

Transcriptomic Analysis of the Dehydration Rate of Mature Rice (Oryza sativa) Seeds

Abstract
:1. Introduction
2. Results
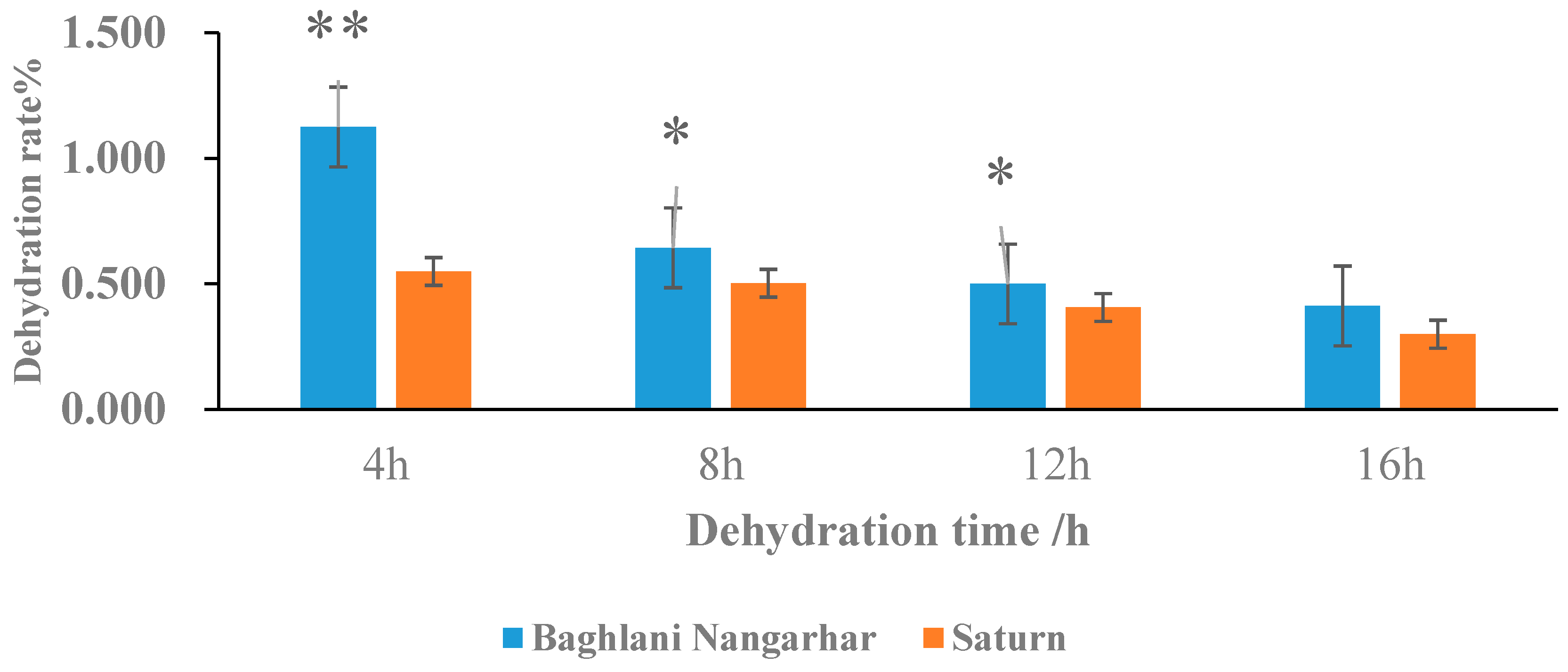
2.1. Dehydration Rate Performance of Rice Core Seed in Relation to Rapid and Slow Dehydration Genotypes
2.2. Transcriptome Sequencing Data Statistics
2.3. Identification of Differentially Expressed Genes
2.4. GO Enrichment Analysis of Differentially Expressed Genes
2.5. KEGG Enrichment Analysis of Differentially Expressed Genes
2.6. Analysis of Dehydration-Related Candidate Genes in Transcriptome
2.6.1. Screening of Candidate Genes in Rapid and Slow Dehydration Materials
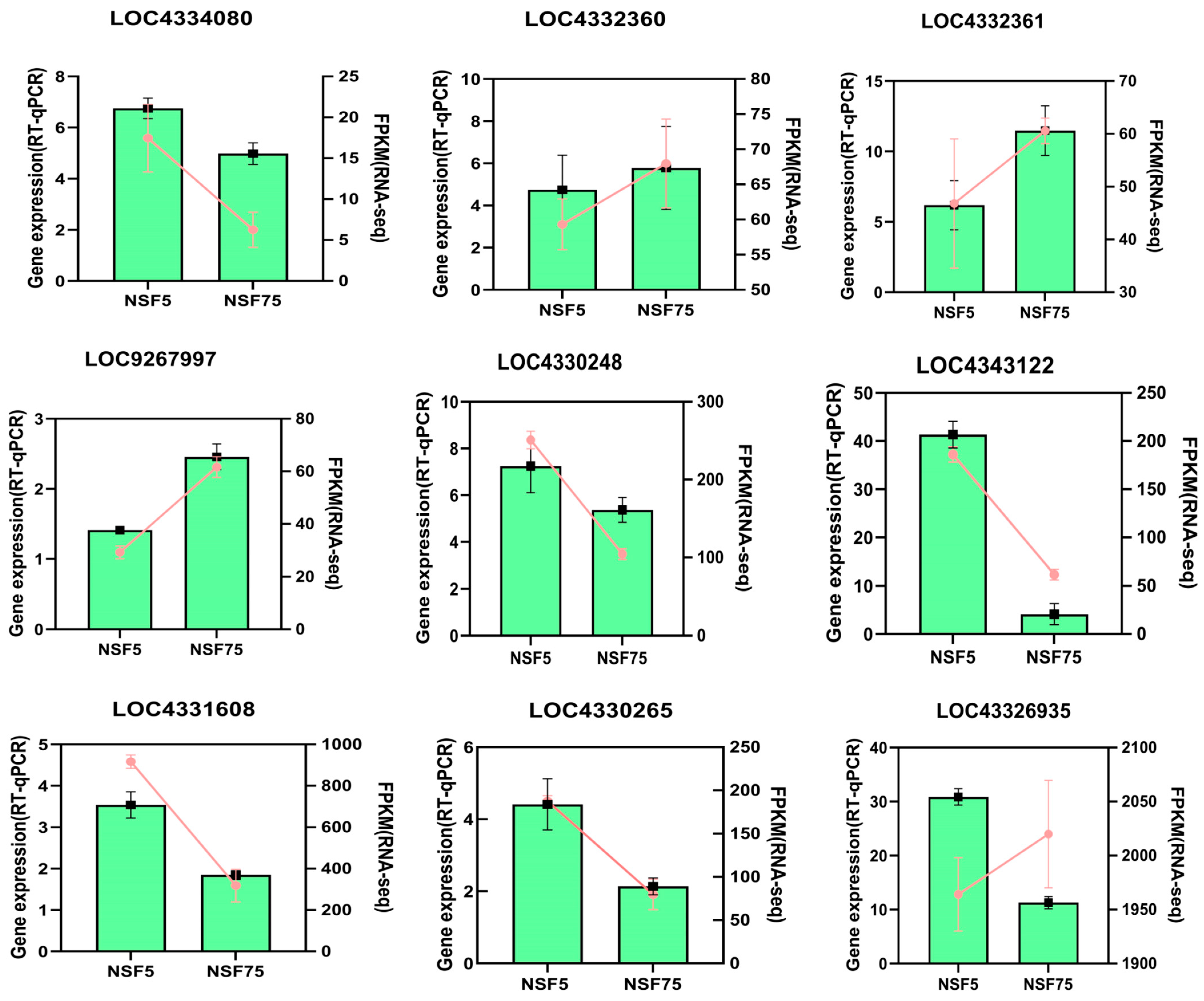
2.6.2. Validation of Transcriptome Sequencing Genes via RT-qPCR
3. Discussion
4. Materials and Methods
4.1. Material Handling
4.2. Methods
4.2.1. Seed RNA Extraction
4.2.2. Transcriptome Sequencing and Data Assembly
4.2.3. Functional Annotation, Classification, and Metabolic Pathway Analysis
- (1)
- Analysis of differentially expressed genes (DEGs). A total of four comparison groups were constructed: CNSF5 vs. NSF5, CNSF5 vs. CNSF75, NSF5 vs. NSF75, and CNSF75 vs. NSF75. The results of the clean reads were compared with the reference genome and stored in binary files. Gene FPKM (FPKM = total exon fragments/mapped reads (Millions) × exon length (kb)) [47] was quantified using Cufflinks [48]. The number of reads of genes in the samples was obtained using HTSeq-count [49] (California Institute of Technology, Pasadena, CA, USA) software. The data were normalized using the software DESeq2 [50], the R package was used to estimate the size factor function for normalization, and the nbinom test function was used to calculate fold-change values and p-values for comparative differences to control the false discovery rate. DEGs were selected with p-values < 0.05. After corrections were performed, a rigorous threshold (Q-value 0.05) was utilized based on the method of Bonferroni [51] to correct the significance levels of terms and pathways.
- (2)
- Enrichment analysis of DEGs via GO and KEGG. GO and KEGG enrichment analyses were performed on the screened differentially expressed genes using GO (http://www.geneontology.org accessed on 1 May 2023) [52] and KEGG (https://www.kegg.jp accessed on 1 May 2023) databases [53]. Term and pathway significance was assessed using the corrected Q-value < 0.05 [51].
4.2.4. RT-qPCR Validation of Differentially Expressed Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Manfre, A.J.; LaHatte, G.A.; Climer, C.R.; Marcotte, W.R. Seed Dehydration and the Establishment of Desiccation Tolerance during Seed Maturation is Altered in the Arabidopsis thaliana Mutant atem6-1. Plant Cell Physiol. 2009, 50, 243–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.H.; Guo, J.L.; Yu, T.; Shi, Z.S. Comparative Study on Dehydration Rate of Kernel among Maize Hybrids and Parents with Different Maturity Periods. J. Maize Sci. 2012, 20, 17–24. [Google Scholar]
- Zhang, W.J.; Wang, Y.H.; Wang, K.R.; Zhao, J.; Zhao, R.L.; Li, S.K. Grain Dehydration Rate of Different Maize Varieties. Crops 2016, 1, 76–81. [Google Scholar]
- Zhu, D.M.; Hu, W.J.; Bie, T.D.; Lu, C.B.; Zhao, R.H.; Gao, D.R. QTL Mapping for Kernel Dehydration Rate after Physiological Maturity Using Four Way RIL Populations of Wheat. J. Triticeae Crops 2020, 40, 49–54. [Google Scholar]
- Xue, P.; Wen, B. Effects of Drying Rates on the Desiccation Tolerance of Citrus maxima‘Feizhouyou’Seeds. Plant Divers. Resour. 2015, 37, 293–300. [Google Scholar]
- Yang, Q.H.; Yin, S.H.; Xia, Y.M.; Lan, Q.Y. Preliminary Study on Germination Capacity and Desiccation-tolerance of Cassia hirsuta Seeds at Various Developmental Stages. J. Wuhan Bot. Res. 2002, 20, 288–292. [Google Scholar]
- Kong, D.J.; Hao, L.Z.; Zhang, F.L.; Zhao, P.; Yang, Z.R.; Buren, J.Y. Desiccation Tolerance of Pugionium Cornutum Seeds during the Development. Plant Physiol. J. 2014, 50, 324–330. [Google Scholar]
- Liu, X.J.; Wang, Z.H.; Wang, X.; Li, T.F.; Zhang, L. Primary Mapping of QTL for Dehydration Rate of Maize Kernel after Physiological Maturing. Acta Agron. Sin. 2010, 36, 47–52. [Google Scholar] [CrossRef]
- Huang, H.; Moller, I.M.; Song, S.Q. Proteomics of desiccation tolerance during development and germination of maize embryos. J. Proteom. 2011, 75, 1247–1262. [Google Scholar] [CrossRef] [PubMed]
- Farrant, J.M.; Pammenter, N.W.; Berjak, P.; Farnsworth, E.J.; Vertucci, C.W. Presence of dehydrin-like proteins and levels of abscisic acid in recalcitrant (desiccation sensitive) seeds may be related to habitat. Seed Sci. Res. 1996, 6, 175–182. [Google Scholar] [CrossRef]
- Li, Y.L.; Dong, Y.B.; Yang, M.L.; Wang, Q.L.; Shi, Q.L.; Zhou, Q.; Deng, F.; Ma, Z.Y.; Qiao, D.H.; Xu, H. QTL Detection for Grain Water Relations and Genetic Correlations with Grain Matter Accumulation at Four Stages after Pollination in Maize. J. Plant Biochem. Physiol. 2014, 2, 1000121–1000129. [Google Scholar]
- Valérie, C.; Carine, R.; Laurence, M.; Agnès, R.; Aline, M.; Agnès, M.; Matthieu, F.; Alain, C.; Claudine, T.; Peter, R.; et al. QTLs and candidate genes for desiccation and abscisic acid content in maize kernels. BMC Plant Biol. 2010, 10, 2. [Google Scholar]
- Li, W.Q.; Yu, Y.H.; Wang, L.X.; Luo, Y.; Peng, Y.; Xu, Y.C.; Liu, X.G.; Wu, S.S.; Jian, L.M.; Xu, J.T.; et al. The genetic architecture of the dynamic changes in grain moisture in maize. Plant Biotechnol. J. 2021, 19, 1195–1205. [Google Scholar] [CrossRef]
- Costa, M.C.; Harm, N.; Ligterink, W.; Buitink, J.; Hilhorst, H.W. Time-series analysis of the transcriptome of the re-establishment of desiccation tolerance by ABA in germinated Arabidopsis thaliana seeds. Genom. Data 2015, 5, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Cecília, C.M.; Karima, R.; Harm, N.; Farzaneh, Y.; Wilco, L.; Julia, B.; Hilhorst, H.W. A gene co-expression network predicts functional genes controlling the re-establishment of desiccation tolerance in germinated Arabidopsis thaliana seeds. Planta 2015, 242, 435–449. [Google Scholar]
- Sandra, I.G.; Ricardo, A.C.; Corina, H.K.; Gerardo, A.J.; Thelma, Y.R.; Stefan, D.F.; Luis, H.E. Regulatory network analysis reveals novel regulators of seed desiccation tolerance in Arabidopsis thaliana. Pnas 2016, 113, E5232–E5241. [Google Scholar]
- Farrant, J.M.; Cooper, K.; Hilgart, A.; Abdalla, K.O.; Bentley, J.; Thomson, J.A.; Dace, H.J.; Peton, N.; Mundree, S.G.; Rafudeen, M.S. A molecular physiological review of vegetative desiccation tolerance in the resurrection plant Xerophytaviscosa (Baker). Planta 2015, 242, 407–426. [Google Scholar] [CrossRef] [Green Version]
- Stephane, D.; Julien, S.; Aldecinei, B.S.; Fabienne, M.; Eveline, D.; Valérie, R.; Philippe, L.; Hervé, E.; Thierry, J. Integrative analysis of the late maturation programme and desiccation tolerance mechanisms in intermediate coffee seeds. J. Exp. Bot. 2018, 69, 1583–1597. [Google Scholar]
- Robert, V.B.; Wai, C.M.; Zhang, Q.W.; Song, X.M.; Edger, P.P.; Bryan, D.; Michael, T.P.; Mockler, T.C.; Bartels, D. Seed desiccation mechanisms coopted for vegetative desiccation in the resurrection grass Oropetium thomaeum. Plant Cell Environ. 2017, 40, 2292–2306. [Google Scholar]
- Li, D.; Li, Y.; Qian, J.; Liu, X.; Xu, H.; Zhang, G.; Ren, J.; Wang, L.; Zhang, L.; Yu, H. Comparative Transcriptome Analysis Revealed Candidate Genes Potentially Related to Desiccation Sensitivity of Recalcitrant Quercus variabilis Seeds. Front. Plant Sci. 2021, 12, 717563. [Google Scholar] [CrossRef]
- Li, C.; Huang, L.; Zhang, M.W.; Liu, J.B.; Niu, J.; Qiao, J.F. Transcriptome Analysis Revealing the Mechanisms of Kernel Dehydration Rate Responding to ABA Application in Maize. Acta Agric. Boreali-Sin. 2020, 35, 15–26. [Google Scholar]
- Qu, J.Z.; Xu, S.T.; Tian, X.K.; Li, T.; Wang, L.C.; Zhong, Y.Y.; Xue, J.Q.; Guo, D.M. Comparative transcriptomics reveals the difference in early endosperm development between maize with different amylose contents. PeerJ 2019, 7, e7528. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, S.; Satoh, K.; Nagata, T.; Kawagashira, N.; Doi, K.; Kishimoto, N.; Yazaki, J.; Ishikawa, M.; Yamada, H.; Ooka, H.; et al. Collection, mapping, and annotation of over 28,000 cDNA clones from japonica rice. Science 2003, 301, 376–379. [Google Scholar] [CrossRef]
- Biswal, A.K.; McConnell, E.W.; Werth, E.G.; Lo, S.F.; Yu, S.M.; Hicks, L.M.; Jones, A.M. The Nucleotide-Dependent Interactome of Rice Heterotrimeric G-Proteinα–Subunit. Proteomics 2019, 19, e1800385. [Google Scholar] [CrossRef]
- Nordquist, K.A.; Dimitrova, Y.N.; Brzovic, P.S.; Ridenour, W.B.; Munro, K.A.; Soss, S.E.; Caprioli, R.M.; Klevit, R.E.; Chazin, W.J. Structural and functional characterization of the monomeric U-box domain from E4B. Biochemistry 2010, 49, 347–355. [Google Scholar] [CrossRef] [Green Version]
- Wiborg, J.; O’Shea, C.; Skriver, K. Biochemical function of typical and variant Arabidopsis thaliana U-box E3 ubiquitin-protein ligases. Biochem. J. 2008, 413, 447–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Cheng, H.H.; Zhou, R.J. Molecular mechanisms and functions of autophagy and the ubiquitin-proteasome pathway. Hereditas 2012, 34, 5–18. [Google Scholar] [PubMed]
- Chen, L.T.; Hamada, S.; Fujiwara, M.; Zhu, T.H.; Thao, N.P.; Wong, H.L.; Krishna, P.; Ueda, T.; Kaku, H.; Shibuya, N.; et al. The Hop/Sti1-Hsp90 Chaperone Complex Facilitates the Maturation and Transport of a PAMP Receptor in Rice Innate Immunity. Cell Host Microbe 2010, 7, 185–196. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.L. Research on the Changes of Seed Vigor and Protection Mechanisms of Mature Dehydration during Wheat Seeds Development; Shandong Agricultural University: Tai’an, China, 2017. [Google Scholar]
- Liu, D. Proteomics and Transcriptomics Analysis of Desiccation Tolerance of Mandshurica Ash Seeds in Response to Re-Dehydration; Northeast Forestry University: Harbin, China, 2013. [Google Scholar]
- Scandalios, J.G. Oxygen stress and superoxide dismutases. Plant Physiol. 1993, 101, 7–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.L.; Wang, X.F.; Jing, X.M.; Lin, J. The Effect of Moisture Content on Storage Life of Rice Seeds. Sci. Agric. Sin. 2005, 38, 1480–1486. [Google Scholar]
- Chen, Q.Z.; Mao, P.S. Advances in heat shock proteins of seed. Pratacultural Sci. 2016, 33, 136–143. [Google Scholar]
- Yu, Y.; Xia, Y.X.; Cai, S.X. The small molecule heat-shock protein in plants. China Biotechnol. 2003, 23, 38–41. [Google Scholar]
- Bai, G.X. Studies on Ultradry Storage and Mechanisms of Desiccation Tolerance of Rice Seeds; Beijing Forestry University: Beijing, China, 2005. [Google Scholar]
- Kermode, A.R. Approaches to elucidate the basis of desiccation-tolerance in seeds. Seed Sci. Res. 1997, 7, 75–95. [Google Scholar] [CrossRef]
- Li, G.L.; Wu, H.X.; Sun, Y.Q. Proteome and its applied Advances in plant Drought stress response. Acta Agric. Boreali-Sin. 2015, 35, 2132–2140. [Google Scholar]
- Waters, E.R. The evolution, function, structure, and expression of the plant sHSPs. J. Exp. Bot. 2013, 64, 391–403. [Google Scholar] [CrossRef] [Green Version]
- Hilton, G.R.; Lioe, H.; Stengel, F.; Baldwin, A.J.; Benesch, J.L. Small heat-shock proteins: Paramedics of the cell. Top. Curr. Chem. 2013, 328, 69–98. [Google Scholar] [PubMed]
- Harmeet, K.; Petla, B.P.; Kamble, N.U.; Singh, A.; Rao, V.; Ghosh, S.; Majee, M. Differentially expressed seed aging responsive heat shock protein OsHSP18.2 implicates in seed vigor, longevity and improves germination and seedling establishment under abiotic stress. Front. Plant Sci. 2015, 6, 713. [Google Scholar]
- Sluse, F.E.; Jarmuszkiewicz, W. Alternative oxidase in the branched mitochondrial respiratory network: An overview on structure, function, regulation, and role. J. Med. Biol. Res. 1998, 31, 733–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Li, Y.; Kristiansen, K.; Wang, J. SOAP: Short oligonucleotide alignment program. Bioinformatics 2008, 24, 713–714. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raivo, K. Package ‘pheatmap’, version 1.0.12; R foundation: Indianapolis, IN, USA, 2019.
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, A.; Trapnell, C.; Donaghey, J.; Rinn, J.L.; Pachter, L. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 2011, 12, R22. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Abdi, H. The Bonferonni and Sˇidák. Corrections for Multiple Comparisons. Encycl. Meas. Stat. 2007, 1, 2007. [Google Scholar]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, 480–484. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NCBI Gene No. | MSU Gene No. | Variance Multiplier | Gene Annotation | |
|---|---|---|---|---|
| log2 (NSF5/CNSF5) | log2 (NSF75/ CNSF75) | |||
| LOC4341326 | LOC_Os06g36930 | 4.3997 | 4.3444 | Putative heat-stress transcription factor A-6a |
| LOC4334080 | LOC_Os03g53340 | 1.1557 | 1.7690 | Similar to heat-stress transcription factor A-2a |
| LOC9268751 | 2.2410 | 1.7960 | Ethylene-responsive transcription factor ABR1 | |
| LOC107279585 | 1.4887 | 1.1606 | ATP synthase subunit 9, mitochondrial | |
| LOC4332361 | LOC_Os03g16030 | 1.9368 | 2.3599 | Low-molecular-mass heat-shock protein Oshsp18.0 |
| LOC9267997 | LOC_Os04g01740 | 1.5389 | 1.7422 | Similar to heat-shock protein 82 |
| Heat-shock protein 81-1 (HSP81-1) (Heat-shock protein 83) | ||||
| Similar to heat-shock protein 80 | ||||
| Non-protein-coding transcript | ||||
| LOC4332360 | LOC_Os03g16020 | 1.3314 | 1.2284 | Low-molecular-mass heat-shock protein Oshsp17.3 |
| NCBI Gene No. | MSU No. or RAP No. | Function Comments | Molecular Function | Biological Process |
|---|---|---|---|---|
| LOC4334080 | LOC_Os03g53340 | Similar to heat-stress transcription factor A-2a | GO:0000978 RNA polymerase II proximal promoter sequence-specific DNA binding; GO:0003677 DNA binding; GO:0003700 DNA-binding transcription factor activity; GO:0043565 sequence-specific DNA binding | GO:0006355 regulation of transcription, DNA-templated; GO:0034605 cellular response to heat; GO:0043618 regulation of transcription from RNA polymerase II promoter in response to stress; GO:0061408 positive regulation of transcription from RNA polymerase II promoter in response to heat stress |
| LOC4332360 | LOC_Os03g16020 | 17.4 kDa class I heat-shock protein-like | GO:0043621 protein self-association; GO:0051082 unfolded protein binding | GO:0000302 response to reactive oxygen species; GO:0006457 protein folding; GO:0009408 response to heat; GO:0009651 response to salt stress; GO:0042542 response to hydrogen peroxide |
| LOC4332361 | LOC_Os03g16030 | 18.1 kDa class I heat-shock protein-like | GO:0043621 protein self-association; GO:0051082 unfolded protein binding | GO:0000302 response to reactive oxygen species; GO:0006457 protein folding; GO:0009408 response to heat; GO:0009651 response to salt stress; GO:0042542 response to hydrogen peroxide |
| LOC9267997 | LOC_Os04g01740 | Heat-shock protein 82 | GO:0005524 ATP binding; GO:0051082 unfolded protein binding; | GO:0006457 protein folding; GO:0034605 cellular response to heat; GO:0050821 protein stabilization |
| LOC4331608 | LOC_Os03g05290 | Probable aquaporin TIP1-1 | GO:0015250 water channel activity; GO:0015267 channel activity | GO:0006833 water transport; GO:0055085 transmembrane transport |
| LOC4330265 | LOC_Os02g44870 | Dehydrin DHN1-like | Unknown | GO:0006950 response to stress; GO:0009414 response to water deprivation; GO:0009415 response to water; GO:0009631 cold acclimation; GO:0009737 response to abscisic acid |
| LOC4326935 | LOC_Os01g50700 | Dehydrin Rab25-like | Unknown | GO:0009414 response to water deprivation; GO:0009415 response to water; GO:0009631 cold acclimation; GO:0009737 response to abscisic acid |
| LOC4330248 | LOC_Os02g44630 | Aquaporin PIP1-1-like | GO:0015250 water channel activity; GO:0015267 channel activity | GO:0006833 water transport; GO:0009414 response to water deprivation; GO:0055085 transmembrane transport |
| LOC4343122 | LOC_Os07g26690 | Probable aquaporin PIP2-1 | GO:0005215 transporter activity; GO:0015250 water channel activity; GO:0015267 channel activity | GO:0006810 transport; GO:0006833 water transport; GO:0055085 transmembrane transport |
| Serial Number | NCBI Login Number | Candidate Genes | Primer Sequences |
|---|---|---|---|
| 1 | LOC4343122 | LOC_Os07g26690 | F: TGTTTAGCCTGTACTCCCATTT |
| R: ACGGAGGGAGTATATTCCAGAT | |||
| 2 | LOC4332360 | LOC_Os03g16020 | F: GCATTGGGCTAATCTAAAACGA |
| R: GCACACCAAAAACACCAGTAAT | |||
| 3 | LOC4332361 | LOC_Os03g16030 | F: GGTTACCGGCTAGTAAGAAACT |
| R: TACTGCAATTGATCACAAACCG | |||
| 4 | LOC4334080 | LOC_Os03g53340 | F: CTACGAAGGTCGATCCGGATAG |
| R: CTTGATCGTCTTCAGGAGCTC | |||
| 5 | LOC9267997 | LOC_Os04g01740 | F: GGAGGAGGTGGACTGAATTAAA |
| R: ACTTTCTCAACGATGGCTTAGA | |||
| 6 | LOC4330248 | LOC_Os02g44630 | F: CATTCAAGAGCAGGTCTTAAGC |
| R: AGTTGTTCAGGGTTCAGATAGG | |||
| 7 | LOC4331608 | LOC_Os03g05290 | F: GAGTCCCAGTGGGTGTACT |
| R: GAGATGAAGAGGACCTCGTAGA | |||
| 8 | LOC4330265 | LOC_Os02g44870 | F: GAGAAGATCGAGGGTGATCAC |
| R: GCTTCTCCTTGATCTTGTCGAG | |||
| 9 | LOC4326935 | LOC_Os01g50700 | F: CAGTCGTGTTTCAGTTCGTTAA |
| R: GGATACACCGTACATGCATAGA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Z.; Gui, J.; Yan, Y.; Zhang, H.; He, J. Transcriptomic Analysis of the Dehydration Rate of Mature Rice (Oryza sativa) Seeds. Int. J. Mol. Sci. 2023, 24, 11527. https://doi.org/10.3390/ijms241411527
Liu Z, Gui J, Yan Y, Zhang H, He J. Transcriptomic Analysis of the Dehydration Rate of Mature Rice (Oryza sativa) Seeds. International Journal of Molecular Sciences. 2023; 24(14):11527. https://doi.org/10.3390/ijms241411527
Chicago/Turabian StyleLiu, Zhongqi, Jinxin Gui, Yuntao Yan, Haiqing Zhang, and Jiwai He. 2023. "Transcriptomic Analysis of the Dehydration Rate of Mature Rice (Oryza sativa) Seeds" International Journal of Molecular Sciences 24, no. 14: 11527. https://doi.org/10.3390/ijms241411527
APA StyleLiu, Z., Gui, J., Yan, Y., Zhang, H., & He, J. (2023). Transcriptomic Analysis of the Dehydration Rate of Mature Rice (Oryza sativa) Seeds. International Journal of Molecular Sciences, 24(14), 11527. https://doi.org/10.3390/ijms241411527