
A Genome-Wide Screen for the Exonisation of Reference SINE-VNTR-Alus and Their Expression in CNS Tissues of Individuals with Amyotrophic Lateral Sclerosis



Abstract
:1. Introduction
2. Results
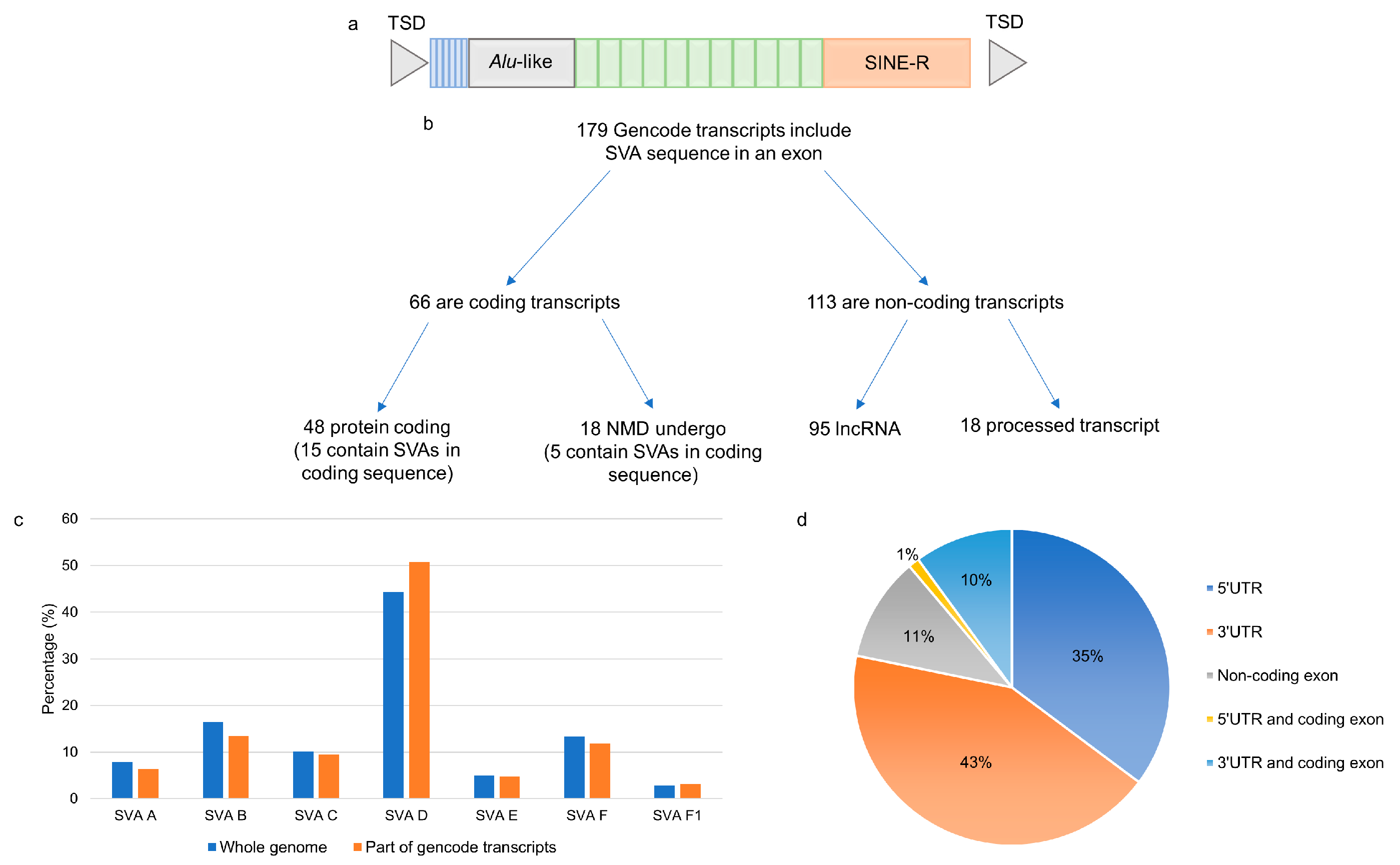
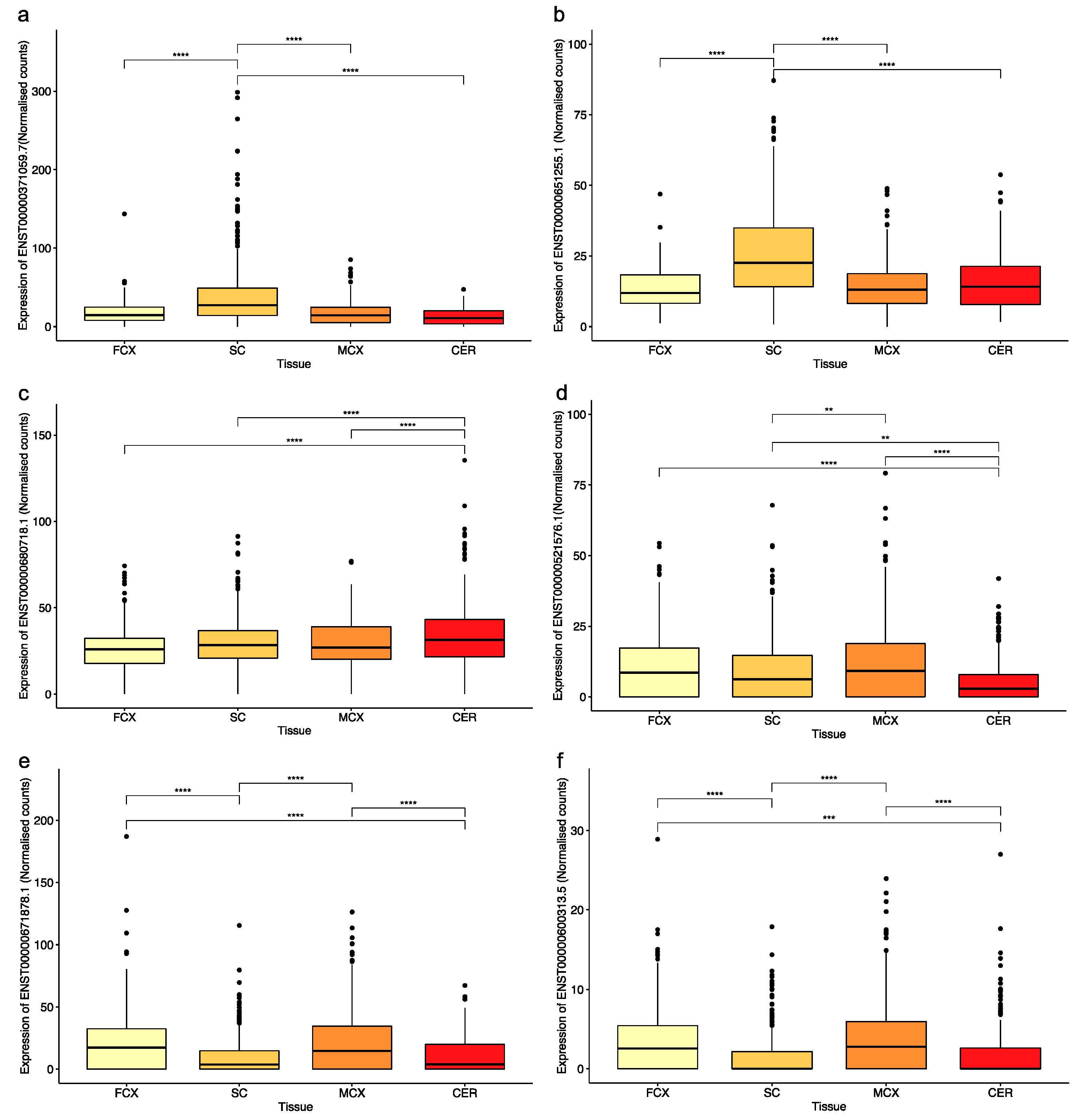
2.1. Exonisation of Reference SVAs in Transcripts That are Differentially Expressed in CNS Tissues
2.2. Identification of Novel Exonised Reference SVAs in the Motor Cortex
2.3. A Polymorphic SVA in an Intron of the CASP8 Gene Alters Transcript Sequence and Expression Levels
2.4. Association Analysis of Polymorphic Reference SVAs with ALS and Their Functional Effects
3. Discussion
4. Materials and Methods
4.1. Identification of Gencode Transcripts Containing Exonised Reference SVAs
4.2. DNA and RNA Samples from NIH NeuroBioBank
4.3. RNA Sequencing and Identification of Novel Exonised Reference SVAs
4.4. Quantification of Transcript Expression in the Target ALS Cohort
4.5. Amplification of Novel Exonised SVA Sequences and Transcripts Using RT-PCR
4.6. Genotyping of SVA Located in CASP8 in the NIH NeuroBioBank Cohort
4.7. Genotyping of Polymorphic Reference SVAs and Disease Association Analysis in Whole Genome Sequencing Data
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, H.; Xing, J.; Grover, D.; Hedges, D.J.; Han, K.; Walker, J.A.; Batzer, M.A. SVA Elements: A Hominid-specific Retroposon Family. J. Mol. Biol. 2005, 354, 994–1007. [Google Scholar] [CrossRef]
- Garcia-Perez, J.L.; Widmann, T.J.; Adams, I.R. The impact of transposable elements on mammalian development. Development 2016, 143, 4101–4114. [Google Scholar] [CrossRef] [Green Version]
- Raiz, J.; Damert, A.; Chira, S.; Held, U.; Klawitter, S.; Hamdorf, M.; Löwer, J.; Strätling, W.H.; Löwer, R.; Schumann, G.G. The non-autonomous retrotransposon SVA is trans -mobilized by the human LINE-1 protein machinery. Nucleic Acids Res. 2012, 40, 1666–1683. [Google Scholar] [CrossRef] [Green Version]
- Feusier, J.; Watkins, W.S.; Thomas, J.; Farrell, A.; Witherspoon, D.J.; Baird, L.; Ha, H.; Xing, J.; Jorde, L.B. Pedigree-based estimation of human mobile element retrotransposition rates. Genome Res. 2019, 29, 1567–1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sudmant, P.H.; Rausch, T.; Gardner, E.J.; Handsaker, R.E.; Abyzov, A.; Huddleston, J.; Zhang, Y.; Ye, K.; Jun, G.; Fritz, M.H.-Y.; et al. An integrated map of structural variation in 2,504 human genomes. Nature 2015, 526, 75–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hancks, D.C.; Kazazian, H.H., Jr. Roles for retrotransposon insertions in human disease. Mob. DNA 2016, 7, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfaff, A.L.; Bubb, V.J.; Quinn, J.P.; Koks, S. Reference SVA insertion polymorphisms are associated with Parkinson’s Disease progression and differential gene expression. npj Park. Dis. 2021, 7, 44. [Google Scholar] [CrossRef]
- Wang, L.; Norris, E.T.; Jordan, I.K. Human Retrotransposon Insertion Polymorphisms Are Associated with Health and Disease via Gene Regulatory Phenotypes. Front. Microbiol. 2017, 8, 1418. [Google Scholar] [CrossRef]
- Koks, S.; Pfaff, A.L.; Bubb, V.J.; Quinn, J.P. Expression Quantitative Trait Loci (eQTLs) Associated with Retrotransposons Demonstrate their Modulatory Effect on the Transcriptome. Int. J. Mol. Sci. 2021, 22, 6319. [Google Scholar] [CrossRef]
- Vialle, R.A.; Lopes, K.D.P.; Bennett, D.A.; Crary, J.F.; Raj, T. Integrating whole-genome sequencing with multi-omic data reveals the impact of structural variants on gene regulation in the human brain. Nat. Neurosci. 2022, 25, 504–514. [Google Scholar] [CrossRef]
- Wang, L.; Rishishwar, L.; Mariño-Ramírez, L.; Jordan, I.K. Human population-specific gene expression and transcriptional network modification with polymorphic transposable elements. Nucleic Acids Res. 2017, 45, 2318–2328. [Google Scholar] [CrossRef]
- De la Morena-Barrio, B.; Stephens, J.; de la Morena-Barrio, M.E.; Stefanucci, L.; Padilla, J.; Miñano, A.; Gleadall, N.; García, J.L.; López-Fernández, M.F.; Morange, P.-E.; et al. Long-Read Sequencing Identifies the First Retrotransposon Insertion and Resolves Structural Variants Causing Antithrombin Deficiency. Thromb. Haemost. 2022, 122, 1369–1378. [Google Scholar] [CrossRef] [PubMed]
- Pfaff, A.L.; Singleton, L.M.; Kõks, S. Mechanisms of disease-associated SINE-VNTR-Alus. Exp. Biol. Med. 2022, 247, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Vezain, M.; Thauvin-Robinet, C.; Vial, Y.; Coutant, S.; Drunat, S.; Urtizberea, J.A.; Rolland, A.; Jacquin-Piques, A.; Fehrenbach, S.; Nicolas, G.; et al. Retrotransposon insertion as a novel mutational cause of spinal muscular atrophy. Hum. Genet. 2023, 142, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Sorek, R.; Ast, G.; Graur, D. Alu-Containing Exons are Alternatively Spliced. Genome Res. 2002, 12, 1060–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Rheenen, W.; van der Spek, R.A.A.; Bakker, M.K.; van Vugt, J.; Hop, P.J.; Zwamborn, R.A.J.; de Klein, N.; Westra, H.-J.; Bakker, O.B.; Deelen, P.; et al. Common and rare variant association analyses in amyotrophic lateral sclerosis identify 15 risk loci with distinct genetic architectures and neuron-specific biology. Nat. Genet. 2021, 53, 1636–1648. [Google Scholar] [CrossRef] [PubMed]
- Hancks, D.C.; Ewing, A.D.; Chen, J.E.; Tokunaga, K.; Kazazian, H.H., Jr. Exon-trapping mediated by the human retrotransposon SVA. Genome Res. 2009, 19, 1983–1991. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, A.B.; Patwardhan, R.P.; Shendure, J.; Seelig, G. Learning the Sequence Determinants of Alternative Splicing from Millions of Random Sequences. Cell 2015, 163, 698–711. [Google Scholar] [CrossRef] [Green Version]
- Payer, L.M.; Steranka, J.P.; Ardeljan, D.; Walker, J.; Fitzgerald, K.C.; Calabresi, P.A.; Cooper, T.A.; Burns, K.H. Aluinsertion variants alter mRNA splicing. Nucleic Acids Res. 2019, 47, 421–431. [Google Scholar] [CrossRef] [Green Version]
- Stacey, S.N.; Kehr, B.; Gudmundsson, J.; Zink, F.; Jonasdottir, A.; Gudjonsson, S.A.; Sigurdsson, A.; Halldorsson, B.V.; Agnarsson, B.A.; Benediktsdottir, K.R.; et al. Insertion of an SVA-E retrotransposon into the CASP8 gene is associated with protection against prostate cancer. Hum. Mol. Genet. 2016, 25, 1008–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, Y.-J.; Choi, Y.; Eo, J.; Noh, Y.-N.; Gim, J.-A.; Jung, Y.-D.; Lee, J.-R.; Kim, H.-S. Structure and Expression Analyses of SVA Elements in Relation to Functional Genes. Genom. Inform. 2013, 11, 142–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savage, A.L.; Iacoangeli, A.; Schumann, G.G.; Rubio-Roldan, A.; Garcia-Perez, J.L.; Al Khleifat, A.; Koks, S.; Bubb, V.J.; Al-Chalabi, A.; Quinn, J.P. Characterisation of retrotransposon insertion polymorphisms in whole genome sequencing data from individuals with amyotrophic lateral sclerosis. Gene 2022, 843, 146799. [Google Scholar] [CrossRef]
- Savage, A.L.; Schumann, G.G.; Breen, G.; Bubb, V.J.; Al-Chalabi, A.; Quinn, J.P. Retrotransposons in the development and progression of amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2019, 90, 284–293. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.J.; Zucca, F.A.; Duyn, J.H.; Crichton, R.R.; Zecca, L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 2014, 13, 1045–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Li, C.; Chen, X.; Li, S.; Shang, H. Abnormal Serum Iron-Status Indicator Changes in Amyotrophic Lateral Sclerosis (ALS) Patients: A Meta-Analysis. Front. Neurol. 2020, 11, 380. [Google Scholar] [CrossRef]
- Van Bree, E.J.; Guimarães, R.L.; Lundberg, M.; Blujdea, E.R.; Rosenkrantz, J.L.; White, F.T.; Poppinga, J.; Ferrer-Raventós, P.; Schneider, A.-F.E.; Clayton, I.; et al. A hidden layer of structural variation in transposable elements reveals potential genetic modifiers in human disease-risk loci. Genome Res. 2022, 32, 656–670. [Google Scholar] [CrossRef]
- Brown, A.-L.; Wilkins, O.G.; Keuss, M.J.; Hill, S.E.; Zanovello, M.; Lee, W.C.; Bampton, A.; Lee, F.C.Y.; Masino, L.; Qi, Y.A.; et al. TDP-43 loss and ALS-risk SNPs drive mis-splicing and depletion of UNC13A. Nature 2022, 603, 131–137. [Google Scholar] [CrossRef]
- Klim, J.R.; Williams, L.A.; Limone, F.; San Juan, I.G.; Davis-Dusenbery, B.N.; Mordes, D.A.; Burberry, A.; Steinbaugh, M.J.; Gamage, K.K.; Kirchner, R.; et al. ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat. Neurosci. 2019, 22, 167–179. [Google Scholar] [CrossRef]
- Ma, X.R.; Prudencio, M.; Koike, Y.; Vatsavayai, S.C.; Kim, G.; Harbinski, F.; Briner, A.; Rodriguez, C.M.; Guo, C.; Akiyama, T.; et al. TDP-43 represses cryptic exon inclusion in the FTD–ALS gene UNC13A. Nature 2022, 603, 124–130. [Google Scholar] [CrossRef]
- Tollervey, J.R.; Curk, T.; Rogelj, B.; Briese, M.; Cereda, M.; Kayikci, M.; König, J.; Hortobágyi, T.; Nishimura, A.L.; Župunski, V.; et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat. Neurosci. 2011, 14, 452–458. [Google Scholar] [CrossRef] [Green Version]
- Savage, A.L.; Bubb, V.J.; Breen, G.; Quinn, J.P. Characterisation of the potential function of SVA retrotransposons to modulate gene expression patterns. BMC Evol. Biol. 2013, 13, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 2019, 47, e47. [Google Scholar] [CrossRef] [Green Version]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NNC (n = 322) | ALS/ALSND (n = 2663) | |
|---|---|---|
| Gender | ||
| Male | 157 (48.8%) | 1601 (60.1%) |
| Female | 165 (51.2%) | 1062 (39.9%) |
| Age * (years) | ||
| Mean (min-max) | 57.4 (17–90) | 59.1 (12–90) |
| Chr | Coordinates | ID | Minor Allele | MAF | OR (95% CI) | Unadj p Value | Bonferroni p Value | |
|---|---|---|---|---|---|---|---|---|
| NNC | ALS/ ALSND | |||||||
| 6 | 29,932,007–29,933,750 | SVA_28 | P | 0.29 | 0.24 | 0.78 (0.65–0.94) | 0.008 | 0.50 |
| 19 | 40,107,200–40,109,854 | SVA_82 | A | 0.15 | 0.19 | 1.37 (1.07–1.75) | 0.012 | 0.77 |
| 3 | 133,784,939–133,786,238 | SVA_20 | A | 0.29 | 0.33 | 1.27 (1.04–1.55) | 0.018 | 1 |
| 9 | 33,423,379–33,424,657 | SVA_51 | A | 0.25 | 0.22 | 0.80 (0.65–0.97) | 0.027 | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pfaff, A.L.; Bubb, V.J.; Quinn, J.P.; Koks, S. A Genome-Wide Screen for the Exonisation of Reference SINE-VNTR-Alus and Their Expression in CNS Tissues of Individuals with Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2023, 24, 11548. https://doi.org/10.3390/ijms241411548
Pfaff AL, Bubb VJ, Quinn JP, Koks S. A Genome-Wide Screen for the Exonisation of Reference SINE-VNTR-Alus and Their Expression in CNS Tissues of Individuals with Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences. 2023; 24(14):11548. https://doi.org/10.3390/ijms241411548
Chicago/Turabian StylePfaff, Abigail L., Vivien J. Bubb, John P. Quinn, and Sulev Koks. 2023. "A Genome-Wide Screen for the Exonisation of Reference SINE-VNTR-Alus and Their Expression in CNS Tissues of Individuals with Amyotrophic Lateral Sclerosis" International Journal of Molecular Sciences 24, no. 14: 11548. https://doi.org/10.3390/ijms241411548
APA StylePfaff, A. L., Bubb, V. J., Quinn, J. P., & Koks, S. (2023). A Genome-Wide Screen for the Exonisation of Reference SINE-VNTR-Alus and Their Expression in CNS Tissues of Individuals with Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences, 24(14), 11548. https://doi.org/10.3390/ijms241411548