
A FRET-Based Assay for the Identification of PCNA Inhibitors



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Development of a FRET Assay to Analyze the PCNA–p15 Interaction
2.2. Screening for PCNA Inhibitors
2.3. Analyzing the Relevance of the Mutation C148S for PCNA Function
2.3.1. C148S Reduces the Apparent Binding of PCNA to p15
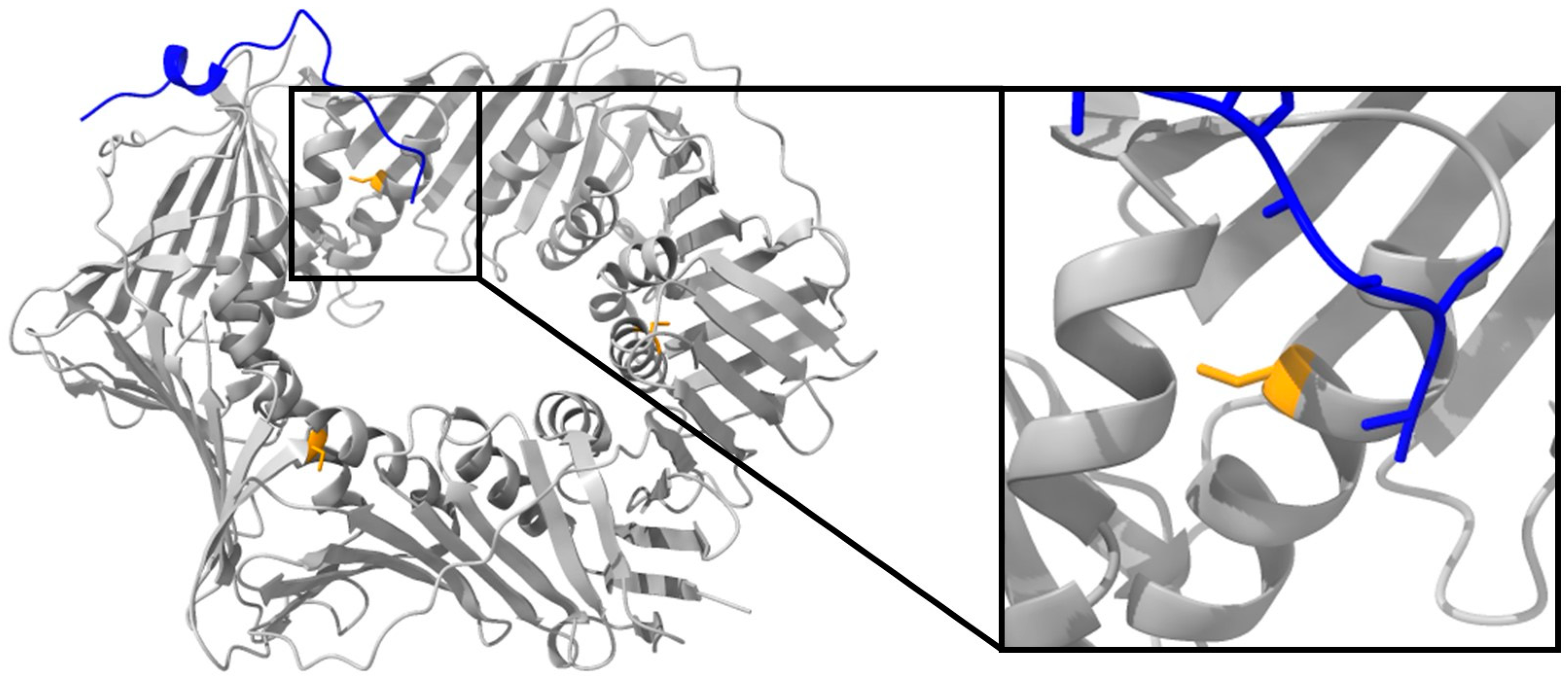
2.3.2. Alchemical Free-Energy Calculations Predict a Destabilizing Effect of the C148S Mutation on PCNA
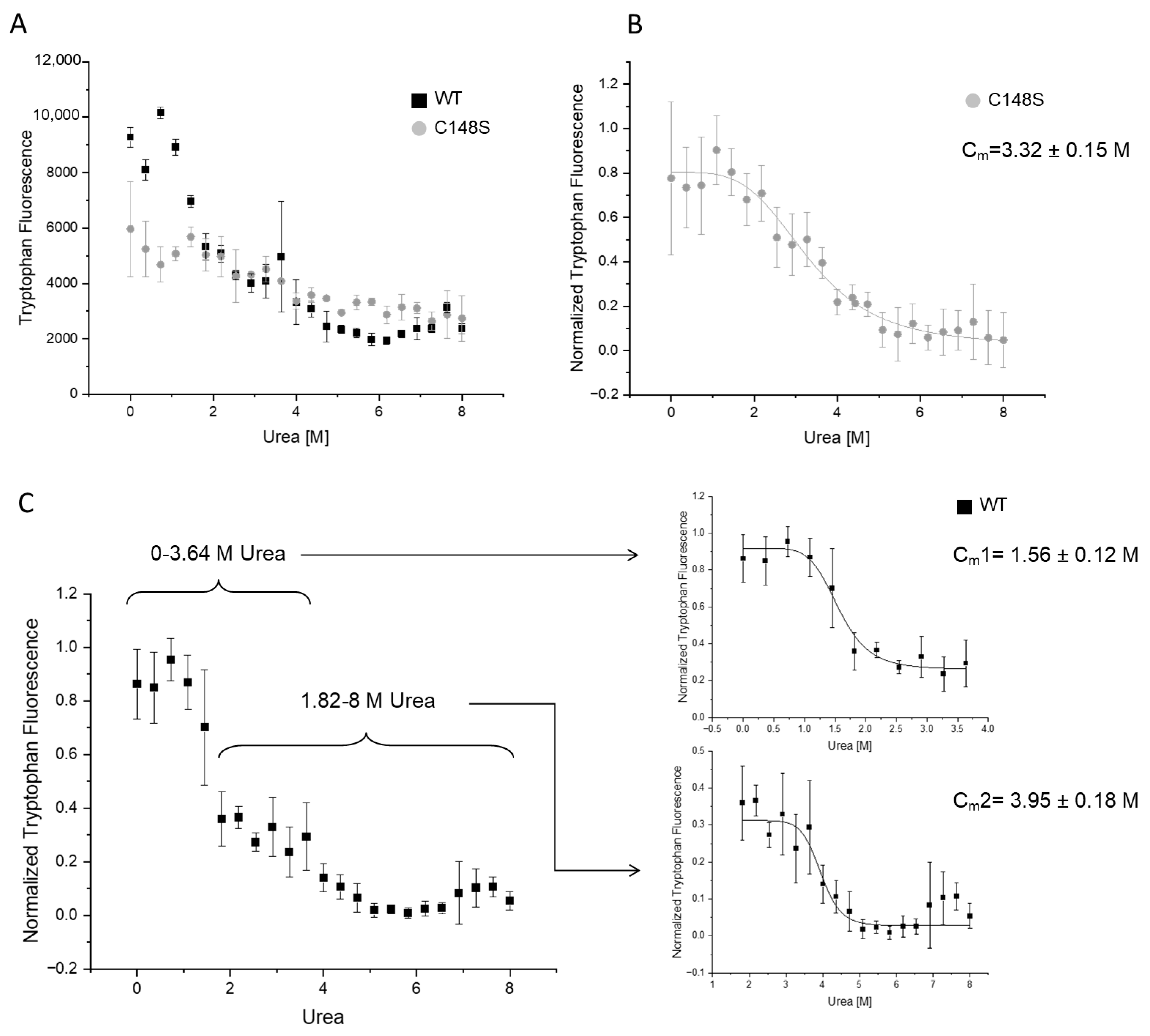
2.3.3. The Amino Acid Substitution C148S Reduces the Chemical Stability of PCNA
2.3.4. C148S Affects the Thermal Stability of PCNA
2.3.5. PCNAC148S Exhibits an Enhanced Aggregation Behavior
2.3.6. The Impaired Thermal Stability of PCNAC148S Is Responsible for Its Reduced Binding Signal to p15 Measured by FRET
3. Discussion
3.1. Development of a FRET-Based Assay to Analyze the PCNA–p15 Interaction
3.2. The Amino Acid Substitution C148S Reduces the Amount of Functional PCNA
3.3. Consequences of the Reduced PCNAC148S Stability for the Cell
3.4. PCNA Stabilizing Molecules as Therapeutic Option
4. Materials and Methods
4.1. Plasmid Construction and Mutagenesis
4.2. Protein Purification
4.3. FRET Binding Assay
4.4. Differential Scanning Fluorimetry (DSF)
4.5. Asymmetrical Flow Field-Flow Fractionation (AF4)
4.6. Intrinsic Tryptophan Fluorescence
4.7. Alchemical Free Energy Predictions
4.7.1. System Preparation
4.7.2. Free Energy Calculations and Hybrid Topologies
4.7.3. MD Simulations
4.8. Glutaraldehyde Crosslinking of PCNA
4.9. Western Blot Analysis
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Indiani, C.; O’Donnell, M. The replication clamp-loading machine at work in the three domains of life. Nat. Rev. Mol. Cell Biol. 2006, 7, 751–761. [Google Scholar] [CrossRef]
- Krishna, T.S.; Kong, X.P.; Gary, S.; Burgers, P.M.; Kuriyan, J. Crystal structure of the eukaryotic DNA polymerase processivity factor PCNA. Cell 1994, 79, 1233–1243. [Google Scholar] [CrossRef]
- Matsumiya, S.; Ishino, Y.; Morikawa, K. Crystal structure of an archaeal DNA sliding clamp: Proliferating cell nuclear antigen from Pyrococcus furiosus. Protein Sci. 2001, 10, 17–23. [Google Scholar] [CrossRef]
- Mulye, M.; Singh, M.I.; Jain, V. From Processivity to Genome Maintenance: The Many Roles of Sliding Clamps. Genes 2022, 13, 2058. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.J.; Johnson, K.; Rudolf, J.; McMahon, S.A.; Carter, L.; Oke, M.; Liu, H.; Taylor, G.L.; White, M.F.; Naismith, J.H. Structure of the heterotrimeric PCNA from Sulfolobus solfataricus. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2006, 62, 944–948. [Google Scholar] [CrossRef]
- Stukenberg, P.T.; Studwell-Vaughan, P.S.; O’Donnell, M. Mechanism of the sliding beta-clamp of DNA polymerase III holoenzyme. J. Biol. Chem. 1991, 266, 11328–11334. [Google Scholar] [CrossRef] [PubMed]
- Gulbis, J.M.; Kelman, Z.; Hurwitz, J.; O’Donnell, M.; Kuriyan, J. Structure of the C-terminal region of p21(WAF1/CIP1) complexed with human PCNA. Cell 1996, 87, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Moldovan, G.L.; Pfander, B.; Jentsch, S. PCNA, the maestro of the replication fork. Cell 2007, 129, 665–679. [Google Scholar] [CrossRef] [PubMed]
- Choe, K.N.; Moldovan, G.-L. Forging ahead through darkness: PCNA, still the principal conductor at the replication fork. Mol. Cell 2017, 65, 380–392. [Google Scholar] [CrossRef]
- González-Magaña, A.; Blanco, F.J. Human PCNA Structure, Function and Interactions. Biomolecules 2020, 10, 570. [Google Scholar] [CrossRef]
- Havens, C.G.; Walter, J.C. Docking of a specialized PIP Box onto chromatin-bound PCNA creates a degron for the ubiquitin ligase CRL4Cdt2. Mol. Cell 2009, 35, 93–104. [Google Scholar] [CrossRef]
- Prestel, A.; Wichmann, N.; Martins, J.M.; Marabini, R.; Kassem, N.; Broendum, S.S.; Otterlei, M.; Nielsen, O.; Willemoës, M.; Ploug, M.; et al. The PCNA interaction motifs revisited: Thinking outside the PIP-box. Cell. Mol. Life Sci. 2019, 76, 4923–4943. [Google Scholar] [CrossRef]
- Warbrick, E. PCNA binding through a conserved motif. Bioessays 1998, 20, 195–199. [Google Scholar] [CrossRef]
- Xu, H.; Zhang, P.; Liu, L.; Lee, M.Y. A novel PCNA-binding motif identified by the panning of a random peptide display library. Biochemistry 2001, 40, 4512–4520. [Google Scholar] [CrossRef] [PubMed]
- Gilljam, K.M.; Feyzi, E.; Aas, P.A.; Sousa, M.M.; Müller, R.; Vågbø, C.B.; Catterall, T.C.; Liabakk, N.B.; Slupphaug, G.; Drabløs, F.; et al. Identification of a novel, widespread, and functionally important PCNA-binding motif. J. Cell Biol. 2009, 186, 645–654. [Google Scholar] [CrossRef]
- Hara, K.; Uchida, M.; Tagata, R.; Yokoyama, H.; Ishikawa, Y.; Hishiki, A.; Hashimoto, H. Structure of proliferating cell nuclear antigen (PCNA) bound to an APIM peptide reveals the universality of PCNA interaction. Acta Crystallogr. F Struct. Biol. Commun. 2018, 74, 214–221. [Google Scholar] [CrossRef]
- Giannos, P.; Kechagias, K.S.; Bowden, S.; Tabassum, N.; Paraskevaidi, M.; Kyrgiou, M. PCNA in Cervical Intraepithelial Neoplasia and Cervical Cancer: An Interaction Network Analysis of Differentially Expressed Genes. Front. Oncol. 2021, 11, 779042. [Google Scholar] [CrossRef]
- Li, C.; Gao, Y.; Lu, C.; Guo, M. Identification of potential biomarkers for colorectal cancer by clinical database analysis and Kaplan-Meier curves analysis. Medicine 2023, 102, e32877. [Google Scholar] [CrossRef]
- Zheng, C.; Yang, R. RCD24, B7-H4 and PCNA expression and clinical significance in ovarian cancer. J. Buon 2019, 24, 715–719. [Google Scholar]
- Stoimenov, I.; Helleday, T. PCNA on the crossroad of cancer. Biochem. Soc. Trans. 2009, 37, 605–613. [Google Scholar] [CrossRef] [PubMed]
- Cardano, M.; Tribioli, C.; Prosperi, E. Targeting Proliferating Cell Nuclear Antigen (PCNA) as an Effective Strategy to Inhibit Tumor Cell Proliferation. Curr. Cancer Drug Targets 2020, 20, 240–252. [Google Scholar] [CrossRef]
- Wang, S.C. PCNA: A silent housekeeper or a potential therapeutic target? Trends Pharmacol. Sci. 2014, 35, 178–186. [Google Scholar] [CrossRef]
- Bose, S.; Ingham, M.; Singh-Kandah, S.V.; Magana, W.; Schwartz, G.K. A phase II study, with a safety lead-in, to evaluate ATX-101, a peptide drug targeting PCNA, in advanced dedifferentiated liposarcoma and leiomyosarcoma. J. Clin. Oncol. 2022, 40, TPS11587. [Google Scholar] [CrossRef]
- Müller, R.; Misund, K.; Holien, T.; Bachke, S.; Gilljam, K.M.; Våtsveen, T.K.; Rø, T.B.; Bellacchio, E.; Sundan, A.; Otterlei, M. Targeting proliferating cell nuclear antigen and its protein interactions induces apoptosis in multiple myeloma cells. PLoS ONE 2013, 8, e70430. [Google Scholar] [CrossRef] [PubMed]
- Lemech, C.R.; Kichenadasse, G.; Marschner, J.-P.; Alevizopoulos, K.; Otterlei, M.; Millward, M. ATX-101, a cell-penetrating protein targeting PCNA, can be safely administered as intravenous infusion in patients and shows clinical activity in a Phase 1 study. Oncogene 2023, 42, 541–544. [Google Scholar] [CrossRef] [PubMed]
- Inoue, A.; Kikuchi, S.; Hishiki, A.; Shao, Y.; Heath, R.; Evison, B.J.; Actis, M.; Canman, C.E.; Hashimoto, H.; Fujii, N. A small molecule inhibitor of monoubiquitinated Proliferating Cell Nuclear Antigen (PCNA) inhibits repair of interstrand DNA cross-link, enhances DNA double strand break, and sensitizes cancer cells to cisplatin. J. Biol. Chem. 2014, 289, 7109–7120. [Google Scholar] [CrossRef] [PubMed]
- Punchihewa, C.; Inoue, A.; Hishiki, A.; Fujikawa, Y.; Connelly, M.; Evison, B.; Shao, Y.; Heath, R.; Kuraoka, I.; Rodrigues, P.; et al. Identification of small molecule proliferating cell nuclear antigen (PCNA) inhibitor that disrupts interactions with PIP-box proteins and inhibits DNA replication. J. Biol. Chem. 2012, 287, 14289–14300. [Google Scholar] [CrossRef] [PubMed]
- Evison, B.J.; Actis, M.L.; Wu, S.Z.; Shao, Y.; Heath, R.J.; Yang, L.; Fujii, N. A site-selective, irreversible inhibitor of the DNA replication auxiliary factor proliferating cell nuclear antigen (PCNA). Bioorg. Med. Chem. 2014, 22, 6333–6343. [Google Scholar] [CrossRef] [PubMed]
- De Biasio, A.; de Opakua, A.I.; Mortuza, G.B.; Molina, R.; Cordeiro, T.N.; Castillo, F.; Villate, M.; Merino, N.; Delgado, S.; Gil-Carton, D.; et al. Structure of p15(PAF)-PCNA complex and implications for clamp sliding during DNA replication and repair. Nat. Commun. 2015, 6, 6439. [Google Scholar] [CrossRef]
- Xie, C.; Yao, M.; Dong, Q. Proliferating cell unclear antigen-associated factor (PAF15): A novel oncogene. Int. J. Biochem. Cell Biol. 2014, 50, 127–131. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Ong, D.S.T.; Hu, B.; Ho, Y.W.; Sauvé, C.-E.G.; Bristow, C.A.; Wang, Q.; Multani, A.S.; Chen, P.; Nezi, L.; Jiang, S.; et al. PAF promotes stemness and radioresistance of glioma stem cells. Proc. Natl. Acad. Sci. USA 2017, 114, E9086–E9095. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Zhu, K.; Dang, C.; Lan, K.; Wang, H.; Yuan, D.; Chen, W.; Meltzer, S.J.; Li, K. Paf15 expression correlates with rectal cancer prognosis, cell proliferation and radiation response. Oncotarget 2016, 7, 38750–38761. [Google Scholar] [CrossRef] [PubMed]
- Roa, S.; Avdievich, E.; Peled, J.U.; Maccarthy, T.; Werling, U.; Kuang, F.L.; Kan, R.; Zhao, C.; Bergman, A.; Cohen, P.E.; et al. Ubiquitylated PCNA plays a role in somatic hypermutation and class-switch recombination and is required for meiotic progression. Proc. Natl. Acad. Sci. USA 2008, 105, 16248–16253. [Google Scholar] [CrossRef]
- Baple, E.L.; Chambers, H.; Cross, H.E.; Fawcett, H.; Nakazawa, Y.; Chioza, B.A.; Harlalka, G.V.; Mansour, S.; Sreekantan-Nair, A.; Patton, M.A.; et al. Hypomorphic PCNA mutation underlies a human DNA repair disorder. J. Clin. Investig. 2014, 124, 3137–3146. [Google Scholar] [CrossRef] [PubMed]
- Raslan, I.R.; de Assis Pereira Matos, P.C.A.; Boaratti Ciarlariello, V.; Daghastanli, K.H.; Rosa, A.B.R.; Arita, J.H.; Aranda, C.S.; Barsottini, O.G.P.; Pedroso, J.L. Beyond Typical Ataxia Telangiectasia: How to Identify the Ataxia Telangiectasia-Like Disorders. Mov. Disord. Clin. Pract. 2021, 8, 118–125. [Google Scholar] [CrossRef]
- Duffy, C.M.; Hilbert, B.J.; Kelch, B.A. A Disease-Causing Variant in PCNA Disrupts a Promiscuous Protein Binding Site. J. Mol. Biol. 2016, 428, 1023–1040. [Google Scholar] [CrossRef]
- Magrino, J.; Munford, V.; Martins, D.J.; Homma, T.K.; Page, B.; Gaubitz, C.; Freire, B.L.; Lerario, A.M.; Vilar, J.B.; Amorin, A.; et al. A thermosensitive PCNA allele underlies an ataxia-telangiectasia-like disorder. J. Biol. Chem. 2023, 299, 104656. [Google Scholar] [CrossRef] [PubMed]
- Bindels, D.S.; Haarbosch, L.; van Weeren, L.; Postma, M.; Wiese, K.E.; Mastop, M.; Aumonier, S.; Gotthard, G.; Royant, A.; Hink, M.A.; et al. mScarlet: A bright monomeric red fluorescent protein for cellular imaging. Nat. Methods 2017, 14, 53–56. [Google Scholar] [CrossRef]
- Shaner, N.C.; Lambert, G.G.; Chammas, A.; Ni, Y.; Cranfill, P.J.; Baird, M.A.; Sell, B.R.; Allen, J.R.; Day, R.N.; Israelsson, M.; et al. A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nat. Methods 2013, 10, 407–409. [Google Scholar] [CrossRef]
- Song, Y.; Madahar, V.; Liao, J. Development of FRET assay into quantitative and high-throughput screening technology platforms for protein-protein interactions. Ann. Biomed. Eng. 2011, 39, 1224–1234. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.-y.; Song, Y.; Liu, Y. A new trend to determine biochemical parameters by quantitative FRET assays. Acta Pharmacol. Sin. 2015, 36, 1408–1415. [Google Scholar] [CrossRef] [PubMed]
- Lazareno, S. Quantification of receptor interactions using binding methods. J. Recept. Signal Transduct. Res. 2001, 21, 139–165. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Prusoff, W.H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [CrossRef]
- Lloyd, M.D. High-Throughput Screening for the Discovery of Enzyme Inhibitors. J. Med. Chem. 2020, 63, 10742–10772. [Google Scholar] [CrossRef]
- De March, M.; Merino, N.; Barrera-Vilarmau, S.; Crehuet, R.; Onesti, S.; Blanco, F.J.; De Biasio, A. Structural basis of human PCNA sliding on DNA. Nat. Commun. 2017, 8, 13935. [Google Scholar] [CrossRef]
- Motulsky, H.J.; Neubig, R.R. Analyzing binding data. Curr. Protoc. Neurosci. 2010, 52, 7.5.1–7.5.65. [Google Scholar] [CrossRef]
- Gapsys, V.; Michielssens, S.; Seeliger, D.; de Groot, B.L. Accurate and Rigorous Prediction of the Changes in Protein Free Energies in a Large-Scale Mutation Scan. Angew. Chem. (Int. Ed. Engl.) 2016, 55, 7364–7368. [Google Scholar] [CrossRef]
- Patel, D.; Patel, J.S.; Ytreberg, F.M. Implementing and Assessing an Alchemical Method for Calculating Protein-Protein Binding Free Energy. J. Chem. Theory Comput. 2021, 17, 2457–2464. [Google Scholar] [CrossRef]
- Crooks, G.E. Entropy production fluctuation theorem and the nonequilibrium work relation for free energy differences. Phys. Rev. E 1999, 60, 2721–2726. [Google Scholar] [CrossRef]
- Jarzynski, C. Nonequilibrium Equality for Free Energy Differences. Phys. Rev. Lett. 1997, 78, 2690–2693. [Google Scholar] [CrossRef]
- Jarzynski, C. Equilibrium free-energy differences from nonequilibrium measurements: A master-equation approach. Phys. Rev. E 1997, 56, 5018–5035. [Google Scholar] [CrossRef]
- Seeliger, D.; de Groot, B.L. Protein Thermostability Calculations Using Alchemical Free Energy Simulations. Biophys. J. 2010, 98, 2309–2316. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Chu, S.K.S.; Frizzo, H.N.; Connolly, M.P.; Caster, R.W.; Siegel, J.B. Evaluating Protein Engineering Thermostability Prediction Tools Using an Independently Generated Dataset. ACS Omega 2020, 5, 6487–6493. [Google Scholar] [CrossRef]
- Wang, W. Protein aggregation and its inhibition in biopharmaceutics. Int. J. Pharm. 2005, 289, 1–30. [Google Scholar] [CrossRef]
- Royer, C.A. Probing protein folding and conformational transitions with fluorescence. Chem. Rev. 2006, 106, 1769–1784. [Google Scholar] [CrossRef]
- Gao, K.; Oerlemans, R.; Groves, M.R. Theory and applications of differential scanning fluorimetry in early-stage drug discovery. Biophys. Rev. 2020, 12, 85–104. [Google Scholar] [CrossRef]
- Leeman, M.; Nilsson, L.; Storm, M.U. Practical applications of asymmetrical flow field-flow fractionation (AF4): A review. LCGC Eur. 2015, 28, 642–651. [Google Scholar]
- Mahler, H.C.; Friess, W.; Grauschopf, U.; Kiese, S. Protein aggregation: Pathways, induction factors and analysis. J. Pharm. Sci. 2009, 98, 2909–2934. [Google Scholar] [CrossRef]
- Jarmoskaite, I.; AlSadhan, I.; Vaidyanathan, P.P.; Herschlag, D. How to measure and evaluate binding affinities. eLife 2020, 9, e57264. [Google Scholar] [CrossRef]
- Sowa, S.T.; Vela-Rodríguez, C.; Galera-Prat, A.; Cázares-Olivera, M.; Prunskaite-Hyyryläinen, R.; Ignatev, A.; Lehtiö, L. A FRET-based high-throughput screening platform for the discovery of chemical probes targeting the scaffolding functions of human tankyrases. Sci. Rep. 2020, 10, 12357. [Google Scholar] [CrossRef] [PubMed]
- Bartolowits, M.D.; Gast, J.M.; Hasler, A.J.; Cirrincione, A.M.; O’Connor, R.J.; Mahmoud, A.H.; Lill, M.A.; Davisson, V.J. Discovery of Inhibitors for Proliferating Cell Nuclear Antigen Using a Computational-Based Linked-Multiple-Fragment Screen. ACS Omega 2019, 4, 15181–15196. [Google Scholar] [CrossRef] [PubMed]
- Wegerer, A.; Sun, T.; Altenbuchner, J. Optimization of an E. coli L-rhamnose-inducible expression vector: Test of various genetic module combinations. BMC Biotechnol. 2008, 8, 2. [Google Scholar] [CrossRef] [PubMed]
- Jahn, T.R.; Radford, S.E. Folding versus aggregation: Polypeptide conformations on competing pathways. Arch. Biochem. Biophys. 2008, 469, 100–117. [Google Scholar] [CrossRef] [PubMed]
- Kontopidis, G.; Wu, S.-Y.; Zheleva, D.I.; Taylor, P.; McInnes, C.; Lane, D.P.; Fischer, P.M.; Walkinshaw, M.D. Structural and biochemical studies of human proliferating cell nuclear antigen complexes provide a rationale for cyclin association and inhibitor design. Proc. Natl. Acad. Sci. USA 2005, 102, 1871–1876. [Google Scholar] [CrossRef]
- González-Mondragón, E.; Zubillaga, R.A.; Saavedra, E.; Chánez-Cárdenas, M.E.; Pérez-Montfort, R.; Hernández-Arana, A. Conserved Cysteine 126 in Triosephosphate Isomerase Is Required Not for Enzymatic Activity but for Proper Folding and Stability. Biochemistry 2004, 43, 3255–3263. [Google Scholar] [CrossRef]
- Pavlin, M.; Qasem, Z.; Sameach, H.; Gevorkyan-Airapetov, L.; Ritacco, I.; Ruthstein, S.; Magistrato, A. Unraveling the Impact of Cysteine-to-Serine Mutations on the Structural and Functional Properties of Cu(I)-Binding Proteins. Int. J. Mol. Sci. 2019, 20, 3462. [Google Scholar] [CrossRef]
- Tsai, Y.-C.; Wang, Y.-H.; Liu, Y.-C. Overexpression of PCNA Attenuates Oxidative Stress-Caused Delay of Gap-Filling during Repair of UV-Induced DNA Damage. J. Nucleic Acids 2017, 2017, 8154646. [Google Scholar] [CrossRef]
- Jaskulski, D.; deRiel, J.K.; Mercer, W.E.; Calabretta, B.; Baserga, R. Inhibition of cellular proliferation by antisense oligodeoxynucleotides to PCNA cyclin. Science 1988, 240, 1544–1546. [Google Scholar] [CrossRef]
- Groehler, A.L.; Lannigan, D.A. A chromatin-bound kinase, ERK8, protects genomic integrity by inhibiting HDM2-mediated degradation of the DNA clamp PCNA. J. Cell Biol. 2010, 190, 575–586. [Google Scholar] [CrossRef]
- Sporbert, A.; Domaing, P.; Leonhardt, H.; Cardoso, M.C. PCNA acts as a stationary loading platform for transiently interacting Okazaki fragment maturation proteins. Nucleic Acids Res. 2005, 33, 3521–3528. [Google Scholar] [CrossRef]
- Kurki, P.; Vanderlaan, M.; Dolbeare, F.; Gray, J.; Tan, E.M. Expression of proliferating cell nuclear antigen (PCNA)/cyclin during the cell cycle. Exp. Cell Res. 1986, 166, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Green, C.M.; Baple, E.L.; Crosby, A.H. PCNA mutation affects DNA repair not replication. Cell Cycle 2014, 13, 3157–3158. [Google Scholar] [CrossRef] [PubMed]
- Invernizzi, G.; Papaleo, E.; Sabate, R.; Ventura, S. Protein aggregation: Mechanisms and functional consequences. Int. J. Biochem. Cell Biol. 2012, 44, 1541–1554. [Google Scholar] [CrossRef]
- De Baets, G.; Van Doorn, L.; Rousseau, F.; Schymkowitz, J. Increased Aggregation Is More Frequently Associated to Human Disease-Associated Mutations Than to Neutral Polymorphisms. PLoS Comput. Biol. 2015, 11, e1004374. [Google Scholar] [CrossRef] [PubMed]
- Gil-Martínez, J.; Bernardo-Seisdedos, G.; Mato, J.M.; Millet, O. The use of pharmacological chaperones in rare diseases caused by reduced protein stability. Proteomics 2022, 22, e2200222. [Google Scholar] [CrossRef]
- Liguori, L.; Monticelli, M.; Allocca, M.; Hay Mele, B.; Lukas, J.; Cubellis, M.V.; Andreotti, G. Pharmacological Chaperones: A Therapeutic Approach for Diseases Caused by Destabilizing Missense Mutations. Int. J. Mol. Sci. 2020, 21, 489. [Google Scholar] [CrossRef]
- Leonhardt, H.; Rahn, H.P.; Weinzierl, P.; Sporbert, A.; Cremer, T.; Zink, D.; Cardoso, M.C. Dynamics of DNA replication factories in living cells. J. Cell Biol. 2000, 149, 271–280. [Google Scholar] [CrossRef]
- Boivin, S.; Kozak, S.; Meijers, R. Optimization of protein purification and characterization using Thermofluor screens. Protein Exp. Purif. 2013, 91, 192–206. [Google Scholar] [CrossRef]
- Wright, T.A.; Stewart, J.M.; Page, R.C.; Konkolewicz, D. Extraction of Thermodynamic Parameters of Protein Unfolding Using Parallelized Differential Scanning Fluorimetry. J. Phys. Chem. Lett. 2017, 8, 553–558. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Aldeghi, M.; de Groot, B.L.; Gapsys, V. Accurate Calculation of Free Energy Changes upon Amino Acid Mutation. Methods Mol. Biol. 2019, 1851, 19–47. [Google Scholar] [CrossRef] [PubMed]
- Gapsys, V.; Michielssens, S.; Seeliger, D.; de Groot, B.L. pmx: Automated protein structure and topology generation for alchemical perturbations. J. Comput. Chem. 2015, 36, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Shirts, M.R.; Bair, E.; Hooker, G.; Pande, V.S. Equilibrium free energies from nonequilibrium measurements using maximum-likelihood methods. Phys. Rev. Lett. 2003, 91, 140601. [Google Scholar] [CrossRef] [PubMed]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hardebeck, S.; Schreiber, S.; Adick, A.; Langer, K.; Jose, J. A FRET-Based Assay for the Identification of PCNA Inhibitors. Int. J. Mol. Sci. 2023, 24, 11858. https://doi.org/10.3390/ijms241411858
Hardebeck S, Schreiber S, Adick A, Langer K, Jose J. A FRET-Based Assay for the Identification of PCNA Inhibitors. International Journal of Molecular Sciences. 2023; 24(14):11858. https://doi.org/10.3390/ijms241411858
Chicago/Turabian StyleHardebeck, Sarah, Sebastian Schreiber, Annika Adick, Klaus Langer, and Joachim Jose. 2023. "A FRET-Based Assay for the Identification of PCNA Inhibitors" International Journal of Molecular Sciences 24, no. 14: 11858. https://doi.org/10.3390/ijms241411858
APA StyleHardebeck, S., Schreiber, S., Adick, A., Langer, K., & Jose, J. (2023). A FRET-Based Assay for the Identification of PCNA Inhibitors. International Journal of Molecular Sciences, 24(14), 11858. https://doi.org/10.3390/ijms241411858