
Inhibition of miR-25 Ameliorates Cardiac Dysfunction and Fibrosis by Restoring Krüppel-like Factor 4 Expression

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
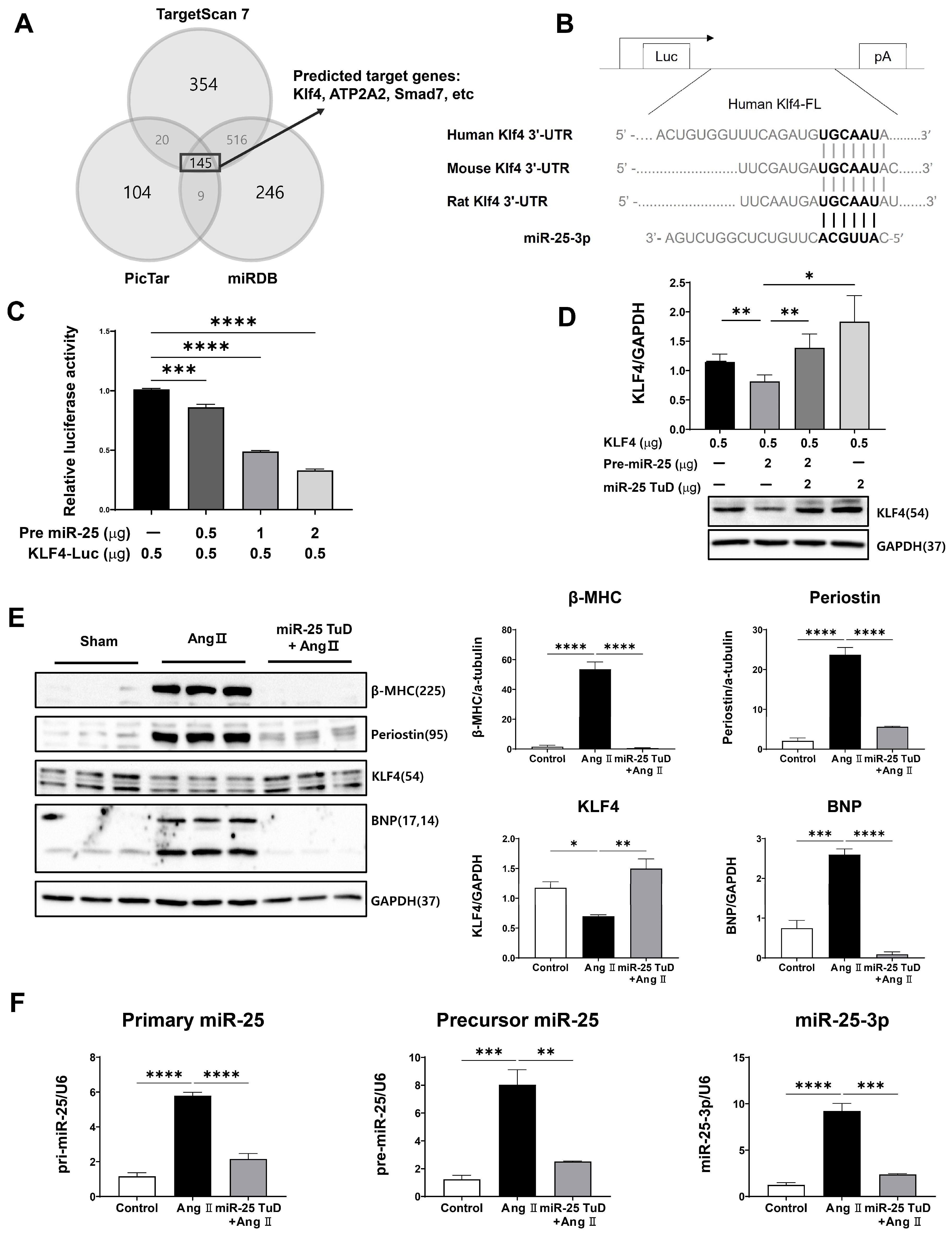
2.1. KLF4 Is a Newly Identified Target of miR-25 in the Heart
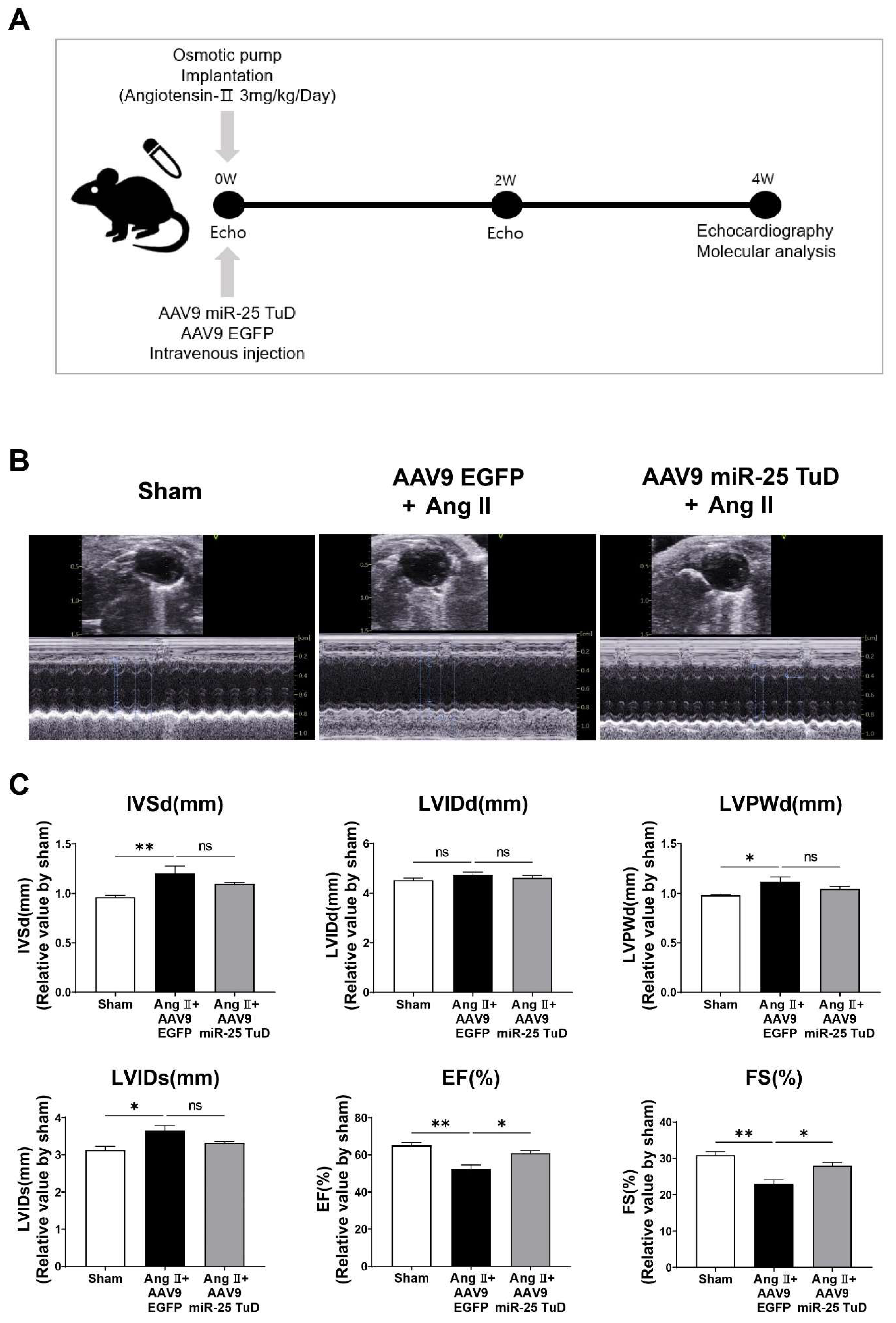
2.2. miR-25 TuD Transfer Prevents Ang II-Induced Cardiac Dysfunction
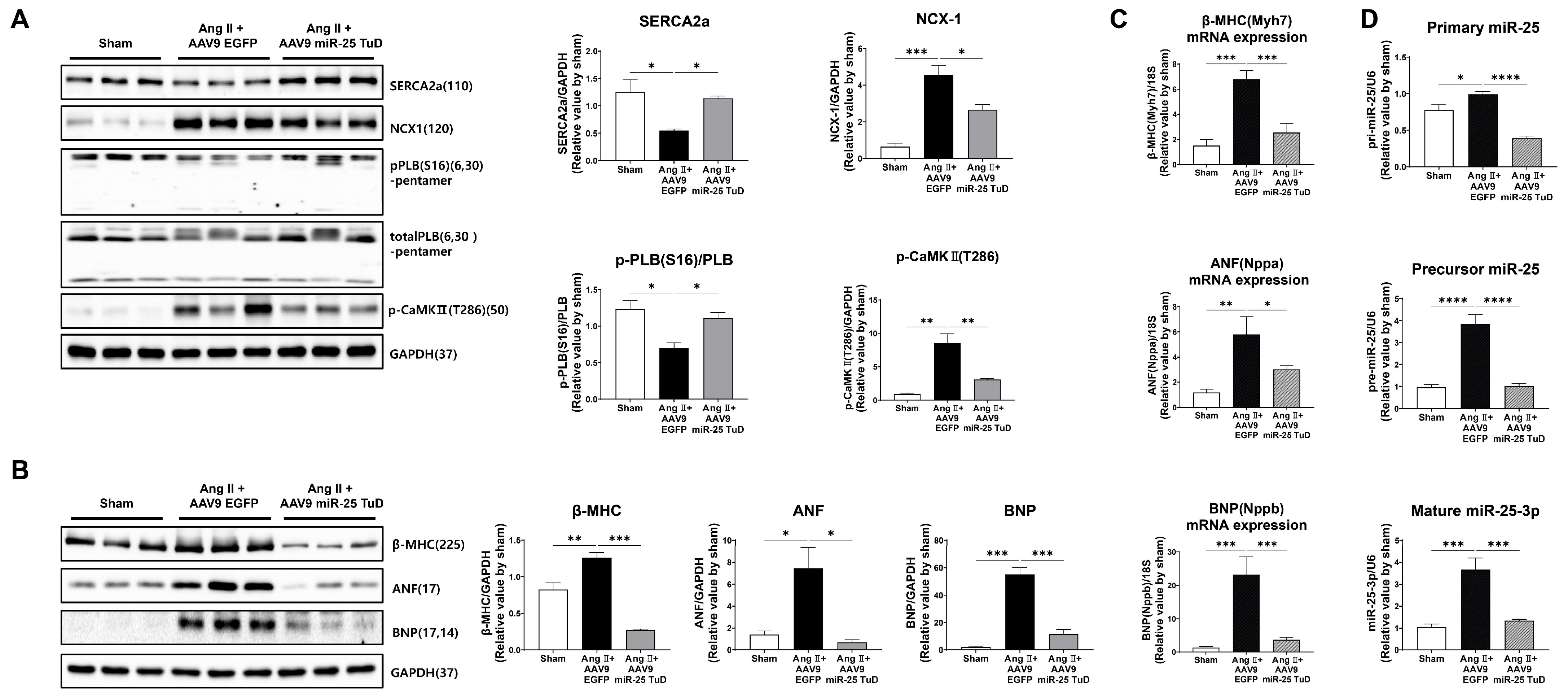
2.3. Inhibition of miR-25 Normalizes Calcium Signaling and Cardiac Stress Markers
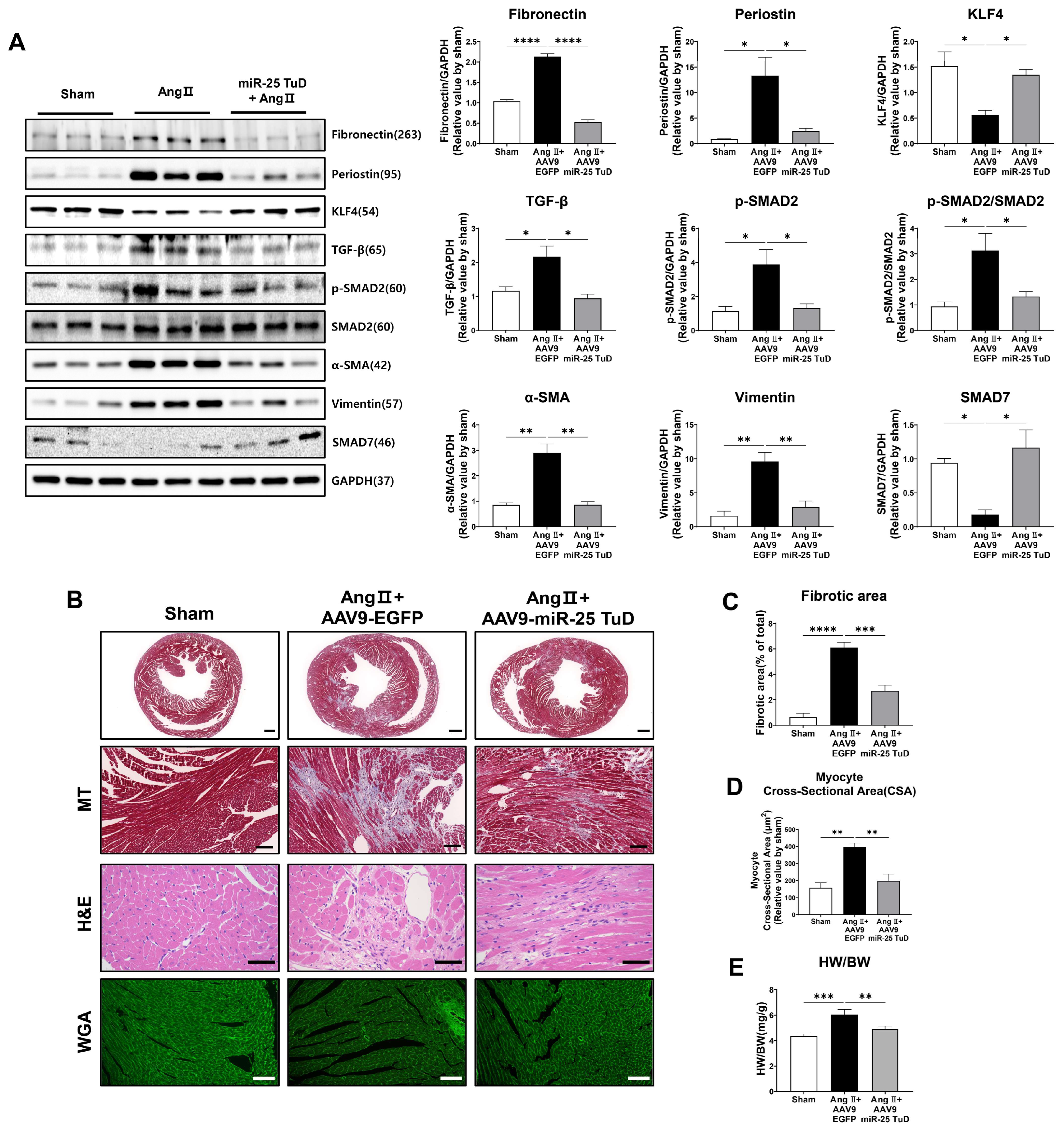
2.4. Restoration of KLF4 Expression via miR-25 TuD Delivery Ameliorates Cardiac Fibrosis
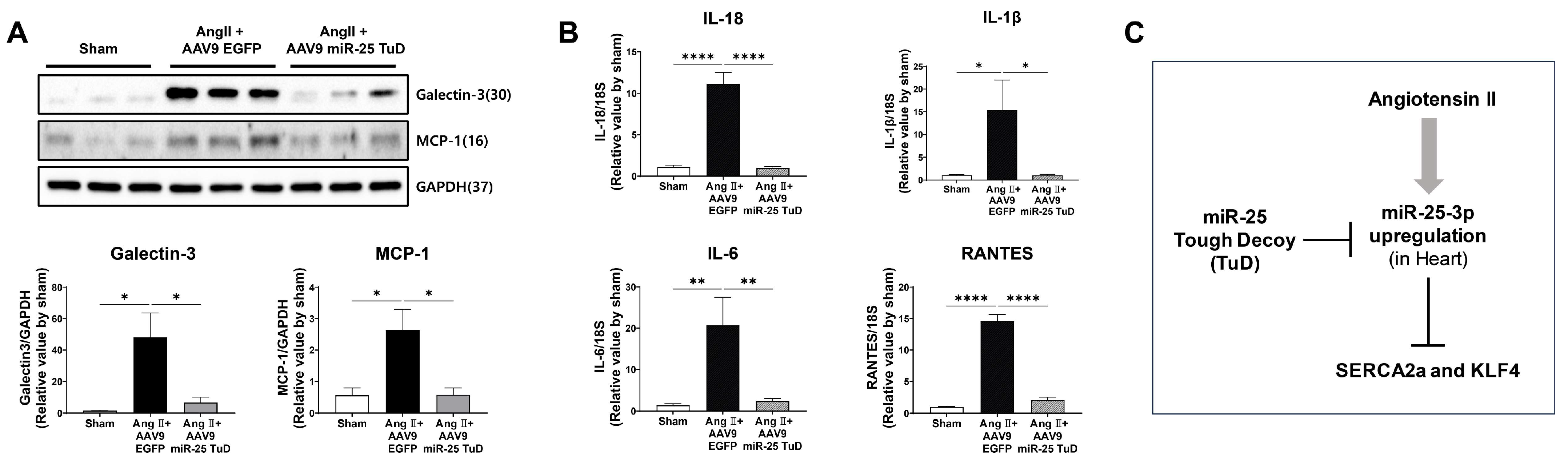
2.5. miR-25 TuD Transfer Prevents Cardiac Inflammation via KLF4 Expression
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatment
4.2. KLF4 3’ UTR Luciferase Assay
4.3. Animals and Experimental Protocol
4.4. Histological Studies
4.5. RNA Extraction and cDNA Synthesis
4.6. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
4.7. Western Blot Analysis
4.8. Echocardiographic Assessment
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fuchs, F.D.; Whelton, P.K. High blood pressure and cardiovascular disease. Hypertension 2020, 75, 285–292. [Google Scholar] [CrossRef]
- Watkins, S.J.; Borthwick, G.M.; Oakenfull, R.; Robson, A.; Arthur, H.M. Angiotensin II-induced cardiomyocyte hypertrophy in vitro is TAK1-dependent and Smad2/3-independent. Hypertens. Res. 2012, 35, 393–398. [Google Scholar] [CrossRef] [Green Version]
- Kagiyama, S.; Eguchi, S.; Frank, G.D.; Inagami, T.; Zhang, Y.C.; Phillips, M.I. Angiotensin II–induced cardiac hypertrophy and hypertension are attenuated by epidermal growth factor receptor antisense. Circulation 2002, 106, 909–912. [Google Scholar] [CrossRef] [PubMed]
- Frey, N.; Katus, H.A.; Olson, E.N.; Hill, J.A. Hypertrophy of the heart: A new therapeutic target? Circulation 2004, 109, 1580–1589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahlquist, C.; Jeong, D.; Rojas-Muñoz, A.; Kho, C.; Lee, A.; Mitsuyama, S.; van Mil, A.; Jin Park, W.; Sluijter, J.P.; Doevendans, P.A. Inhibition of miR-25 improves cardiac contractility in the failing heart. Nature 2014, 508, 531–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyamoto, M.I.; Del Monte, F.; Schmidt, U.; DiSalvo, T.S.; Kang, Z.B.; Matsui, T.; Guerrero, J.L.; Gwathmey, J.K.; Rosenzweig, A.; Hajjar, R.J. Adenoviral gene transfer of SERCA2a improves left-ventricular function in aortic-banded rats in transition to heart failure. Proc. Natl. Acad. Sci. USA 2000, 97, 793–798. [Google Scholar] [CrossRef]
- Oh, J.G.; Jang, S.P.; Yoo, J.; Lee, M.-A.; Lee, S.H.; Lim, T.; Jeong, E.; Kho, C.; Kook, H.; Hajjar, R.J. Role of the PRC2-Six1-miR-25 signaling axis in heart failure. J. Mol. Cell. Cardiol. 2019, 129, 58–68. [Google Scholar] [CrossRef] [Green Version]
- Jeong, D.; Yoo, J.; Lee, P.; Kepreotis, S.V.; Lee, A.; Wahlquist, C.; Brown, B.D.; Kho, C.; Mercola, M.; Hajjar, R.J. miR-25 tough decoy enhances cardiac function in heart failure. Mol. Ther. 2018, 26, 718–729. [Google Scholar] [CrossRef] [Green Version]
- Jackson, R.J.; Standart, N. How do microRNAs regulate gene expression? Science’s STKE 2007, 2007, re1. [Google Scholar] [CrossRef] [Green Version]
- Yap, C.; Mieremet, A.; de Vries, C.J.; Micha, D.; de Waard, V. Six shades of vascular smooth muscle cells illuminated by KLF4 (Krüppel-Like Factor 4). Arterioscler. Thromb. Vasc. Biol. 2021, 41, 2693–2707. [Google Scholar] [CrossRef]
- Aksoy, I.; Giudice, V.; Delahaye, E.; Wianny, F.; Aubry, M.; Mure, M.; Chen, J.; Jauch, R.; Bogu, G.K.; Nolden, T. Klf4 and Klf5 differentially inhibit mesoderm and endoderm differentiation in embryonic stem cells. Nat. Commun. 2014, 5, 3719. [Google Scholar] [CrossRef] [Green Version]
- He, Z.; He, J.; Xie, K. KLF4 transcription factor in tumorigenesis. Cell Death Discov. 2023, 9, 118. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Han, Q.; Xiong, Y.; Li, T.; Liu, Z.; Xu, H.; Wu, Y.; Wang, N.; Liu, X. Krüpple-like-factor 4 attenuates lung fibrosis via inhibiting epithelial-mesenchymal transition. Sci. Rep. 2017, 7, 15847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, D.; Chen, P.-p.; Zheng, P.-q.; Yin, F.; Cheng, Q.; Zhou, Z.-l.; Xie, H.-y.; Li, J.-y.; Ni, J.-y.; Wang, Y.-z. KLF4 initiates sustained YAP activation to promote renal fibrosis in mice after ischemia-reperfusion kidney injury. Acta Pharmacol. Sin. 2021, 42, 436–450. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Wu, Z.; Liu, T.; Ullenbruch, M.R.; Jin, H.; Phan, S.H. Gut-enriched Kruppel-like factor interaction with Smad3 inhibits myofibroblast differentiation. Am. J. Respir. Cell Mol. Biol. 2007, 36, 78–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wang, Y.; Liu, Y.; Wang, N.; Qi, Y.; Du, J. Krüppel-like factor 4 transcriptionally regulates TGF-β1 and contributes to cardiac myofibroblast differentiation. PLoS ONE 2013, 8, e63424. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.-H.; Kim, H.-J.; Na, H.; Nam, M.-W.; Kim, J.-Y.; Kim, J.-S.; Koo, S.-H.; Lee, M.-O. RORα induces KLF4-mediated M2 polarization in the liver macrophages that protect against nonalcoholic steatohepatitis. Cell Rep. 2017, 20, 124–135. [Google Scholar] [CrossRef] [Green Version]
- Liao, X.; Sharma, N.; Kapadia, F.; Zhou, G.; Lu, Y.; Hong, H.; Paruchuri, K.; Mahabeleshwar, G.H.; Dalmas, E.; Venteclef, N. Krüppel-like factor 4 regulates macrophage polarization. J. Clin. Investig. 2011, 121, 2736–2749. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, T.; Yamashita, M.; Horimai, C.; Hayashi, M. Kruppel-like factor 4 protein regulates isoproterenol-induced cardiac hypertrophy by modulating myocardin expression and activity. J. Biol. Chem. 2014, 289, 26107–26118. [Google Scholar] [CrossRef] [Green Version]
- Pollak, N.M.; Hoffman, M.; Goldberg, I.J.; Drosatos, K. Krüppel-like factors: Crippling and uncrippling metabolic pathways. JACC Basic Transl. Sci. 2018, 3, 132–156. [Google Scholar] [CrossRef]
- Li, H.; Li, C.; Zheng, T.; Wang, Y.; Wang, J.; Fan, X.; Zheng, X.; Tian, G.; Yuan, Z.; Chen, T. Cardiac Fibroblast Activation Induced by Oxygen–Glucose Deprivation Depends on the HIF-1α/miR-212-5p/KLF4 Pathway. J. Cardiovasc. Transl. Res. 2023; online ahead of print. [Google Scholar]
- Penke, L.R.; Speth, J.M.; Huang, S.K.; Fortier, S.M.; Baas, J.; Peters-Golden, M. KLF4 is a therapeutically tractable brake on fibroblast activation that promotes resolution of pulmonary fibrosis. JCI Insight 2022, 7, e160688. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Peng, Z.; Tang, H.; Xie, D.; Jia, Z.; Zhong, L.; Zhao, S.; Ma, Z.; Gao, Y.; Zeng, L. Loss of KLF4 and consequential downregulation of Smad7 exacerbate oncogenic TGF-β signaling in and promote progression of hepatocellular carcinoma. Oncogene 2017, 36, 2957–2968. [Google Scholar] [CrossRef] [Green Version]
- Chandran, R.R.; Xie, Y.; Gallardo-Vara, E.; Adams, T.; Garcia-Milian, R.; Kabir, I.; Sheikh, A.Q.; Kaminski, N.; Martin, K.A.; Herzog, E.L. Distinct roles of KLF4 in mesenchymal cell subtypes during lung fibrogenesis. Nat. Commun. 2021, 12, 7179. [Google Scholar] [CrossRef] [PubMed]
- Bulut, G.B.; Alencar, G.F.; Owsiany, K.M.; Nguyen, A.T.; Karnewar, S.; Haskins, R.M.; Waller, L.K.; Cherepanova, O.A.; Deaton, R.A.; Shankman, L.S. KLF4 (Kruppel-Like Factor 4)-dependent perivascular plasticity contributes to adipose tissue inflammation. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 284–301. [Google Scholar] [CrossRef] [PubMed]
- Hamik, A.; Lin, Z.; Kumar, A.; Balcells, M.; Sinha, S.; Katz, J.; Feinberg, M.W.; Gerszten, R.E.; Edelman, E.R.; Jain, M.K. Kruppel-like factor 4 regulates endothelial inflammation. J. Biol. Chem. 2007, 282, 13769–13779. [Google Scholar] [CrossRef] [Green Version]
- Feinberg, M.W.; Cao, Z.; Wara, A.K.; Lebedeva, M.A.; SenBanerjee, S.; Jain, M.K. Kruppel-like factor 4 is a mediator of proinflammatory signaling in macrophages. J. Biol. Chem. 2005, 280, 38247–38258. [Google Scholar] [CrossRef] [Green Version]
- Paulus, W.J.; Zile, M.R. From systemic inflammation to myocardial fibrosis: The heart failure with preserved ejection fraction paradigm revisited. Circ. Res. 2021, 128, 1451–1467. [Google Scholar] [CrossRef]
- Kania, G.; Blyszczuk, P.; Eriksson, U. Mechanisms of cardiac fibrosis in inflammatory heart disease. Trends Cardiovasc. Med. 2009, 19, 247–252. [Google Scholar] [CrossRef]
- Rabkin, S.W. The role of interleukin 18 in the pathogenesis of hypertension-induced vascular disease. Nat. Rev. Cardiol. 2009, 6, 192–199. [Google Scholar] [CrossRef]
- Li, C.; Yu, L.; Mai, C.; Mu, T.; Zeng, Y. KLF4 down-regulation resulting from TLR4 promotion of ERK1/2 phosphorylation underpins inflammatory response in sepsis. J. Cell. Mol. Med. 2021, 25, 2013–2024. [Google Scholar] [CrossRef]
- Lu, Q.; Meng, Q.; Qi, M.; Li, F.; Liu, B. Shear-sensitive lncRNA AF131217. 1 inhibits inflammation in HUVECs via regulation of KLF4. Hypertension 2019, 73, e25–e34. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Lin, S.; Li, C.; Ouyang, Z.; Chen, Z.; Li, S.; Huang, Y.; Luo, W.; Zheng, Z.; Guo, P. MiR-92a/KLF4/p110δ regulates titanium particles-induced macrophages inflammation and osteolysis. Cell Death Discov. 2022, 8, 197. [Google Scholar] [CrossRef] [PubMed]
- Drazner, M.H. The progression of hypertensive heart disease. Circulation 2011, 123, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Dong, D.-L.; Chen, C.; Huo, R.; Wang, N.; Li, Z.; Tu, Y.-J.; Hu, J.-T.; Chu, X.; Huang, W.; Yang, B.-F. Reciprocal repression between microRNA-133 and calcineurin regulates cardiac hypertrophy: A novel mechanism for progressive cardiac hypertrophy. Hypertension 2010, 55, 946–952. [Google Scholar] [CrossRef] [Green Version]
- Duan, Y.; Zhou, B.; Su, H.; Liu, Y.; Du, C. miR-150 regulates high glucose-induced cardiomyocyte hypertrophy by targeting the transcriptional co-activator p300. Exp. Cell Res. 2013, 319, 173–184. [Google Scholar] [CrossRef]
- Wehbe, N.; Nasser, S.A.; Pintus, G.; Badran, A.; Eid, A.H.; Baydoun, E. MicroRNAs in cardiac hypertrophy. Int. J. Mol. Sci. 2019, 20, 4714. [Google Scholar] [CrossRef] [Green Version]
- Heymans, S.; Corsten, M.F.; Verhesen, W.; Carai, P.; Van Leeuwen, R.E.; Custers, K.; Peters, T.; Hazebroek, M.; Stöger, L.; Wijnands, E. Macrophage microRNA-155 promotes cardiac hypertrophy and failure. Circulation 2013, 128, 1420–1432. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Qin, D.; Shi, H.; Zhang, Y.; Li, H.; Han, Q. MiR-195-5p promotes cardiomyocyte hypertrophy by targeting MFN2 and FBXW7. BioMed Res. Int. 2019, 2019, 1580982-10. [Google Scholar] [CrossRef]
- Ding, Y.-q.; Zhang, Y.-h.; Lu, J.; Li, B.; Yu, W.-j.; Yue, Z.-b.; Hu, Y.-h.; Wang, P.-x.; Li, J.-y.; Cai, S.-d. MicroRNA-214 contributes to Ang II-induced cardiac hypertrophy by targeting SIRT3 to provoke mitochondrial malfunction. Acta Pharmacol. Sin. 2021, 42, 1422–1436. [Google Scholar] [CrossRef]
- Eder, P.; Molkentin, J.D. TRPC channels as effectors of cardiac hypertrophy. Circ. Res. 2011, 108, 265–272. [Google Scholar] [CrossRef]
- Díez, J. Mechanisms of cardiac fibrosis in hypertension. J. Clin. Hypertens. 2007, 9, 546–550. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.l.; Xu, H.; Liu, Z.b.; Wu, Q.c.; Zhu, R.r.; Liu, J.c. miR-21 promotes cardiac fibroblast-to-myofibroblast transformation and myocardial fibrosis by targeting Jagged1. J. Cell. Mol. Med. 2018, 22, 3816–3824. [Google Scholar] [CrossRef]
- Surina, S.; Fontanella, R.A.; Scisciola, L.; Marfella, R.; Paolisso, G.; Barbieri, M. miR-21 in human cardiomyopathies. Front. Cardiovasc. Med. 2021, 8, 767064. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Morelli, M.B.; Matarese, A.; Sardu, C.; Santulli, G. Cardiomyocyte-derived exosomal microRNA-92a mediates post-ischemic myofibroblast activation both in vitro and ex vivo. ESC Heart Fail. 2020, 7, 285–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morelli, M.B.; Shu, J.; Sardu, C.; Matarese, A.; Santulli, G. Cardiosomal microRNAs are essential in post-infarction myofibroblast phenoconversion. Int. J. Mol. Sci. 2019, 21, 201. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Qi, Y.; Du, J.-Q.; Zhang, D.-f. MicroRNA-34a regulates cardiac fibrosis after myocardial infarction by targeting Smad4. Expert Opin. Ther. Targets 2014, 18, 1355–1365. [Google Scholar] [CrossRef] [PubMed]
- Lighthouse, J.K.; Small, E.M. Transcriptional control of cardiac fibroblast plasticity. J. Mol. Cell. Cardiol. 2016, 91, 52–60. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, T.; Gan, Q.; Franke, A.S.; Ho, R.; Zhang, J.; Chen, Y.E.; Hayashi, M.; Majesky, M.W.; Somlyo, A.V.; Owens, G.K. Smooth and cardiac muscle-selective knock-out of Krüppel-like factor 4 causes postnatal death and growth retardation. J. Biol. Chem. 2010, 285, 21175–21184. [Google Scholar] [CrossRef] [Green Version]
- Tassone, E.; Bradaschia-Correa, V.; Xiong, X.; Sastre-Perona, A.; Josephson, A.M.; Khodadadi-Jamayran, A.; Melamed, J.; Bu, L.; Kahler, D.J.; Ossowski, L. KLF4 as a rheostat of osteolysis and osteogenesis in prostate tumors in the bone. Oncogene 2019, 38, 5766–5777. [Google Scholar] [CrossRef]
- Di Stefano, B.; Collombet, S.; Jakobsen, J.S.; Wierer, M.; Sardina, J.L.; Lackner, A.; Stadhouders, R.; Segura-Morales, C.; Francesconi, M.; Limone, F. C/EBPα creates elite cells for iPSC reprogramming by upregulating Klf4 and increasing the levels of Lsd1 and Brd4. Nat. Cell Biol. 2016, 18, 371–381. [Google Scholar] [CrossRef]
- Liao, X.; Haldar, S.M.; Lu, Y.; Jeyaraj, D.; Paruchuri, K.; Nahori, M.; Cui, Y.; Kaestner, K.H.; Jain, M.K. Krüppel-like factor 4 regulates pressure-induced cardiac hypertrophy. J. Mol. Cell. Cardiol. 2010, 49, 334–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Samulski, R.J. Engineering adeno-associated virus vectors for gene therapy. Nat. Rev. Genet. 2020, 21, 255–272. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, C.; Cho, S.; Jeong, D. Inhibition of miR-25 Ameliorates Cardiac Dysfunction and Fibrosis by Restoring Krüppel-like Factor 4 Expression. Int. J. Mol. Sci. 2023, 24, 12434. https://doi.org/10.3390/ijms241512434
Lee C, Cho S, Jeong D. Inhibition of miR-25 Ameliorates Cardiac Dysfunction and Fibrosis by Restoring Krüppel-like Factor 4 Expression. International Journal of Molecular Sciences. 2023; 24(15):12434. https://doi.org/10.3390/ijms241512434
Chicago/Turabian StyleLee, Cholong, Sunghye Cho, and Dongtak Jeong. 2023. "Inhibition of miR-25 Ameliorates Cardiac Dysfunction and Fibrosis by Restoring Krüppel-like Factor 4 Expression" International Journal of Molecular Sciences 24, no. 15: 12434. https://doi.org/10.3390/ijms241512434
APA StyleLee, C., Cho, S., & Jeong, D. (2023). Inhibition of miR-25 Ameliorates Cardiac Dysfunction and Fibrosis by Restoring Krüppel-like Factor 4 Expression. International Journal of Molecular Sciences, 24(15), 12434. https://doi.org/10.3390/ijms241512434