, S.A.; Geyer, J. Transport of steroid 3-sulfates and steroid 17-sulfates by the sodium-dependent organic anion transporter SOAT (SLC10A6). J. Steroid Biochem. Mol. Biol. 2018, 179, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Grosser, G.; Fietz, D.; Gunther, S.; Bakhaus, K.; Schweigmann, H.; Ugele, B.; Brehm, R.; Petzinger, E.; Bergmann, M.; Geyer, J. Cloning and functional characterization of the mouse sodium-dependent organic anion transporter Soat (Slc10a6). J. Steroid Biochem. Mol. Biol. 2013, 138, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Strott, C.A. Sulfonation and molecular action. Endocr. Rev. 2002, 23, 703–732. [Google Scholar] [CrossRef]
- Mueller, J.W.; Gilligan, L.C.; Idkowiak, J.; Arlt, W.; Foster, P.A. The Regulation of Steroid Action by Sulfation and Desulfation. Endocr. Rev. 2015, 36, 526–563. [Google Scholar] [CrossRef]
- Fietz, D. Transporter for sulfated steroid hormones in the testis—Expression pattern, biological significance and implications for fertility in men and rodents. J. Steroid Biochem. Mol. Biol. 2018, 179, 8–19. [Google Scholar] [CrossRef]
- Döring, B.; Lutteke, T.; Geyer, J.; Petzinger, E. The SLC10 carrier family: Transport functions and molecular structure. Curr. Top. Membr. 2012, 70, 105–168. [Google Scholar] [CrossRef]
- Geyer, J.; Wilke, T.; Petzinger, E. The solute carrier family SLC10: More than a family of bile acid transporters regarding function and phylogenetic relationships. Naunyn Schmiedebergs Arch. Pharmacol. 2006, 372, 413–431. [Google Scholar] [CrossRef] [Green Version]
- Hagenbuch, B.; Meier, P.J. Molecular cloning, chromosomal localization, and functional characterization of a human liver Na+/bile acid cotransporter. J. Clin. Invest. 1994, 93, 1326–1331. [Google Scholar] [CrossRef] [Green Version]
- Wong, M.H.; Oelkers, P.; Dawson, P.A. Identification of a mutation in the ileal sodium-dependent bile acid transporter gene that abolishes transport activity. J. Biol. Chem. 1995, 270, 27228–27234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagenbuch, B.; Dawson, P. The sodium bile salt cotransport family SLC10. Pflugers Arch. 2004, 447, 566–570. [Google Scholar] [CrossRef] [PubMed]
- Fietz, D.; Bakhaus, K.; Wapelhorst, B.; Grosser, G.; Gunther, S.; Alber, J.; Döring, B.; Kliesch, S.; Weidner, W.; Galuska, C.E.; et al. Membrane transporters for sulfated steroids in the human testis--cellular localization, expression pattern and functional analysis. PLoS ONE 2013, 8, e62638. [Google Scholar] [CrossRef] [PubMed]
- Grosser, G.; Müller, S.F.; Kirstgen, M.; Döring, B.; Geyer, J. Substrate Specificities and Inhibition Pattern of the Solute Carrier Family 10 Members NTCP, ASBT and SOAT. Front. Mol. Biosci. 2021, 8, 689757. [Google Scholar] [CrossRef] [PubMed]
- Claro da Silva, T.; Polli, J.E.; Swaan, P.W. The solute carrier family 10 (SLC10): Beyond bile acid transport. Mol. Aspects Med. 2013, 34, 252–269. [Google Scholar] [CrossRef] [Green Version]
- Burger, S.; Döring, B.; Hardt, M.; Beuerlein, K.; Gerstberger, R.; Geyer, J. Co-expression studies of the orphan carrier protein Slc10a4 and the vesicular carriers VAChT and VMAT2 in the rat central and peripheral nervous system. Neuroscience 2011, 193, 109–121. [Google Scholar] [CrossRef]
- Geyer, J.; Fernandes, C.F.; Döring, B.; Burger, S.; Godoy, J.R.; Rafalzik, S.; Hubschle, T.; Gerstberger, R.; Petzinger, E. Cloning and molecular characterization of the orphan carrier protein Slc10a4: Expression in cholinergic neurons of the rat central nervous system. Neuroscience 2008, 152, 990–1005. [Google Scholar] [CrossRef]
- Schmidt, S.; Moncada, M.; Burger, S.; Geyer, J. Expression, sorting and transport studies for the orphan carrier SLC10A4 in neuronal and non-neuronal cell lines and in Xenopus laevis oocytes. BMC Neurosci. 2015, 16, 35. [Google Scholar] [CrossRef] [Green Version]
- Larhammar, M.; Patra, K.; Blunder, M.; Emilsson, L.; Peuckert, C.; Arvidsson, E.; Ronnlund, D.; Preobraschenski, J.; Birgner, C.; Limbach, C.; et al. SLC10A4 is a vesicular amine-associated transporter modulating dopamine homeostasis. Biol. Psychiatry 2015, 77, 526–536. [Google Scholar] [CrossRef] [Green Version]
- Melief, E.J.; Gibbs, J.T.; Li, X.; Morgan, R.G.; Keene, C.D.; Montine, T.J.; Palmiter, R.D.; Darvas, M. Characterization of cognitive impairments and neurotransmitter changes in a novel transgenic mouse lacking Slc10a4. Neuroscience 2016, 324, 399–406. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, C.F.; Godoy, J.R.; Döring, B.; Cavalcanti, M.C.; Bergmann, M.; Petzinger, E.; Geyer, J. The novel putative bile acid transporter SLC10A5 is highly expressed in liver and kidney. Biochem. Biophys. Res. Commun. 2007, 361, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Godoy, J.R.; Fernandes, C.; Döring, B.; Beuerlein, K.; Petzinger, E.; Geyer, J. Molecular and phylogenetic characterization of a novel putative membrane transporter (SLC10A7), conserved in vertebrates and bacteria. Eur. J. Cell. Biol. 2007, 86, 445–460. [Google Scholar] [CrossRef] [PubMed]
- Ashikov, A.; Abu Bakar, N.; Wen, X.Y.; Niemeijer, M.; Rodrigues Pinto Osorio, G.; Brand-Arzamendi, K.; Hasadsri, L.; Hansikova, H.; Raymond, K.; Vicogne, D.; et al. Integrating glycomics and genomics uncovers SLC10A7 as essential factor for bone mineralization by regulating post-Golgi protein transport and glycosylation. Hum. Mol. Genet. 2018, 27, 3029–3045. [Google Scholar] [CrossRef] [PubMed]
- Dubail, J.; Huber, C.; Chantepie, S.; Sonntag, S.; Tuysuz, B.; Mihci, E.; Gordon, C.T.; Steichen-Gersdorf, E.; Amiel, J.; Nur, B.; et al. SLC10A7 mutations cause a skeletal dysplasia with amelogenesis imperfecta mediated by GAG biosynthesis defects. Nat. Commun. 2018, 9, 3087. [Google Scholar] [CrossRef] [Green Version]
- Laugel-Haushalter, V.; Bar, S.; Schaefer, E.; Stoetzel, C.; Geoffroy, V.; Alembik, Y.; Kharouf, N.; Huckert, M.; Hamm, P.; Hemmerle, J.; et al. A New SLC10A7 Homozygous Missense Mutation Responsible for a Milder Phenotype of Skeletal Dysplasia With Amelogenesis Imperfecta. Front. Genet. 2019, 10, 504. [Google Scholar] [CrossRef] [Green Version]
- Karakus, E.; Wannowius, M.; Müller, S.F.; Leiting, S.; Leidolf, R.; Noppes, S.; Oswald, S.; Diener, M.; Geyer, J. The orphan solute carrier SLC10A7 is a novel negative regulator of intracellular calcium signaling. Sci. Rep. 2020, 10, 7248. [Google Scholar] [CrossRef]
- Wannowius, M.; Karakus, E.; Geyer, J. Functional Analysis of Rare Genetic Variants in the Negative Regulator of Intracellular Calcium Signaling RCAS/SLC10A7. Front. Mol. Biosci. 2021, 8, 741946. [Google Scholar] [CrossRef]
- Hu, N.J.; Iwata, S.; Cameron, A.D.; Drew, D. Crystal structure of a bacterial homologue of the bile acid sodium symporter ASBT. Nature 2011, 478, 408–411. [Google Scholar] [CrossRef]
- Zhou, X.; Levin, E.J.; Pan, Y.; McCoy, J.G.; Sharma, R.; Kloss, B.; Bruni, R.; Quick, M.; Zhou, M. Structural basis of the alternating-access mechanism in a bile acid transporter. Nature 2014, 505, 569–573. [Google Scholar] [CrossRef] [Green Version]
- Asami, J.; Kimura, K.T.; Fujita-Fujiharu, Y.; Ishida, H.; Zhang, Z.; Nomura, Y.; Liu, K.; Uemura, T.; Sato, Y.; Ono, M.; et al. Structure of the bile acid transporter and HBV receptor NTCP. Nature 2022, 606, 1021–1026. [Google Scholar] [CrossRef]
- Goutam, K.; Ielasi, F.S.; Pardon, E.; Steyaert, J.; Reyes, N. Structural basis of sodium-dependent bile salt uptake into the liver. Nature 2022, 606, 1015–1020. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Irobalieva, R.N.; Bang-Sorensen, R.; Nosol, K.; Mukherjee, S.; Agrawal, P.; Stieger, B.; Kossiakoff, A.A.; Locher, K.P. Structure of human NTCP reveals the basis of recognition and sodium-driven transport of bile salts into the liver. Cell. Res. 2022, 32, 773–776. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Iwamoto, M.; Yun, J.H.; Uchikubo-Kamo, T.; Son, D.; Jin, Z.; Yoshida, H.; Ohki, M.; Ishimoto, N.; Mizutani, K.; et al. Structural insights into the HBV receptor and bile acid transporter NTCP. Nature 2022, 606, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef] [PubMed]
- Bakhaus, K.; Fietz, D.; Kliesch, S.; Weidner, W.; Bergmann, M.; Geyer, J. The polymorphism L204F affects transport and membrane expression of the sodium-dependent organic anion transporter SOAT (SLC10A6). J. Steroid Biochem. Mol. Biol. 2018, 179, 36–44. [Google Scholar] [CrossRef]
- Bennien, J.; Fischer, T.; Geyer, J. Rare genetic variants in the sodium-dependent organic anion transporter SOAT (SLC10A6): Effects on transport function and membrane expression. J. Steroid Biochem. Mol. Biol. 2018, 179, 26–35. [Google Scholar] [CrossRef]
- Al-Ansari, N.; Xu, G.; Kollman-Bauerly, K.; Coppola, C.; Shefer, S.; Ujhazy, P.; Ortiz, D.; Ma, L.; Yang, S.; Tsai, R.; et al. Analysis of the effect of intestinal resection on rat ileal bile Acid transporter expression and on bile Acid and cholesterol homeostasis. Pediatr. Res. 2002, 52, 286–291. [Google Scholar] [CrossRef]
- Bijsmans, I.T.; Bouwmeester, R.A.; Geyer, J.; Faber, K.N.; Van de Graaf, S.F. Homo- and hetero-dimeric architecture of the human liver Na(+)-dependent taurocholate co-transporting protein. Biochem. J. 2012, 441, 1007–1015. [Google Scholar] [CrossRef] [Green Version]
- Chothe, P.P.; Czuba, L.C.; Moore, R.H.; Swaan, P.W. Human bile acid transporter ASBT (SLC10A2) forms functional non-covalent homodimers and higher order oligomers. Biochim. Biophys. Acta Biomembr. 2018, 1860, 645–653. [Google Scholar] [CrossRef]
- Kramer, W.; Girbig, F.; Gutjahr, U.; Kowalewski, S.; Jouvenal, K.; Muller, G.; Tripier, D.; Wess, G. Intestinal bile acid absorption. Na(+)-dependent bile acid transport activity in rabbit small intestine correlates with the coexpression of an integral 93-kDa and a peripheral 14-kDa bile acid-binding membrane protein along the duodenum-ileum axis. J. Biol. Chem. 1993, 268, 18035–18046. [Google Scholar] [CrossRef]
- Noppes, S.; Müller, S.F.; Bennien, J.; Holtemeyer, M.; Palatini, M.; Leidolf, R.; Alber, J.; Geyer, J. Homo- and heterodimerization is a common feature of the solute carrier family SLC10 members. Biol. Chem. 2019, 400, 1371–1384. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T. New method for fast and accurate binding-site identification and analysis. Chem. Biol. Drug. Des. 2007, 69, 146–148. [Google Scholar] [CrossRef]
- Schrodinger, LLC. Schrödinger Release 2022-4: LigPrep; Schrödinger, LLC: New York, NY, USA, 2021. [Google Scholar]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [Green Version]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Schrodinger, LLC. The PyMOL Molecular Graphics System; Version 2.5.4; Schrödinger, LLC: New York, NY, USA, 2022. [Google Scholar]
- Grosser, G.; Baringhaus, K.H.; Döring, B.; Kramer, W.; Petzinger, E.; Geyer, J. Identification of novel inhibitors of the steroid sulfate carrier ‘sodium-dependent organic anion transporter’ SOAT (SLC10A6) by pharmacophore modelling. Mol. Cell. Endocrinol. 2016, 428, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Schweigmann, H.; Sanchez-Guijo, A.; Ugele, B.; Hartmann, K.; Hartmann, M.F.; Bergmann, M.; Pfarrer, C.; Döring, B.; Wudy, S.A.; Petzinger, E.; et al. Transport of the placental estriol precursor 16alpha-hydroxy-dehydroepiandrosterone sulfate (16alpha-OH-DHEAS) by stably transfected OAT4-, SOAT-, and NTCP-HEK293 cells. J. Steroid Biochem. Mol. Biol. 2014, 143, 259–265. [Google Scholar] [CrossRef]
- Stieger, B.; Geier, A. Genetic variations of bile salt transporters as predisposing factors for drug-induced cholestasis, intrahepatic cholestasis of pregnancy and therapeutic response of viral hepatitis. Expert. Opin. Drug. Metab. Toxicol. 2011, 7, 411–425. [Google Scholar] [CrossRef] [Green Version]
- Karakus, E.; Zahner, D.; Grosser, G.; Leidolf, R.; Gundogdu, C.; Sanchez-Guijo, A.; Wudy, S.A.; Geyer, J. Estrone-3-Sulfate Stimulates the Proliferation of T47D Breast Cancer Cells Stably Transfected With the Sodium-Dependent Organic Anion Transporter SOAT (SLC10A6). Front. Pharmacol. 2018, 9, 941. [Google Scholar] [CrossRef]
- Geyer, J.; Bakhaus, K.; Bernhardt, R.; Blaschka, C.; Dezhkam, Y.; Fietz, D.; Grosser, G.; Hartmann, K.; Hartmann, M.F.; Neunzig, J.; et al. The role of sulfated steroid hormones in reproductive processes. J. Steroid Biochem. Mol. Biol. 2017, 172, 207–221. [Google Scholar] [CrossRef]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Berglund, L.; Bjorling, E.; Oksvold, P.; Fagerberg, L.; Asplund, A.; Szigyarto, C.A.; Persson, A.; Ottosson, J.; Wernerus, H.; Nilsson, P.; et al. A genecentric Human Protein Atlas for expression profiles based on antibodies. Mol. Cell. Proteomics 2008, 7, 2019–2027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhlen, M.; Bjorling, E.; Agaton, C.; Szigyarto, C.A.; Amini, B.; Andersen, E.; Andersson, A.C.; Angelidou, P.; Asplund, A.; Asplund, C.; et al. A human protein atlas for normal and cancer tissues based on antibody proteomics. Mol. Cell. Proteomics 2005, 4, 1920–1932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponten, F.; Jirstrom, K.; Uhlen, M. The Human Protein Atlas--a tool for pathology. J. Pathol. 2008, 216, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, K.; Bennien, J.; Wapelhorst, B.; Bakhaus, K.; Schumacher, V.; Kliesch, S.; Weidner, W.; Bergmann, M.; Geyer, J.; Fietz, D. Current insights into the sulfatase pathway in human testis and cultured Sertoli cells. Histochem. Cell. Biol. 2016, 146, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Rizner, T.L.; Thalhammer, T.; Ozvegy-Laczka, C. The Importance of Steroid Uptake and Intracrine Action in Endometrial and Ovarian Cancers. Front. Pharmacol. 2017, 8, 346. [Google Scholar] [CrossRef] [Green Version]
- Koenen, A.; Kock, K.; Keiser, M.; Siegmund, W.; Kroemer, H.K.; Grube, M. Steroid hormones specifically modify the activity of organic anion transporting polypeptides. Eur. J. Pharm. Sci. 2012, 47, 774–780. [Google Scholar] [CrossRef]
- Kullak-Ublick, G.A.; Fisch, T.; Oswald, M.; Hagenbuch, B.; Meier, P.J.; Beuers, U.; Paumgartner, G. Dehydroepiandrosterone sulfate (DHEAS): Identification of a carrier protein in human liver and brain. FEBS Lett. 1998, 424, 173–176. [Google Scholar] [CrossRef] [Green Version]
- Bossuyt, X.; Muller, M.; Meier, P.J. Multispecific amphipathic substrate transport by an organic anion transporter of human liver. J. Hepatol. 1996, 25, 733–738. [Google Scholar] [CrossRef]
- Lee, W.; Glaeser, H.; Smith, L.H.; Roberts, R.L.; Moeckel, G.W.; Gervasini, G.; Leake, B.F.; Kim, R.B. Polymorphisms in human organic anion-transporting polypeptide 1A2 (OATP1A2): Implications for altered drug disposition and central nervous system drug entry. J. Biol. Chem. 2005, 280, 9610–9617. [Google Scholar] [CrossRef] [Green Version]
- Meyer zu Schwabedissen, H.E.; Tirona, R.G.; Yip, C.S.; Ho, R.H.; Kim, R.B. Interplay between the nuclear receptor pregnane X receptor and the uptake transporter organic anion transporter polypeptide 1A2 selectively enhances estrogen effects in breast cancer. Cancer Res. 2008, 68, 9338–9347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franke, R.M.; Scherkenbach, L.A.; Sparreboom, A. Pharmacogenetics of the organic anion transporting polypeptide 1A2. Pharmacogenomics 2009, 10, 339–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Y.; Konig, J.; Leier, I.; Buchholz, U.; Keppler, D. Hepatic uptake of bilirubin and its conjugates by the human organic anion transporter SLC21A6. J. Biol. Chem. 2001, 276, 9626–9630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kullak-Ublick, G.A.; Ismair, M.G.; Stieger, B.; Landmann, L.; Huber, R.; Pizzagalli, F.; Fattinger, K.; Meier, P.J.; Hagenbuch, B. Organic anion-transporting polypeptide B (OATP-B) and its functional comparison with three other OATPs of human liver. Gastroenterology 2001, 120, 525–533. [Google Scholar] [CrossRef]
- Tamai, I.; Nozawa, T.; Koshida, M.; Nezu, J.; Sai, Y.; Tsuji, A. Functional characterization of human organic anion transporting polypeptide B (OATP-B) in comparison with liver-specific OATP-C. Pharm. Res. 2001, 18, 1262–1269. [Google Scholar] [CrossRef]
- Zhang, Y.; Hays, A.; Noblett, A.; Thapa, M.; Hua, D.H.; Hagenbuch, B. Transport by OATP1B1 and OATP1B3 enhances the cytotoxicity of epigallocatechin 3-O-gallate and several quercetin derivatives. J. Nat. Prod. 2013, 76, 368–373. [Google Scholar] [CrossRef] [Green Version]
- Hsiang, B.; Zhu, Y.; Wang, Z.; Wu, Y.; Sasseville, V.; Yang, W.P.; Kirchgessner, T.G. A novel human hepatic organic anion transporting polypeptide (OATP2). Identification of a liver-specific human organic anion transporting polypeptide and identification of rat and human hydroxymethylglutaryl-CoA reductase inhibitor transporters. J. Biol. Chem. 1999, 274, 37161–37168. [Google Scholar] [CrossRef] [Green Version]
- Gui, C.; Wahlgren, B.; Lushington, G.H.; Hagenbuch, B. Identification, Ki determination and CoMFA analysis of nuclear receptor ligands as competitive inhibitors of OATP1B1-mediated estradiol-17beta-glucuronide transport. Pharmacol. Res. 2009, 60, 50–56. [Google Scholar] [CrossRef] [Green Version]
- Ismair, M.G.; Stieger, B.; Cattori, V.; Hagenbuch, B.; Fried, M.; Meier, P.J.; Kullak-Ublick, G.A. Hepatic uptake of cholecystokinin octapeptide by organic anion-transporting polypeptides OATP4 and OATP8 of rat and human liver. Gastroenterology 2001, 121, 1185–1190. [Google Scholar] [CrossRef] [Green Version]
- Vavricka, S.R.; Van Montfoort, J.; Ha, H.R.; Meier, P.J.; Fattinger, K. Interactions of rifamycin SV and rifampicin with organic anion uptake systems of human liver. Hepatology 2002, 36, 164–172. [Google Scholar] [CrossRef]
- Van de Steeg, E.; Stranecky, V.; Hartmannova, H.; Noskova, L.; Hrebicek, M.; Wagenaar, E.; Van Esch, A.; De Waart, D.R.; Oude Elferink, R.P.; Kenworthy, K.E.; et al. Complete OATP1B1 and OATP1B3 deficiency causes human Rotor syndrome by interrupting conjugated bilirubin reuptake into the liver. J. Clin. Invest. 2012, 122, 519–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pizzagalli, F.; Varga, Z.; Huber, R.D.; Folkers, G.; Meier, P.J.; St-Pierre, M.V. Identification of steroid sulfate transport processes in the human mammary gland. J. Clin. Endocrinol. Metab. 2003, 88, 3902–3912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nozawa, T.; Suzuki, M.; Takahashi, K.; Yabuuchi, H.; Maeda, T.; Tsuji, A.; Tamai, I. Involvement of estrone-3-sulfate transporters in proliferation of hormone-dependent breast cancer cells. J. Pharmacol. Exp. Ther. 2004, 311, 1032–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sai, Y.; Kaneko, Y.; Ito, S.; Mitsuoka, K.; Kato, Y.; Tamai, I.; Artursson, P.; Tsuji, A. Predominant contribution of organic anion transporting polypeptide OATP-B (OATP2B1) to apical uptake of estrone-3-sulfate by human intestinal Caco-2 cells. Drug. Metab. Dispos. 2006, 34, 1423–1431. [Google Scholar] [CrossRef] [Green Version]
- Lai, Y. Transporters in Drug Discovery and Development: Detailed Concepts and Best Practice, 1st ed.; Woodhead Publishing: Shaxton, UK, 2013; 734p. [Google Scholar]
- Yamaguchi, H.; Sugie, M.; Okada, M.; Mikkaichi, T.; Toyohara, T.; Abe, T.; Goto, J.; Hishinuma, T.; Shimada, M.; Mano, N. Transport of estrone 3-sulfate mediated by organic anion transporter OATP4C1: Estrone 3-sulfate binds to the different recognition site for digoxin in OATP4C1. Drug. Metab. Pharmacokinet. 2010, 25, 314–317. [Google Scholar] [CrossRef]
- Mikkaichi, T.; Suzuki, T.; Onogawa, T.; Tanemoto, M.; Mizutamari, H.; Okada, M.; Chaki, T.; Masuda, S.; Tokui, T.; Eto, N.; et al. Isolation and characterization of a digoxin transporter and its rat homologue expressed in the kidney. Proc. Natl. Acad. Sci. USA 2004, 101, 3569–3574. [Google Scholar] [CrossRef] [Green Version]
- Nozaki, Y.; Kusuhara, H.; Kondo, T.; Hasegawa, M.; Shiroyanagi, Y.; Nakazawa, H.; Okano, T.; Sugiyama, Y. Characterization of the uptake of organic anion transporter (OAT) 1 and OAT3 substrates by human kidney slices. J. Pharmacol. Exp. Ther. 2007, 321, 362–369. [Google Scholar] [CrossRef]
- Burckhardt, G.; Burckhardt, B.C. In vitro and in vivo evidence of the importance of organic anion transporters (OATs) in drug therapy. Handb. Exp. Pharmacol. 2011, 201, 29–104. [Google Scholar] [CrossRef]
- Mathialagan, S.; Costales, C.; Tylaska, L.; Kimoto, E.; Vildhede, A.; Johnson, J.; Johnson, N.; Sarashina, T.; Hashizume, K.; Isringhausen, C.D.; et al. In vitro studies with two human organic anion transporters: OAT2 and OAT7. Xenobiotica 2018, 48, 1037–1049. [Google Scholar] [CrossRef]
- Bakhaus, K.; Bennien, J.; Fietz, D.; Sanchez-Guijo, A.; Hartmann, M.; Serafini, R.; Love, C.C.; Golovko, A.; Wudy, S.A.; Bergmann, M.; et al. Sodium-dependent organic anion transporter (Slc10a6(−/−)) knockout mice show normal spermatogenesis and reproduction, but elevated serum levels for cholesterol sulfate. J. Steroid Biochem. Mol. Biol. 2018, 179, 45–54. [Google Scholar] [CrossRef]
- Raeside, J.I. A Brief Account of the Discovery of the Fetal/Placental Unit for Estrogen Production in Equine and Human Pregnancies: Relation to Human Medicine. Yale J. Biol. Med. 2017, 90, 449–461. [Google Scholar] [PubMed]
- Chatuphonprasert, W.; Jarukamjorn, K.; Ellinger, I. Physiology and Pathophysiology of Steroid Biosynthesis, Transport and Metabolism in the Human Placenta. Front. Pharmacol. 2018, 9, 1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morel, Y.; Roucher, F.; Plotton, I.; Goursaud, C.; Tardy, V.; Mallet, D. Evolution of steroids during pregnancy: Maternal, placental and fetal synthesis. Ann. Endocrinol. 2016, 77, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Pasqualini, J.R. Enzymes involved in the formation and transformation of steroid hormones in the fetal and placental compartments. J. Steroid Biochem. Mol. Biol. 2005, 97, 401–415. [Google Scholar] [CrossRef]
- Fokina, V.M.; Patrikeeva, S.; Wang, X.M.; Noguchi, S.; Tomi, M.; Konig, J.; Ahmed, M.S.; Nanovskaya, T. Role of Uptake Transporters OAT4, OATP2A1, and OATP1A2 in Human Placental Bio-disposition of Pravastatin. J. Pharm. Sci. 2022, 111, 505–516. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, M.V.; Hagenbuch, B.; Ugele, B.; Meier, P.J.; Stallmach, T. Characterization of an organic anion-transporting polypeptide (OATP-B) in human placenta. J. Clin. Endocrinol. Metab. 2002, 87, 1856–1863. [Google Scholar] [CrossRef]
- Ugele, B.; Bahn, A.; Rex-Haffner, M. Functional differences in steroid sulfate uptake of organic anion transporter 4 (OAT4) and organic anion transporting polypeptide 2B1 (OATP2B1) in human placenta. J. Steroid Biochem. Mol. Biol. 2008, 111, 1–6. [Google Scholar] [CrossRef]
- Ugele, B.; St-Pierre, M.V.; Pihusch, M.; Bahn, A.; Hantschmann, P. Characterization and identification of steroid sulfate transporters of human placenta. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E390–E398. [Google Scholar] [CrossRef]
- Frandsen, V.A.; Stakemann, G. The Site of Production of Oestrogenic Hormones in Human Pregnancy. 3. Further Observations on the Hormone Excretion in Pregnancy with Anencephalic Foetus. Acta Endocrinol. 1964, 47, 265–276. [Google Scholar]
- Newby, D.; Aitken, D.A.; Howatson, A.G.; Connor, J.M. Placental synthesis of oestriol in Down’s syndrome pregnancies. Placenta 2000, 21, 263–267. [Google Scholar] [CrossRef]
- Conner, P.; Lundstrom, E.; Von Schoultz, B. Breast cancer and hormonal therapy. Clin. Obstet. Gynecol. 2008, 51, 592–606. [Google Scholar] [CrossRef] [PubMed]
- Osborne, C.K. Tamoxifen in the treatment of breast cancer. N. Engl. J. Med. 1998, 339, 1609–1618. [Google Scholar] [CrossRef] [PubMed]
- Aoki, K.; Terauchi, Y. Effect of Dehydroepiandrosterone (DHEA) on Diabetes Mellitus and Obesity. Vitam. Horm. 2018, 108, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Cleary, M.P.; Seidenstat, R.; Tannen, R.H.; Schwartz, A.G. The effect of dehydroepiandrosterone on adipose tissue cellularity in mice. Proc. Soc. Exp. Biol. Med. 1982, 171, 276–284. [Google Scholar] [CrossRef]
- Coleman, D.L.; Schwizer, R.W.; Leiter, E.H. Effect of genetic background on the therapeutic effects of dehydroepiandrosterone (DHEA) in diabetes-obesity mutants and in aged normal mice. Diabetes 1984, 33, 26–32. [Google Scholar] [CrossRef]
- Granholm, N.H.; Staber, L.D.; Wilkin, P.J. Effects of dehydroepiandrosterone on obesity and glucose-6-phosphate dehydrogenase activity in the lethal yellow mouse (strain 129/Sv-Ay/Aw). J. Exp. Zool. 1987, 242, 67–74. [Google Scholar] [CrossRef]
- Corona, G.; Rastrelli, G.; Giagulli, V.A.; Sila, A.; Sforza, A.; Forti, G.; Mannucci, E.; Maggi, M. Dehydroepiandrosterone supplementation in elderly men: A meta-analysis study of placebo-controlled trials. J. Clin. Endocrinol. Metab. 2013, 98, 3615–3626. [Google Scholar] [CrossRef] [Green Version]
- Kimura, M.; Tanaka, S.; Yamada, Y.; Kiuchi, Y.; Yamakawa, T.; Sekihara, H. Dehydroepiandrosterone decreases serum tumor necrosis factor-alpha and restores insulin sensitivity: Independent effect from secondary weight reduction in genetically obese Zucker fatty rats. Endocrinology 1998, 139, 3249–3253. [Google Scholar] [CrossRef]
- De Pergola, G.; Giagulli, V.A.; Garruti, G.; Cospite, M.R.; Giorgino, F.; Cignarelli, M.; Giorgino, R. Low dehydroepiandrosterone circulating levels in premenopausal obese women with very high body mass index. Metabolism 1991, 40, 187–190. [Google Scholar] [CrossRef]
- Hernandez-Morante, J.J.; Perez-de-Heredia, F.; Lujan, J.A.; Zamora, S.; Garaulet, M. Role of DHEA-S on body fat distribution: Gender- and depot-specific stimulation of adipose tissue lipolysis. Steroids 2008, 73, 209–215. [Google Scholar] [CrossRef]
- Sayin, S.; Kutlu, R.; Kulaksizoglu, M. The relationship between sex steroids, insulin resistance and body compositions in obese women: A case-control study. J. Med. Biochem. 2020, 39, 25–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tchernof, A.; Despres, J.P.; Belanger, A.; Dupont, A.; Prud’homme, D.; Moorjani, S.; Lupien, P.J.; Labrie, F. Reduced testosterone and adrenal C19 steroid levels in obese men. Metabolism 1995, 44, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Morales, A.J.; Haubrich, R.H.; Hwang, J.Y.; Asakura, H.; Yen, S.S. The effect of six months treatment with a 100 mg daily dose of dehydroepiandrosterone (DHEA) on circulating sex steroids, body composition and muscle strength in age-advanced men and women. Clin. Endocrinol. 1998, 49, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Nestler, J.E.; Barlascini, C.O.; Clore, J.N.; Blackard, W.G. Dehydroepiandrosterone reduces serum low density lipoprotein levels and body fat but does not alter insulin sensitivity in normal men. J. Clin. Endocrinol. Metab. 1988, 66, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Villareal, D.T.; Holloszy, J.O. Effect of DHEA on abdominal fat and insulin action in elderly women and men: A randomized controlled trial. JAMA 2004, 292, 2243–2248. [Google Scholar] [CrossRef] [Green Version]
- Yen, S.S.; Morales, A.J.; Khorram, O. Replacement of DHEA in aging men and women. Potential remedial effects. Ann. N. Y Acad. Sci. 1995, 774, 128–142. [Google Scholar] [CrossRef]
- Kawai, T.; Autieri, M.V.; Scalia, R. Adipose tissue inflammation and metabolic dysfunction in obesity. Am. J. Physiol. Cell. Physiol. 2021, 320, C375–C391. [Google Scholar] [CrossRef]
- Kosters, A.; Abebe, D.F.; Felix, J.C.; Dawson, P.A.; Karpen, S.J. Inflammation-associated upregulation of the sulfated steroid transporter Slc10a6 in mouse liver and macrophage cell lines. Hepatol. Res. 2016, 46, 794–803. [Google Scholar] [CrossRef] [Green Version]
- Karakus, E.; Schmid, A.; Leiting, S.; Fuhler, B.; Schaffler, A.; Jakob, T.; Geyer, J. Role of the Steroid Sulfate Uptake Transporter Soat (Slc10a6) in Adipose Tissue and 3T3-L1 Adipocytes. Front. Mol. Biosci. 2022, 9, 863912. [Google Scholar] [CrossRef]
- Galuska, C.E.; Hartmann, M.F.; Sanchez-Guijo, A.; Bakhaus, K.; Geyer, J.; Schuler, G.; Zimmer, K.P.; Wudy, S.A. Profiling intact steroid sulfates and unconjugated steroids in biological fluids by liquid chromatography-tandem mass spectrometry (LC-MS-MS). Analyst 2013, 138, 3792–3801. [Google Scholar] [CrossRef]





| Gene | Chromosome | Protein | Protein Length | Transport Substrate | |||
|---|---|---|---|---|---|---|---|
| Steroid Sulfates | Bile Acids | TLC | |||||
 | SLC10A1 | 14q24 | NTCP | 349 aa | + | + | + |
| SLC10A4 | 4p11 | P4 | 437 aa | − | − | − | |
| SLC10A2 | 13q33 | ASBT | 348 aa | − | + | + | |
| SLC10A6 | 4q21 | SOAT | 377 aa | + | − | + | |
| SLC10A5 | 8q21 | P5 | 438 aa | − | − | − | |
| SLC10A3 | Xq28 | P3 | 477 aa | − | − | − | |
| SLC10A7 | 4q31 | RCAS | 340 aa | − | − | − | |
| Cell Line | Organ | Origin | Cell Type | Category |
|---|---|---|---|---|
| ASC diff | Mesenchymal | Adipose tissue | Adipose stromal cells | Uncategorized |
| BJ | Mesenchymal | Foreskin | Fibroblast | Finite |
| BJ hTERT+ | Mesenchymal | Foreskin | Fibroblast | Telomerase immortalized |
| HaCaT | Skin | Skin | Keratinocyte | Spontaneously immortalized |
| HBEC3-KT | Lung | Central lung bronchiole | Bronchial epithelial cell | Telomerase immortalized |
| HeLa | Female reproductive system | Cervix | Epithelial cells derived from cervical cancer cells | Cancer |
| HSkMC | Muscle | Trapezius and erector spinae muscles | Skeletal muscle cells | Uncategorized |
| hTCEpi | Eye | Cornea | Corneal epithelial cells | Telomerase immortalized |
| U-266/70 | Lymphoid | Peripheral blood | Cells derived from multiple myeloma cells | Cancer |
| U-266/84 | Lymphoid | Peripheral blood | Cells derived from multiple myeloma cells | Cancer |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wannowius, M.; Karakus, E.; Aktürk, Z.; Breuer, J.; Geyer, J. Role of the Sodium-Dependent Organic Anion Transporter (SOAT/SLC10A6) in Physiology and Pathophysiology. Int. J. Mol. Sci. 2023, 24, 9926. https://doi.org/10.3390/ijms24129926
Wannowius M, Karakus E, Aktürk Z, Breuer J, Geyer J. Role of the Sodium-Dependent Organic Anion Transporter (SOAT/SLC10A6) in Physiology and Pathophysiology. International Journal of Molecular Sciences. 2023; 24(12):9926. https://doi.org/10.3390/ijms24129926
Chicago/Turabian StyleWannowius, Marie, Emre Karakus, Zekeriya Aktürk, Janina Breuer, and Joachim Geyer. 2023. "Role of the Sodium-Dependent Organic Anion Transporter (SOAT/SLC10A6) in Physiology and Pathophysiology" International Journal of Molecular Sciences 24, no. 12: 9926. https://doi.org/10.3390/ijms24129926
APA StyleWannowius, M., Karakus, E., Aktürk, Z., Breuer, J., & Geyer, J. (2023). Role of the Sodium-Dependent Organic Anion Transporter (SOAT/SLC10A6) in Physiology and Pathophysiology. International Journal of Molecular Sciences, 24(12), 9926. https://doi.org/10.3390/ijms24129926