
Calpain Regulation and Dysregulation—Its Effects on the Intercalated Disk

Abstract
:1. Introduction
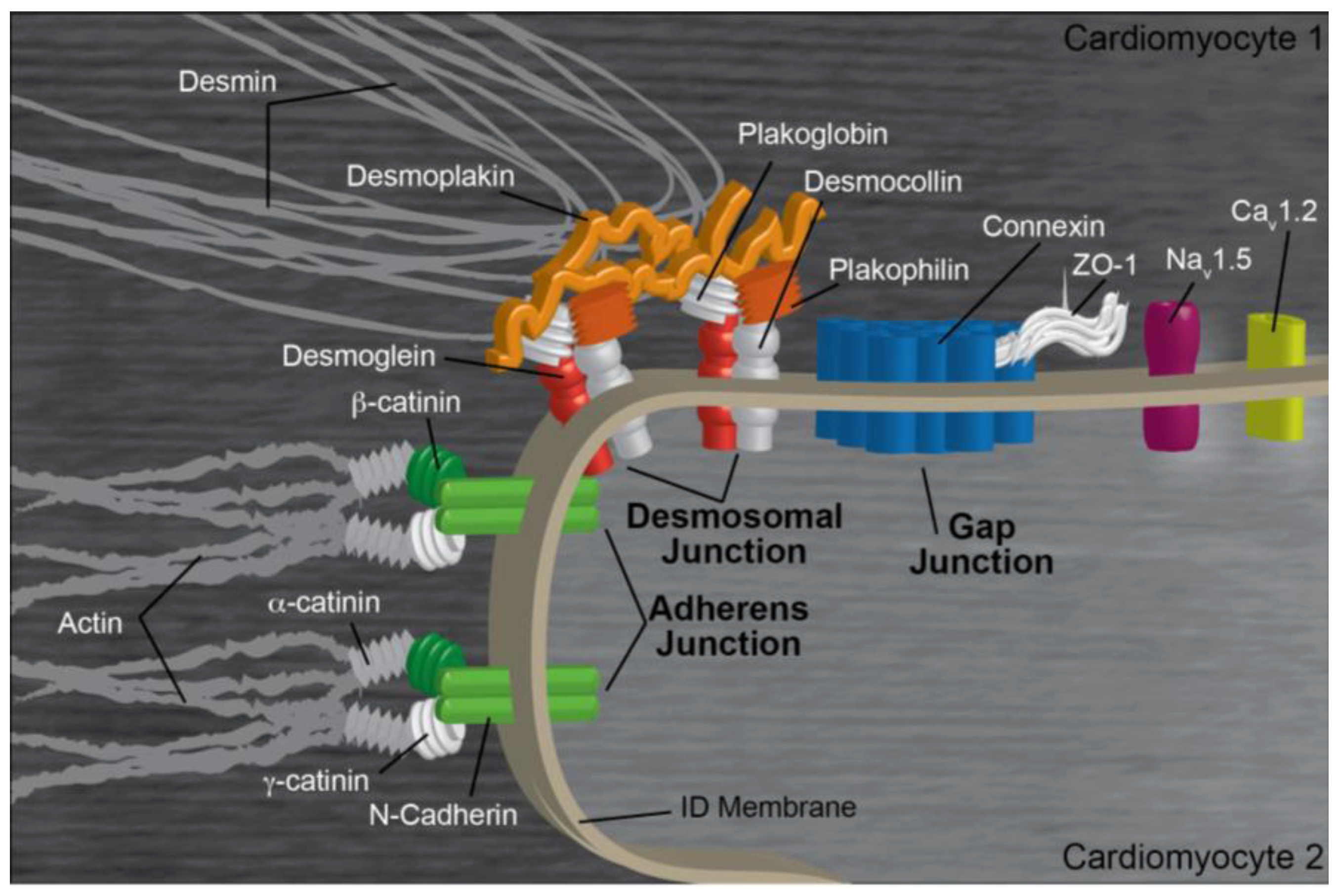
2. Intercalated Disk Components, Functions, and Regulation
2.1. Gap Junctions
2.2. Adherens Junctions
2.3. Desmosomes
3. Calpain
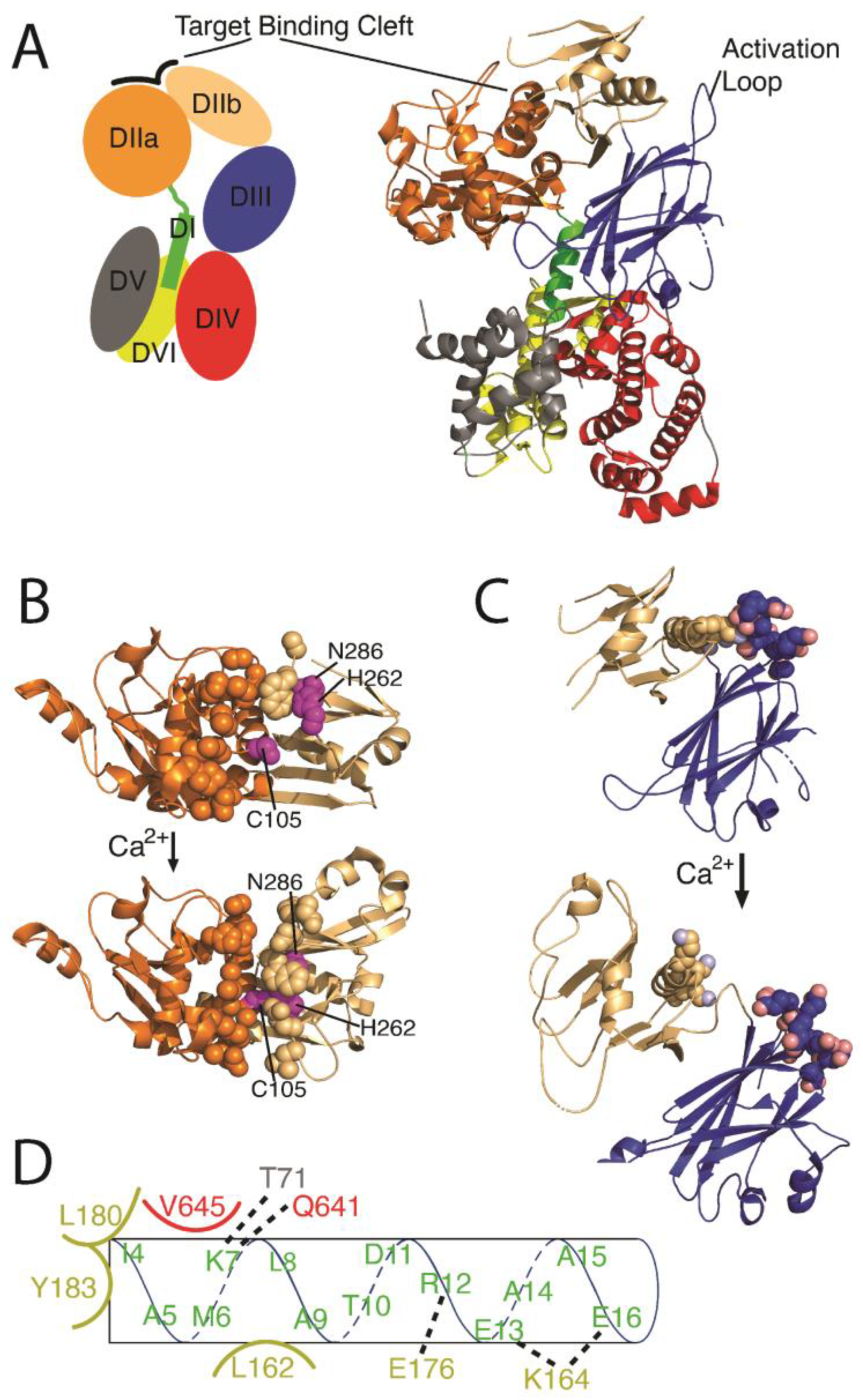
3.1. Calpain Isoforms and Structural Organization
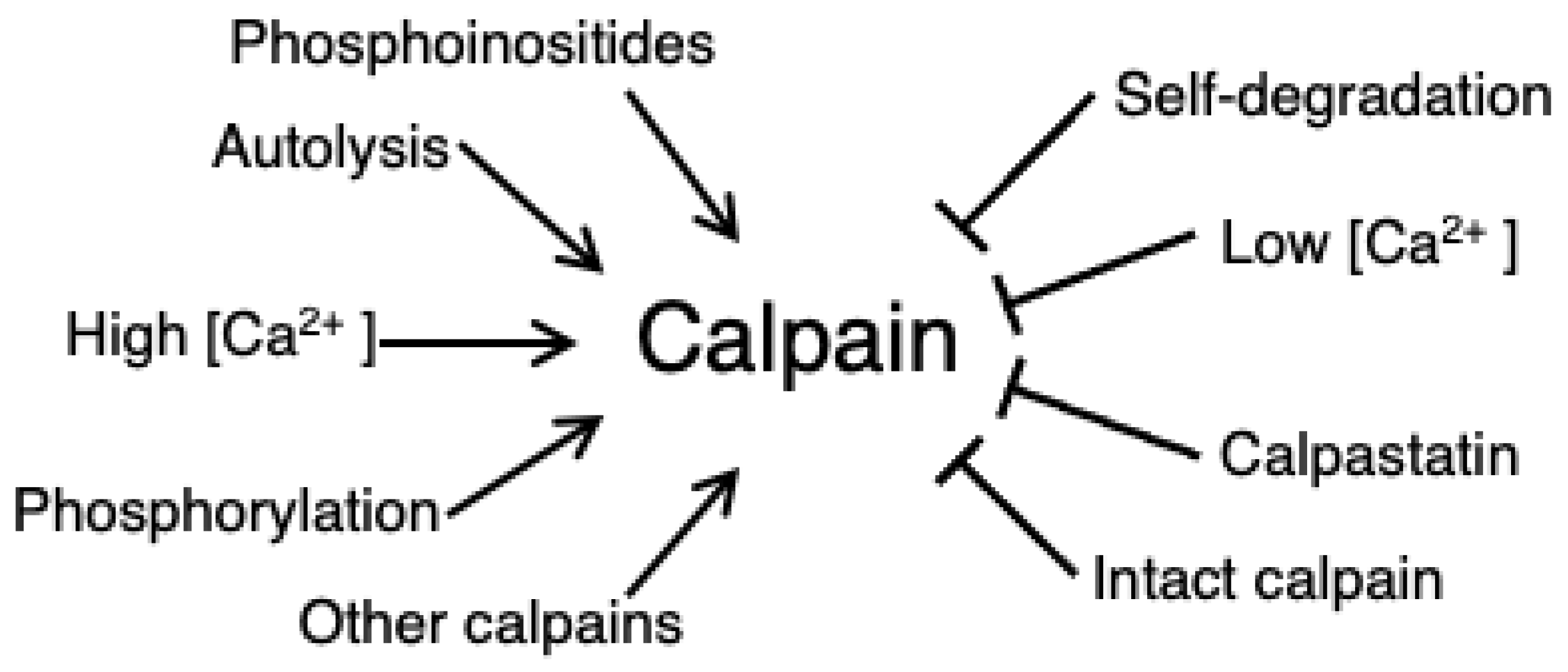
3.2. Calpain Activation and Regulation
4. Calpain in Cardiac Pathophysiology
4.1. Normal Calpain Function
4.2. Calpain-Mediated Pathophysiology in Non-Cardiac Tissue—What Can This Teach Us about Calpain-Linked Function within the ICD?
4.3. Calpain-Mediated Pathophysiology in the Heart
4.3.1. Calpain-Mediated ICD Targeting and Arrhythmogenic Cardiomyopathy
4.3.2. Calpain-Linked ICD Targeting and Atrial Fibrillation
4.3.3. Calpain-Linked ICD Targeting and Myocardial Infarction
4.3.4. Calpain-Linked ICD Targeting and Hyperhomocysteinemia
4.3.5. Calpain-Linked ICD Targeting and Other Heart Dysfunctions
4.4. Inhibiting Calpain in Heart Disease
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shaffer, F.; McCraty, R.; Zerr, C.L. A healthy heart is not a metronome: An integrative review of the heart’s anatomy and heart rate variability. Front. Psychol. 2014, 5, 1040. [Google Scholar] [CrossRef] [Green Version]
- Yin, S.; Zhang, X.; Zhan, C.; Wu, J.; Xu, J.; Cheung, J. Measuring Single Cardiac Myocyte Contractile Force via Moving a Magnetic Bead. Biophys. J. 2005, 88, 1489–1495. [Google Scholar] [CrossRef] [Green Version]
- Lin, G.; Pister, K.S.; Roos, K.P. Surface micromachined polysilicon heart cell force transducer. J. Microelectromechan. Syst. 2000, 9, 9–17. [Google Scholar] [CrossRef]
- Gordon, E.; Schimmel, L.; Frye, M. The Importance of Mechanical Forces for in vitro Endothelial Cell Biology. Front. Physiol. 2020, 11, 684. [Google Scholar] [CrossRef]
- Estigoy, C.B.; Pontén, F.; Odeberg, J.; Herbert, B.; Guilhaus, M.; Charleston, M.; Ho, J.W.K.; Cameron, D.; dos Remedios, C.G. Intercalated discs: Multiple proteins perform multiple functions in non-failing and failing human hearts. Biophys. Rev. 2009, 1, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Hervé, J.-C.; Derangeon, M. Gap-junction-mediated cell-to-cell communication. Cell Tissue Res. 2012, 352, 21–31. [Google Scholar] [CrossRef]
- Harris, T.J.C. An Introduction to Adherens Junctions: From Molecular Mechanisms to Tissue Development and Disease. In Adherens Junctions: From Molecular Mechanisms to Tissue Development and Disease; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2012; pp. 1–5. [Google Scholar]
- Delva, E.; Tucker, D.K.; Kowalczyk, A.P. The Desmosome. Cold Spring Harb. Perspect. Biol. 2009, 1, a002543. [Google Scholar] [CrossRef] [Green Version]
- Manring, H.R.; Dorn, L.E.; Ex-Willey, A.; Accornero, F.; Ackermann, M.A. At the heart of inter- and intracellular signaling: The intercalated disc. Biophys. Rev. 2018, 10, 961–971. [Google Scholar] [CrossRef]
- Kleber, A.G.; Saffitz, J.E. Role of the intercalated disc in cardiac propagation and arrhythmogenesis. Front. Physiol. 2014, 5, 404. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Lee, S.-M.; Ku, B.-J.; Moon, M.-J. Fine structure of the intercalated disc and cardiac junctions in the black widow spider Latrodectus mactans. Appl. Microsc. 2020, 50, 20. [Google Scholar] [CrossRef]
- Zhang, W.; Ma, X.; Zhong, M.; Zheng, Z.; Li, L.; Wang, Z.; Zhang, Y. Role of the Calpain System in Pulmonary Vein Connexin Remodeling in Dogs with Atrial Fibrillation. Cardiology 2008, 112, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Sinovas, A.; Sánchez, J.A.; Valls-Lacalle, L.; Consegal, M.; Ferreira-González, I. Connexins in the Heart: Regulation, Function and Involvement in Cardiac Disease. Int. J. Mol. Sci. 2021, 22, 4413. [Google Scholar] [CrossRef]
- Van Campenhout, R.; Gomes, A.R.; De Groof, T.W.M.; Muyldermans, S.; Devoogdt, N.; Vinken, M. Mechanisms Underlying Connexin Hemichannel Activation in Disease. Int. J. Mol. Sci. 2021, 22, 3503. [Google Scholar] [CrossRef] [PubMed]
- Pitts, J.D.; Finbow, M.E. The Gap Junction. J. Cell Sci. 1986, 239–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, M.V.; Malhotra, S.K. The gap junction. Adv. Struct. Biol. 1996, 4, 41–74. [Google Scholar] [CrossRef]
- Shimizu, K.; Stopfer, M. Gap Junctions. Curr. Biol. 2013, 23, R1026–R1031. [Google Scholar] [CrossRef] [Green Version]
- Severs, N.J.; Bruce, A.F.; Dupont, E.; Rothery, S. Remodelling of gap junctions and connexin expression in diseased myocardium. Cardiovasc. Res. 2008, 80, 9–19. [Google Scholar] [CrossRef] [Green Version]
- De Smet, M.A.; Lissoni, A.; Nezlobinsky, T.; Wang, N.; Dries, E.; Pérez-Hernández, M.; Lin, X.; Amoni, M.; Vervliet, T.; Witschas, K.; et al. Cx43 hemichannel microdomain signaling at the intercalated disc enhances cardiac excitability. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Shaw, R.M.; Colecraft, H.M. L-type calcium channel targeting and local signalling in cardiac myocytes. Cardiovasc. Res. 2013, 98, 177–186. [Google Scholar] [CrossRef] [Green Version]
- Jørgensen, N.R.; Teilmann, S.C.; Henriksen, Z.; Civitelli, R.; Sørensen, O.H.; Steinberg, T.H. Activation of L-type Calcium Channels Is Required for Gap Junction-mediated Intercellular Calcium Signaling in Osteoblastic Cells. J. Biol. Chem. 2003, 278, 4082–4086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hichri, E.; Abriel, H.; Kucera, J.P. Distribution of cardiac sodium channels in clusters potentiates ephaptic interactions in the intercalated disc. J. Physiol. 2018, 596, 563–589. [Google Scholar] [CrossRef] [PubMed]
- Charras, G.; Yap, A.S. Tensile Forces and Mechanotransduction at Cell–Cell Junctions. Curr. Biol. 2018, 28, R445–R457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mège, R.M.; Ishiyama, N. Integration of Cadherin Adhesion and Cytoskeleton at Adherens Junctions. Cold Spring Harb. Perspect. Biol. 2017, 9, a028738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanslembrouck, B.; Chen, J.-H.; Larabell, C.; van Hengel, J. Microscopic Visualization of Cell-Cell Adhesion Complexes at Micro and Nanoscale. Front. Cell Dev. Biol. 2022, 10, 745. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Qiu, Y.; Zhang, H.M.; Yang, D. Intercalated discs: Cellular adhesion and signaling in heart health and diseases. Heart Fail. Rev. 2018, 24, 115–132. [Google Scholar] [CrossRef] [PubMed]
- Ishiyama, N.; Lee, S.-H.; Liu, S.; Li, G.-Y.; Smith, M.J.; Reichardt, L.F.; Ikura, M. Dynamic and Static Interactions between p120 Catenin and E-Cadherin Regulate the Stability of Cell-Cell Adhesion. Cell 2010, 141, 117–128. [Google Scholar] [CrossRef] [Green Version]
- Hong, J.Y.; Oh, I.-H.; McCrea, P.D. Phosphorylation and isoform use in p120-catenin during development and tumorigenesis. Biochim. Biophys. Acta BBA Mol. Cell Res. 2016, 1863, 102–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, K.A.; Biggins, L.; Sharpe, H.J. Protein tyrosine phosphatases in cell adhesion. Biochem. J. 2021, 478, 1061–1083. [Google Scholar] [CrossRef]
- Kowalczyk, A.P.; Nanes, B.A. Adherens Junction Turnover: Regulating Adhesion Through Cadherin Endocytosis, Degradation, and Recycling. In Adherens Junctions: From Molecular Mechanisms to Tissue Development and Disease; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2012; pp. 197–222. [Google Scholar]
- Kreplak, L.; Herrmann, H.; Aebi, U. Tensile Properties of Single Desmin Intermediate Filaments. Biophys. J. 2008, 94, 2790–2799. [Google Scholar] [CrossRef] [Green Version]
- Cheng, F.; Eriksson, J.E. Intermediate Filaments and the Regulation of Cell Motility during Regeneration and Wound Healing. Cold Spring Harb. Perspect. Biol. 2017, 9, a022046. [Google Scholar] [CrossRef]
- McLendon, P.M.; Robbins, J. Desmin-related cardiomyopathy: An unfolding story. Am. J. Physiol. Circ. Physiol. 2011, 301, H1220–H1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, L.; Hatzfeld, M.; Keil, R. Desmosomes as Signaling Hubs in the Regulation of Cell Behavior. Front. Cell Dev. Biol. 2021, 9, 745670. [Google Scholar] [CrossRef] [PubMed]
- Sato, P.Y.; Coombs, W.; Lin, X.; Nekrasova, O.; Green, K.J.; Isom, L.L.; Taffet, S.M.; Delmar, M. Interactions Between Ankyrin-G, Plakophilin-2, and Connexin43 at the Cardiac Intercalated Disc. Circ. Res. 2011, 109, 193–201. [Google Scholar] [CrossRef]
- Ono, Y.; Sorimachi, H.; Suzuki, K. Structure and Physiology of Calpain, an Enigmatic Protease. Biochem. Biophys. Res. Commun. 1998, 245, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Ono, Y.; Sorimachi, H. Calpains—An elaborate proteolytic system. Biochim. Biophys. Acta BBA Proteins Proteom. 2012, 1824, 224–236. [Google Scholar] [CrossRef] [Green Version]
- Davis, T.L.; Walker, J.R.; Finerty, P.J.; Mackenzie, F.; Newman, E.M.; Dhe-Paganon, S. The Crystal Structures of Human Calpains 1 and 9 Imply Diverse Mechanisms of Action and Auto-inhibition. J. Mol. Biol. 2007, 366, 216–229. [Google Scholar] [CrossRef]
- Zheng, P.; Chen, X.; Zhu, H.; Zheng, W.; Mao, L.; Lin, C.; Liu, J.; Zheng, M. Capn4 is a marker of poor clinical outcomes and promotes nasopharyngeal carcinoma metastasis via nuclear factor-κB-induced matrix metalloproteinase 2 expression. Cancer Sci. 2014, 105, 630–638. [Google Scholar] [CrossRef] [Green Version]
- Sorimachi, H.; Ono, Y. Regulation and physiological roles of the calpain system in muscular disorders. Cardiovasc. Res. 2012, 96, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Campbell, R.L.; Davies, P.L. Structure–function relationships in calpains1. Biochem. J. 2012, 447, 335–351. [Google Scholar] [CrossRef]
- Hosfield, C.M.; Elce, J.S.; Davies, P.L.; Jia, Z. Crystal structure of calpain reveals the structural basis for Ca2+-dependent protease activity and a novel mode of enzyme activation. EMBO J. 1999, 18, 6880–6889. [Google Scholar] [CrossRef] [Green Version]
- Goll, D.E.; Thompson, V.F.; Li, H.; Wei, W.; Cong, J. The Calpain System. Physiol. Rev. 2003, 83, 731–801. [Google Scholar] [CrossRef]
- Strobl, S.; Fernandez-Catalan, C.; Braun, M.; Huber, R.; Masumoto, H.; Nakagawa, K.; Irie, A.; Sorimachi, H.; Bourenkow, G.; Bartunik, H.; et al. The crystal structure of calcium-free human m-calpain suggests an electrostatic switch mechanism for activation by calcium. Proc. Natl. Acad. Sci. USA 2000, 97, 588–592. [Google Scholar] [CrossRef]
- Bozóky, Z.; Alexa, A.; Tompa, P.; Friedrich, P. Multiple interactions of the ‘transducer’ govern its function in calpain activation by Ca2+. Biochem. J. 2005, 388, 741–744. [Google Scholar] [CrossRef] [Green Version]
- Alexa, A.; Bozóky, Z.; Farkas, A.; Tompa, P.; Friedrich, P. Contribution of Distinct Structural Elements to Activation of Calpain by Ca2+ Ions. J. Biol. Chem. 2004, 279, 20118–20126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cong, J.; E Goll, D.; Peterson, A.M.; Kapprell, H.P. The role of autolysis in activity of the Ca2+-dependent proteinases (μ-calpain and m-calpain). J. Biol. Chem. 1989, 264, 10096–10103. [Google Scholar] [CrossRef] [PubMed]
- Benyamin, Y. The structural basis of calpain behavior. FEBS J. 2006, 273, 3413–3414. [Google Scholar] [CrossRef] [PubMed]
- Khorchid, A.; Ikura, M. How calpain is activated by calcium. Nat. Struct. Biol. 2002, 9, 239–241. [Google Scholar] [CrossRef]
- Ye, Q.; Campbell, R.L.; Davies, P.L. Structures of human calpain-3 protease core with and without bound inhibitor reveal mechanisms of calpain activation. J. Biol. Chem. 2018, 293, 4056–4070. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Cao, J.; Gao, X.; Ma, Q.; Ren, J.; Xue, Y. GPS-CCD: A Novel Computational Program for the Prediction of Calpain Cleavage Sites. PLoS ONE 2011, 6, e19001. [Google Scholar] [CrossRef]
- Liu, Z.-X.; Yu, K.; Dong, J.; Zhao, L.; Liu, Z.; Zhang, Q.; Li, S.; Du, Y.; Cheng, H. Precise Prediction of Calpain Cleavage Sites and Their Aberrance Caused by Mutations in Cancer. Front. Genet. 2019, 10, 715. [Google Scholar] [CrossRef] [Green Version]
- Duverle, D.A.; Mamitsuka, H. CalCleaveMKL: A Tool for Calpain Cleavage Prediction. Calpain Methods Protoc. 2019, 1915, 121–147. [Google Scholar] [CrossRef]
- Shinkai-Ouchi, F.; Koyama, S.; Ono, Y.; Hata, S.; Ojima, K.; Shindo, M.; Duverle, D.; Ueno, M.; Kitamura, F.; Doi, N.; et al. Predictions of Cleavability of Calpain Proteolysis by Quantitative Structure-Activity Relationship Analysis Using Newly Determined Cleavage Sites and Catalytic Efficiencies of an Oligopeptide Array. Mol. Cell. Proteom. 2016, 15, 1262–1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tompa, P.; Buzder-Lantos, P.; Tantos, A.; Farkas, A.; Szilágyi, A.; Bánóczi, Z.; Hudecz, F.; Friedrich, P. On the Sequential Determinants of Calpain Cleavage. J. Biol. Chem. 2004, 279, 20775–20785. [Google Scholar] [CrossRef] [Green Version]
- Michetti, M.; Salamino, F.; Minafra, R.; Melloni, E.; Pontremoli, S. Calcium-binding properties of human erythrocyte calpain. Biochem. J. 1997, 325, 721–726. [Google Scholar] [CrossRef] [Green Version]
- Markowitz, J.; Rustandi, R.R.; Varney, K.M.; Wilder, P.T.; Udan, R.; Wu, S.L.; Horrocks, W.D.; Weber, D.J. Calcium-Binding Properties of Wild-Type and EF-Hand Mutants of S100B in the Presence and Absence of a Peptide Derived from the C-Terminal Negative Regulatory Domain of p53. Biochemistry 2005, 44, 7305–7314. [Google Scholar] [CrossRef] [PubMed]
- Immadisetty, K.; Sun, B.; Kekenes-Huskey, P.M. Structural Changes beyond the EF-Hand Contribute to Apparent Calcium Binding Affinities: Insights from Parvalbumins. J. Phys. Chem. B 2021, 125, 6390–6405. [Google Scholar] [CrossRef]
- Liriano, M.A.; Varney, K.M.; Wright, N.T.; Hoffman, C.L.; Toth, E.A.; Ishima, R.; Weber, D.J. Target Binding to S100B Reduces Dynamic Properties and Increases Ca2+-Binding Affinity for Wild Type and EF-Hand Mutant Proteins. J. Mol. Biol. 2012, 423, 365–385. [Google Scholar] [CrossRef] [Green Version]
- Wright, N.T.; Varney, K.M.; Ellis, K.C.; Markowitz, J.; Gitti, R.K.; Zimmer, D.B.; Weber, D.J. The Three-dimensional Solution Structure of Ca2+-bound S100A1 as Determined by NMR Spectroscopy. J. Mol. Biol. 2005, 353, 410–426. [Google Scholar] [CrossRef] [PubMed]
- Taveau, M.; Bourg, N.; Sillon, G.; Roudaut, C.; Bartoli, M.; Richard, I. Calpain 3 Is Activated through Autolysis within the Active Site and Lyses Sarcomeric and Sarcolemmal Components. Mol. Cell. Biol. 2003, 23, 9127–9135. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Thompson, V.F.; Goll, D.E. Effects of autolysis on properties of μ- and m-calpain. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2004, 1691, 91–103. [Google Scholar] [CrossRef] [Green Version]
- Saido, T.; Shibata, M.; Takenawa, T.; Murofushi, H.; Suzuki, K. Positive regulation of mu-calpain action by polyphosphoinositides. J. Biol. Chem. 1992, 267, 24585–24590. [Google Scholar] [CrossRef]
- Shao, H.; Chou, J.; Baty, C.J.; Burke, N.A.; Watkins, S.C.; Stolz, D.B.; Wells, A. Spatial Localization of m-Calpain to the Plasma Membrane by Phosphoinositide Biphosphate Binding during Epidermal Growth Factor Receptor-Mediated Activation. Mol. Cell. Biol. 2006, 26, 5481–5496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tompa, P.; Emori, Y.; Sorimachi, H.; Suzuki, K.; Friedrich, P. Domain III of Calpain Is a Ca2+-Regulated Phospholipid-Binding Domain. Biochem. Biophys. Res. Commun. 2001, 280, 1333–1339. [Google Scholar] [CrossRef]
- Lu, H.-T.; Feng, R.-Q.; Tang, J.-K.; Zhou, J.-J.; Gao, F.; Ren, J. CaMKII/calpain interaction mediates ischemia/reperfusion injury in isolated rat hearts. Cell Death Dis. 2020, 11, 388. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.-H.; Gu, X.-M.; Wu, F.; Jin, Z.-X.; Zhou, J.-J. CaMKII inhibition mitigates ischemia/reperfusion-elicited calpain activation and the damage to membrane skeleton proteins in isolated rat hearts. Biochem. Biophys. Res. Commun. 2017, 491, 687–692. [Google Scholar] [CrossRef]
- Lohse, M.J.; Engelhardt, S.; Eschenhagen, T. What Is the Role of β-Adrenergic Signaling in Heart Failure? Circ. Res. 2003, 93, 896–906. [Google Scholar] [CrossRef]
- Glading, A.; Bodnar, R.J.; Reynolds, I.J.; Shiraha, H.; Satish, L.; Potter, D.A.; Blair, H.C.; Wells, A. Epidermal Growth Factor Activates m-Calpain (Calpain II), at Least in Part, by Extracellular Signal-Regulated Kinase-Mediated Phosphorylation. Mol. Cell. Biol. 2004, 24, 2499–2512. [Google Scholar] [CrossRef] [Green Version]
- Glading, A.; Lauffenburger, D.A.; Wells, A. Cutting to the chase: Calpain proteases in cell motility. Trends Cell Biol. 2002, 12, 46–54. [Google Scholar] [CrossRef]
- Elce, J.S.; Hegadorn, C.; Arthur, S. Autolysis, Ca2+ Requirement, and Heterodimer Stability in m-Calpain. J. Biol. Chem. 1997, 272, 11268–11275. [Google Scholar] [CrossRef] [Green Version]
- Geesink, G.H.; Koohmaraie, M. Ionic strength-induced inactivation of mu-calpain in postmortem muscle. J. Anim. Sci. 2000, 78, 2336–2343. [Google Scholar] [CrossRef]
- Yang, H.; Ma, H.; Takano, E.; Hatanaka, M.; Maki, M. Analysis of calcium-dependent interaction between amino-terminal conserved region of calpastatin functional domain and calmodulin-like domain of mu-calpain large subunit. J. Biol. Chem. 1994, 269, 18977–18984. [Google Scholar] [CrossRef]
- Melloni, E.; Averna, M.; De Tullio, R.; Capini, P.; Salamino, F.; Pontremoli, S. Changes in calpastatin localization and expression during calpain activation: A new mechanism for the regulation of intracellular Ca2+-dependent proteolysis. Cell. Mol. Life Sci. 2003, 60, 2669–2678. [Google Scholar] [CrossRef] [PubMed]
- Melloni, E.; Averna, M.; Stifanese, R.; De Tullio, R.; Defranchi, E.; Salamino, F.; Pontremoli, S. Association of Calpastatin with Inactive Calpain: A Novel Mechanism to Control the Activation of the Protease? J. Biol. Chem. 2006, 281, 24945–24954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Averna, M.; de Tullio, R.; Passalacqua, M.; Salamino, F.; Pontremoli, S.; Melloni, E. Changes in intracellular calpastatin localization are mediated by reversible phosphorylation. Biochem. J. 2001, 354, 25–30. [Google Scholar] [CrossRef]
- Carragher, N.O.; Frame, M.C. Focal adhesion and actin dynamics: A place where kinases and proteases meet to promote invasion. Trends Cell Biol. 2004, 14, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, Y.S.L. The Critical Role of Calpain in Cell Proliferation. J. Biomol. Res. Ther. 2014, 3, 1000112. [Google Scholar] [CrossRef] [Green Version]
- Ariyoshi, H.; Okahara, K.; Sakon, M.; Kambayashi, J.-I.; Kawashima, S.-I.; Kawasaki, T.; Monden, M. Possible Involvement of m-Calpain in Vascular Smooth Muscle Cell Proliferation. Arter. Thromb. Vasc. Biol. 1998, 18, 493–498. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.A.; Schnellmann, R.G. Calpains, mitochondria, and apoptosis. Cardiovasc. Res. 2012, 96, 32–37. [Google Scholar] [CrossRef] [Green Version]
- Ng, R.; Manring, H.; Papoutsidakis, N.; Albertelli, T.; Tsai, N.; See, C.J.; Li, X.; Park, J.; Stevens, T.L.; Bobbili, P.J.; et al. Patient mutations linked to arrhythmogenic cardiomyopathy enhance calpain-mediated desmoplakin degradation. J. Clin. Investig. 2019, 4, e128643. [Google Scholar] [CrossRef] [Green Version]
- Shi, D.; He, T.; Tang, W.; Li, H.; Wang, C.; Zheng, M.; Hu, J.; Song, X.; Ding, Y.; Chen, Y.-Y.; et al. Local application of MDL28170-loaded PCL film improves functional recovery by preserving survival of motor neurons after traumatic spinal cord injury. Neurosci. Lett. 2018, 694, 161–167. [Google Scholar] [CrossRef]
- Ma, M.; Li, L.; Wang, X.; Bull, D.L.; Shofer, F.S.; Meaney, D.F.; Neumar, R.W.; Bramlett, H.M.; Dietrich, W.D.; Smith, D.H.; et al. Short-Duration Treatment with the Calpain Inhibitor MDL-28170 Does Not Protect Axonal Transport in an in Vivo Model of Traumatic Axonal Injury. J. Neurotrauma 2012, 29, 445–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedrich, E.E.; Hong, Z.; Xiong, S.; Zhong, M.; Di, A.; Rehman, J.; Komarova, Y.A.; Malik, A.B. Endothelial cell Piezo1 mediates pressure-induced lung vascular hyperpermeability via disruption of adherens junctions. Proc. Natl. Acad. Sci. USA 2019, 116, 12980–12985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Q.; Zhou, H.; Li, X.; Xiao, B. The mechanosensitive Piezo1 channel: A three-bladed propeller-like structure and a lever-like mechanogating mechanism. FEBS J. 2018, 286, 2461–2470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoda, T.; Kaufman, K.M.; Wen, T.; Caldwell, J.M.; Osswald, G.A.; Purnima, P.; Zimmermann, N.; Collins, M.H.; Rehn, K.; Foote, H.; et al. Desmoplakin and periplakin genetically and functionally contribute to eosinophilic esophagitis. Nat. Commun. 2021, 12, 6795. [Google Scholar] [CrossRef]
- Rios-Doria, J.; Day, K.C.; Kuefer, R.; Rashid, M.G.; Chinnaiyan, A.M.; Rubin, M.A.; Day, M.L. The Role of Calpain in the Proteolytic Cleavage of E-cadherin in Prostate and Mammary Epithelial Cells. J. Biol. Chem. 2003, 278, 1372–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, Y.-N.; Jung, Y.-S.; Lee, S.H.; Moon, C.-H.; Kim, C.-H.; Baik, E.J. Calpain-Mediated N-Cadherin Proteolytic Processing in Brain Injury. J. Neurosci. 2009, 29, 5974–5984. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Takeichi, M. NMDA-Receptor Activation Induces Calpain-Mediated β-Catenin Cleavages for Triggering Gene Expression. Neuron 2007, 53, 387–397. [Google Scholar] [CrossRef] [Green Version]
- von Reyn, C.R.; Spaethling, J.M.; Mesfin, M.N.; Ma, M.; Neumar, R.W.; Smith, D.H.; Siman, R.; Meaney, D.F. Calpain Mediates Proteolysis of the Voltage-Gated Sodium Channel α-Subunit. J. Neurosci. 2009, 29, 10350–10356. [Google Scholar] [CrossRef] [Green Version]
- Lin, Z.; Zhao, J.; Nitoiu, D.; Scott, C.A.; Plagnol, V.; Smith, F.J.; Wilson, N.J.; Cole, C.; Schwartz, M.E.; McLean, W.I.; et al. Loss-of-Function Mutations in CAST Cause Peeling Skin, Leukonychia, Acral Punctate Keratoses, Cheilitis, and Knuckle Pads. Am. J. Hum. Genet. 2015, 96, 440–447. [Google Scholar] [CrossRef] [Green Version]
- Vite, A.; Gandjbakhch, E.; Prost, C.; Fressart, V.; Fouret, P.; Neyroud, N.; Gary, F.; Donal, E.; Varnous, S.; Fontaine, G.; et al. Desmosomal Cadherins Are Decreased in Explanted Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy Patient Hearts. PLoS ONE 2013, 8, e75082. [Google Scholar] [CrossRef]
- Kirchner, F.; Schuetz, A.; Boldt, L.-H.; Martens, K.; Dittmar, G.; Haverkamp, W.; Thierfelder, L.; Heinemann, U.; Gerull, B. Molecular Insights into Arrhythmogenic Right Ventricular Cardiomyopathy Caused by Plakophilin-2 Missense Mutations. Circ. Cardiovasc. Genet. 2012, 5, 400–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoover, C.A.; Ott, K.L.; Manring, H.R.; Dew, T.; Borzok, M.A.; Wright, N.T. Creating a ‘Molecular Band-Aid’; Blocking an Exposed Protease Target Site in Desmoplakin. J. Pers. Med. 2021, 11, 401. [Google Scholar] [CrossRef] [PubMed]
- Brundel, B.J.J.M.; Henning, R.H.; Kampinga, H.H.; Van Gelder, I.C.; Crijns, H.J.G.M. Molecular Mechanisms of Remodeling in Human Atrial Fibrillation. Cardiovasc. Res. 2002, 54, 315–324. [Google Scholar] [CrossRef] [Green Version]
- Al-Makhamreh, H.; Alrabadi, N.; Haikal, L.; Krishan, M.; Al-Badaineh, N.; Odeh, O.; Barqawi, T.; Nawaiseh, M.; Shaban, A.; Abdin, B.; et al. Paroxysmal and Non-Paroxysmal Atrial Fibrillation in Middle Eastern Patients: Clinical Features and the Use of Medications. Analysis of the Jordan Atrial Fibrillation (JoFib) Study. Int. J. Environ. Res. Public Health 2022, 19, 6173. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, H.; An, Y.; Ikeda, S.; Aono, Y.; Doi, K.; Ishii, M.; Iguchi, M.; Masunaga, N.; Esato, M.; Tsuji, H.; et al. Progression From Paroxysmal to Sustained Atrial Fibrillation Is Associated With Increased Adverse Events. Stroke 2018, 49, 2301–2308. [Google Scholar] [CrossRef] [PubMed]
- Woodcock, E.A.; Kistler, P.M.; Ju, Y.-K. Phosphoinositide signalling and cardiac arrhythmias. Cardiovasc. Res. 2008, 82, 286–295. [Google Scholar] [CrossRef] [Green Version]
- Elvira, M.; Díez, J.; Wang, K.; Villalobo, A. Phosphorylation of connexin-32 by protein kinase C prevents its proteolysis by mu-calpain and m-calpain. J. Biol. Chem. 1993, 268, 14294–14300. [Google Scholar] [CrossRef]
- Abele, K.; Yang, J. Regulation of voltage-gated calcium channels by proteolysis. Sheng Li Xue Bao Acta Physiol. Sin. 2012, 64, 504–514. [Google Scholar]
- Murphy, R.M.; Dutka, T.L.; Horvath, D.; Bell, J.R.; Delbridge, L.M.; Lamb, G.D. Ca2+-dependent proteolysis of junctophilin-1 and junctophilin-2 in skeletal and cardiac muscle. J. Physiol. 2012, 591, 719–729. [Google Scholar] [CrossRef]
- Orchard, C.; Brette, F. t-tubules and sarcoplasmic reticulum function in cardiac ventricular myocytes. Cardiovasc. Res. 2007, 77, 237–244. [Google Scholar] [CrossRef]
- Neuhof, C.; Neuhof, H. Calpain system and its involvement in myocardial ischemia and reperfusion injury. World J. Cardiol. 2014, 6, 638. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, M.L.; Escobar, G.P.; Mukherjee, R.; Goshorn, D.K.; Sheats, N.J.; Bruce, J.A.; Mains, I.M.; Hendrick, J.K.; Hewett, K.W.; Gourdie, R.G.; et al. Matrix Metalloproteinase-7 Affects Connexin-43 Levels, Electrical Conduction, and Survival After Myocardial Infarction. Circulation 2006, 113, 2919–2928. [Google Scholar] [CrossRef] [PubMed]
- Kwak, H.-I.; Kang, H.; Dave, J.M.; Mendoza, E.A.; Su, S.-C.; Maxwell, S.A.; Bayless, K.J. Calpain-mediated vimentin cleavage occurs upstream of MT1-MMP membrane translocation to facilitate endothelial sprout initiation. Angiogenesis 2012, 15, 287–303. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.; Kwak, H.-I.; Kaunas, R.; Bayless, K.J. Fluid Shear Stress and Sphingosine 1-Phosphate Activate Calpain to Promote Membrane Type 1 Matrix Metalloproteinase (MT1-MMP) Membrane Translocation and Endothelial Invasion into Three-dimensional Collagen Matrices. J. Biol. Chem. 2011, 286, 42017–42026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Postovit, L.-M.; Dutt, P.; Dourdin, N.; Park, M.; A Greer, P.; Graham, C.H.; Elce, J.S. Calpain is required for MMP-2 and u-PA expression in SV40 large T-antigen-immortalized cells. Biochem. Biophys. Res. Commun. 2002, 297, 294–301. [Google Scholar] [CrossRef]
- Jang, H.S.; Lal, S.; Greenwood, J.A. Calpain 2 is Required for Glioblastoma Cell Invasion: Regulation of Matrix Metalloproteinase 2. Neurochem. Res. 2010, 35, 1796–1804. [Google Scholar] [CrossRef] [Green Version]
- Averna, M.; Bavestrello, M.; Cresta, F.; Pedrazzi, M.; De Tullio, R.; Minicucci, L.; Sparatore, B.; Salamino, F.; Pontremoli, S.; Melloni, E. Abnormal activation of calpain and protein kinase Cα promotes a constitutive release of matrix metalloproteinase 9 in peripheral blood mononuclear cells from cystic fibrosis patients. Arch. Biochem. Biophys. 2016, 604, 103–112. [Google Scholar] [CrossRef]
- Kudo-Sakamoto, Y.; Akazawa, H.; Ito, K.; Takano, J.; Yano, M.; Yabumoto, C.; Naito, A.T.; Oka, T.; Lee, J.-K.; Sakata, Y.; et al. Calpain-dependent Cleavage of N-cadherin Is Involved in the Progression of Post-myocardial Infarction Remodeling. J. Biol. Chem. 2014, 289, 19408–19419. [Google Scholar] [CrossRef]
- Kunkel, G.H.; Chaturvedi, P.; Tyagi, S.C. Resuscitation of a dead cardiomyocyte. Hear. Fail. Rev. 2015, 20, 709–719. [Google Scholar] [CrossRef]
- Moshal, K.S.; Singh, M.; Sen, U.; Rosenberger, D.S.E.; Henderson, B.; Tyagi, N.; Zhang, H.; Tyagi, S.C. Homocysteine-mediated activation and mitochondrial translocation of calpain regulates MMP-9 in MVEC. Am. J. Physiol. Circ. Physiol. 2006, 291, H2825–H2835. [Google Scholar] [CrossRef]
- Finkel, T.; Menazza, S.; Holmström, K.M.; Parks, R.J.; Liu, J.; Sun, J.; Liu, J.; Pan, X.; Murphy, E. The Ins and Outs of Mitochondrial Calcium. Circ. Res. 2015, 116, 1810–1819. [Google Scholar] [CrossRef]
- Zhao, G.; Zhang, H.M.; Qiu, Y.; Ye, X.; Yang, D. Cleavage of Desmosomal Cadherins Promotes γ-Catenin Degradation and Benefits Wnt Signaling in Coxsackievirus B3-Induced Destruction of Cardiomyocytes. Front. Microbiol. 2020, 11, 767. [Google Scholar] [CrossRef]
- De Bock, M.; Wang, N.; Decrock, E.; Bultynck, G.; Leybaert, L. Intracellular Cleavage of the Cx43 C-Terminal Domain by Matrix-Metalloproteases: A Novel Contributor to Inflammation? Mediat. Inflamm. 2015, 2015, 257471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dwivedi, A.; Slater, S.C.; George, S.J. MMP-9 and -12 cause N-cadherin shedding and thereby β-catenin signalling and vascular smooth muscle cell proliferation. Cardiovasc. Res. 2008, 81, 178–186. [Google Scholar] [CrossRef] [Green Version]
- Olsen, J.J.; Pohl, S.Ö.G.; Deshmukh, A.; Visweswaran, M.; Ward, N.C.; Arfuso, F.; Agostino, M.; Dharmarajan, A.M. The Role of Wnt Signalling in Angiogenesis. Clin. Biochem. Rev. 2017, 38, 131–142. [Google Scholar] [PubMed]
- Bi, S.-H.; Jin, Z.-X.; Zhang, J.-Y.; Chen, T.; Zhang, S.-L.; Yang, Y.; Duan, W.-X.; Yi, D.-H.; Zhou, J.-J.; Ren, J. Calpain inhibitor MDL 28170 protects against the Ca2+ paradox in rat hearts. Clin. Exp. Pharmacol. Physiol. 2012, 39, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, B.; Huang, C.-K.; Guo, A.; Wu, J.; Zhang, X.; Chen, R.; Chen, C.; Kutschke, W.; Weiss, R.M.; et al. Targeting Calpain for Heart Failure Therapy. JACC Basic Transl. Sci. 2018, 3, 503–517. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Chen, L.; Huang, X.; Wu, K.; Ding, S.; Wang, W.; Wang, B.; Smith, C.; Ren, C.; Ni, H.; et al. Calpain inhibitor MDL28170 improves the transplantation-mediated therapeutic effect of bone marrow-derived mesenchymal stem cells following traumatic brain injury. Stem Cell Res. Ther. 2019, 10, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Thompson, J.; Hu, Y.; Dean, J.; Lesnefsky, E.J. Inhibition of the ubiquitous calpains protects complex I activity and enables improved mitophagy in the heart following ischemia-reperfusion. Am. J. Physiol. Physiol. 2019, 317, C910–C921. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Gene | Protein | Tissue Expression | Notes |
|---|---|---|---|
| CAPN1 | Calpain-1 | Ubiquitous expression | Also known as μ-calpain |
| CAPN2 | Calpain-2 | Ubiquitous expression | Also known as m-calpain |
| CAPN3 | Calpain-3 | Expressed in skeletal muscle | |
| CAPN4 or CAPNS | Calpain-4 | Ubiquitous expression | Another name for the small regulatory subunit Also known as calpain-S |
| CAPN5 | Calpain-5 | Ubiquitous expression | Expressed in lower levels compared to calpain-1-2 |
| CAPN6 | Calpain-6 | Expressed in embryonic tissue and the placenta | |
| CAPN7 | Calpain-7 | Ubiquitous expression | Expressed in lower levels compared to Calpain-1-2 |
| CAPN8 | Calpain-8 | Expressed in gastrointestinal tissue | Complex with calpain-9 to make G-calpain |
| CAPN9 | Calpain-9 | Expressed in gastrointestinal tissue | Complex with calpain-8 to make G-calpain |
| CAPN10 | Calpain-10 | Ubiquitous expression | Expressed in lower levels compared to Calpain-1-2 |
| CAPN11 | Calpain-11 | Expressed in testis tissue | |
| CANP12 | Calpain-12 | Expressed in hair follicle tissue | |
| CAPN13 | Calpain-13 | Ubiquitous expression | Expressed in lower levels compared to Calpain-1-2 |
| CAPN14 | Calpain-14 | Ubiquitous expression | Expressed in lower levels compared to Calpain-1-2 |
| CAPN15 | Calpain-15 | Ubiquitous expression | Expressed in lower levels compared to Calpain-1-2 |
| CAPN16 | Calpain-16 | Ubiquitous expression | Expressed in lower levels compared to Calpain-1-2 Truncated protein, may be nonfunctional |
| Disease | Dysregulation Mechanism | Calpain Target | Reference |
|---|---|---|---|
| Hyperpermeability Pulmonary Edema | Hyperactivity | VC-cadherin p120 catenin β-catenin | [84] |
| Breast Cancer Adenocarcinoma | Hyperactivity | E-cadherin | [87] |
| Alzheimer’s Disease/Bipolar Disorder/Neuronal Cancer | Hyperactivity | N-cadherin β -catenin | [88,89] |
| Neuronal Cell Death | Hyperactivity | Nav1.5 | [90] |
| PLACK Syndrome | Hyperactivity | LOF mutations of calpastatin result in speculated proteolytic cleavage of desmosomal proteins | [91] |
| Eosinophilic Esophagitis | Hyperactivity | desmoglein, desmoplakin, periplakin | [86] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoder, M.W.; Wright, N.T.; Borzok, M.A. Calpain Regulation and Dysregulation—Its Effects on the Intercalated Disk. Int. J. Mol. Sci. 2023, 24, 11726. https://doi.org/10.3390/ijms241411726
Yoder MW, Wright NT, Borzok MA. Calpain Regulation and Dysregulation—Its Effects on the Intercalated Disk. International Journal of Molecular Sciences. 2023; 24(14):11726. https://doi.org/10.3390/ijms241411726
Chicago/Turabian StyleYoder, Micah W., Nathan T. Wright, and Maegen A. Borzok. 2023. "Calpain Regulation and Dysregulation—Its Effects on the Intercalated Disk" International Journal of Molecular Sciences 24, no. 14: 11726. https://doi.org/10.3390/ijms241411726
APA StyleYoder, M. W., Wright, N. T., & Borzok, M. A. (2023). Calpain Regulation and Dysregulation—Its Effects on the Intercalated Disk. International Journal of Molecular Sciences, 24(14), 11726. https://doi.org/10.3390/ijms241411726