
Compromised Mitotic Fidelity in Human Pluripotent Stem Cells


Abstract
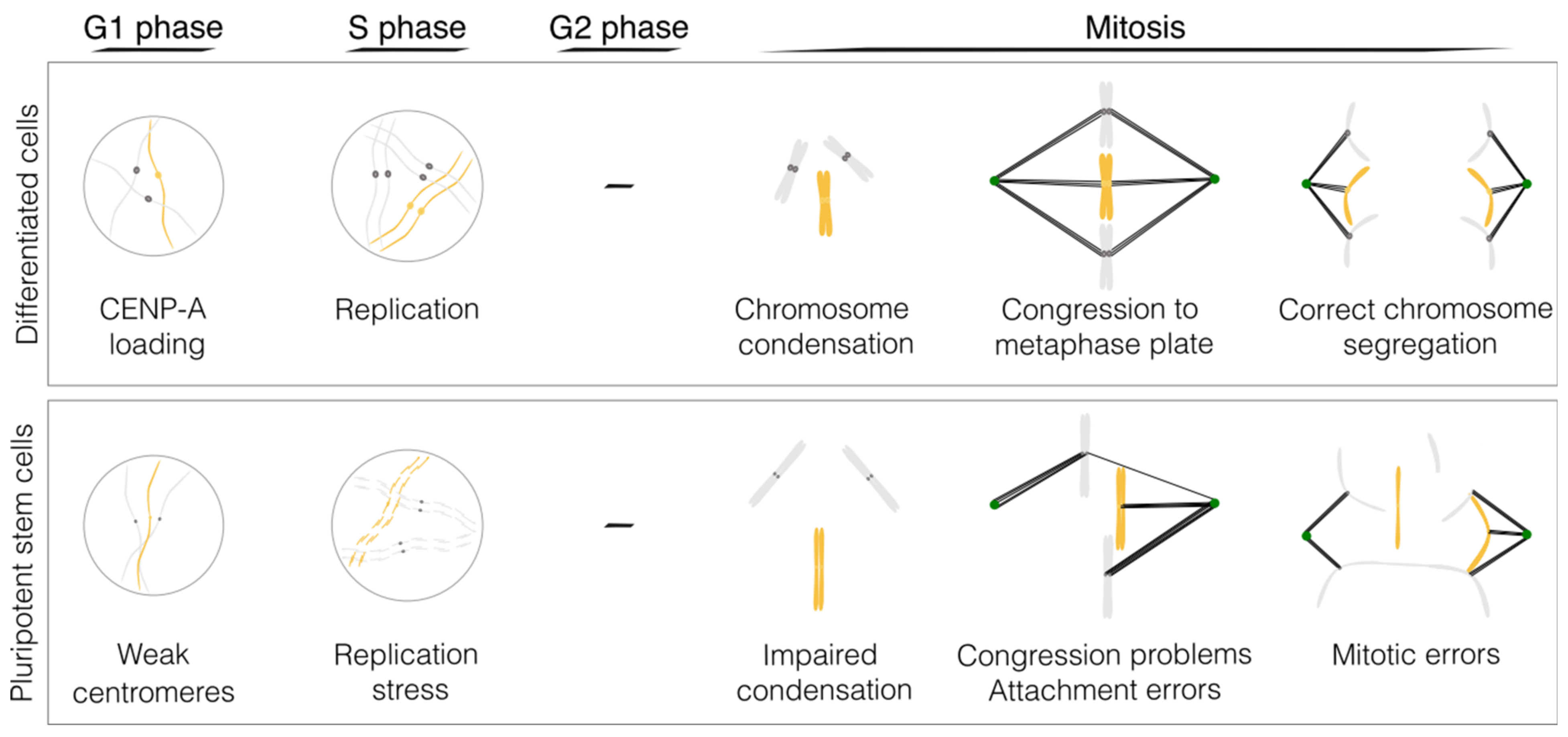
:1. Human Pluripotent Stem Cells Present Frequent Karyotypic Abnormalities
2. Molecular Mechanisms Underlying the Compromised Mitotic Fidelity in Pluripotent Stem Cells
2.1. DNA Replication
2.2. Chromosome Condensation
2.3. The Centromere
2.4. Kinetochore–Microtubule Dynamics
2.5. Spindle Assembly Checkpoint
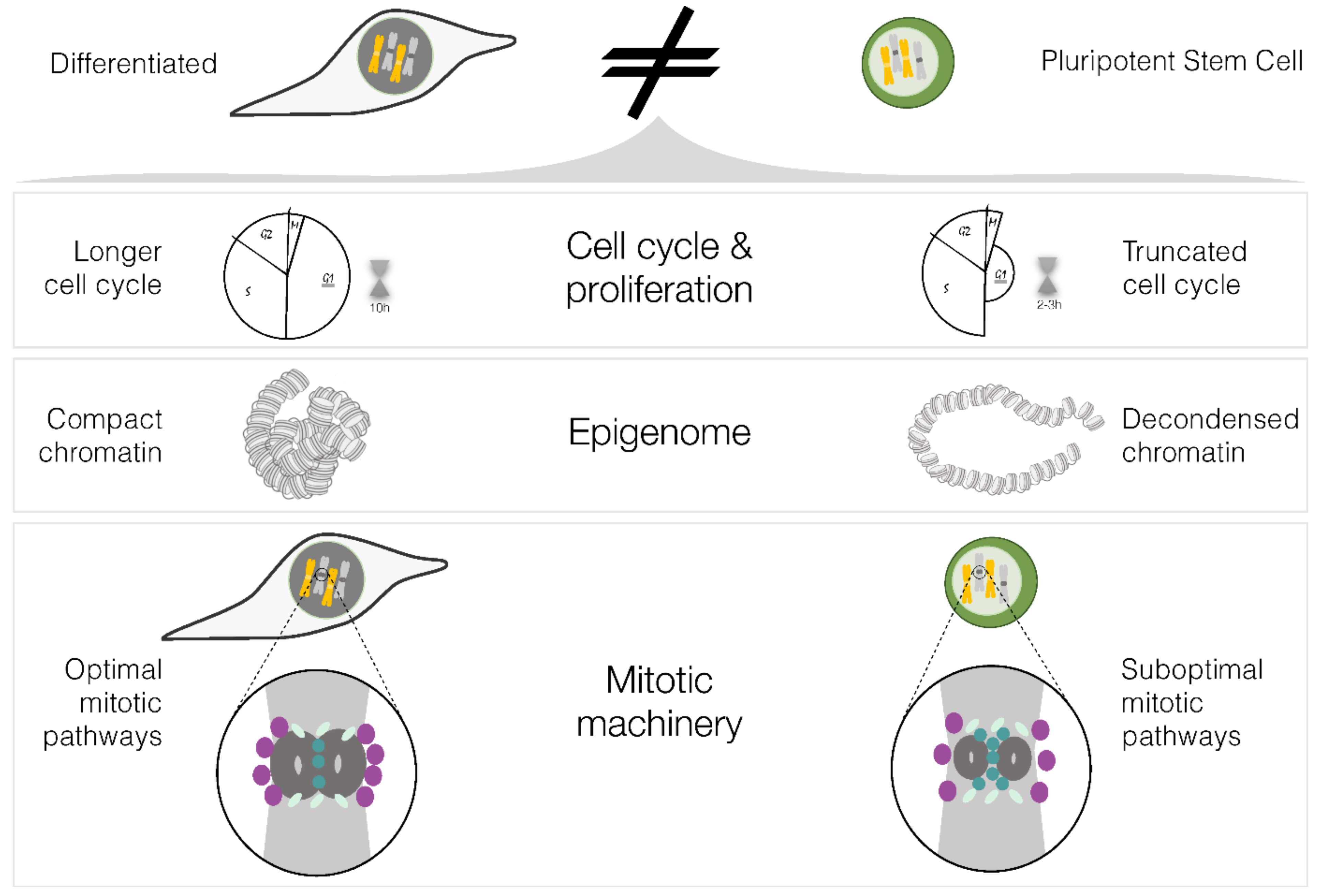
3. Physiological Properties of PSCs Which Can Affect Mitotic Fidelity
3.1. Cell Cycle
3.2. Histone Post-Translational Modifications
3.3. Mitotic Machinery of PSCs
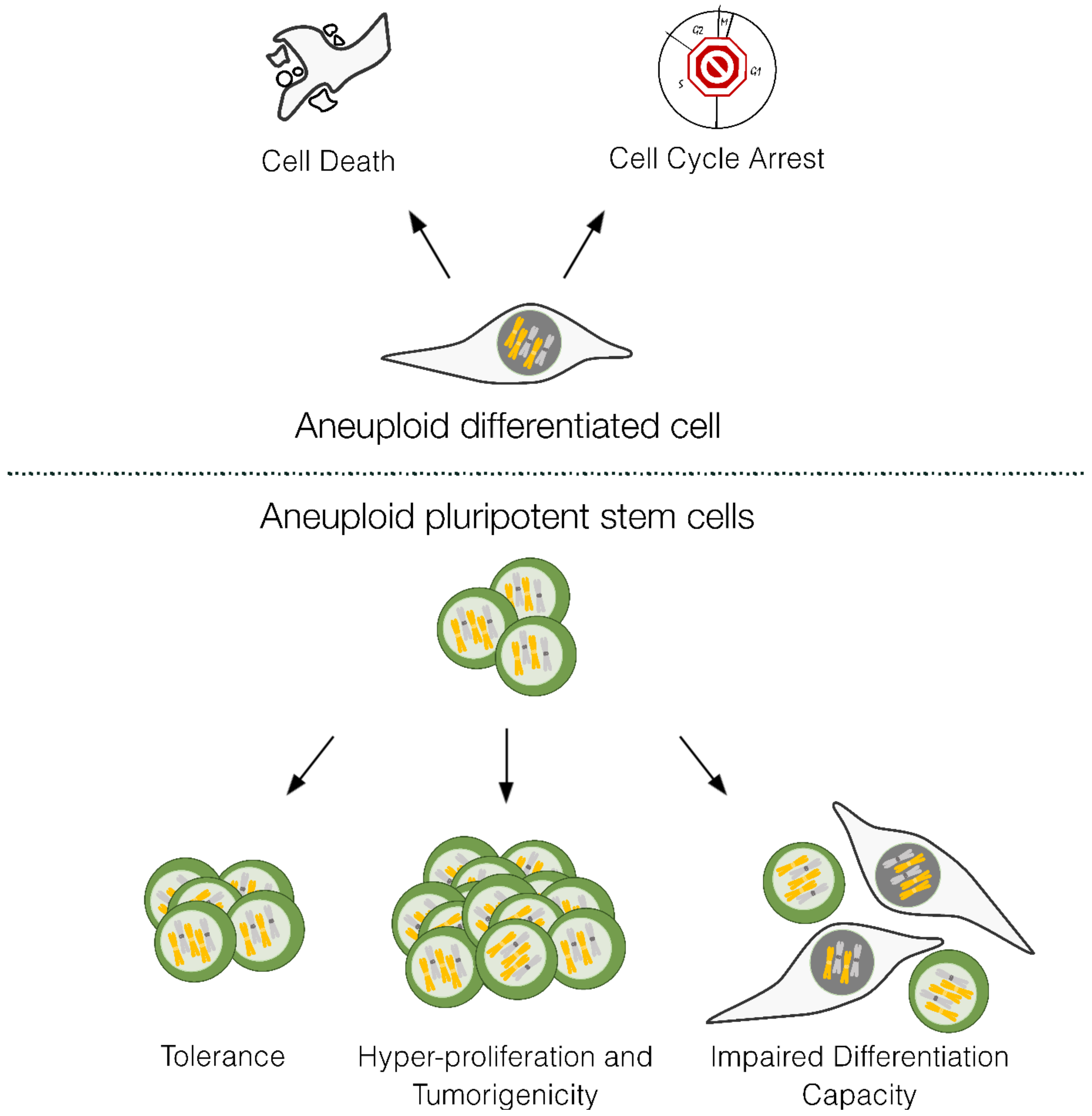
4. Consequences of Decreased Mitotic Fidelity in Pluripotent Stem Cells
5. Can We Increase the Mitotic Fidelity of PSCs?
6. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellin, M.; Marchetto, M.C.; Gage, F.H.; Mummery, C.L. Induced Pluripotent Stem Cells: The New Patient? Nat. Rev. Mol. Cell Biol. 2012, 13, 713–726. [Google Scholar] [CrossRef] [PubMed]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic Stem Cell Lines Derived from Human Blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wernig, M.; Meissner, A.; Foreman, R.; Brambrink, T.; Ku, M.; Hochedlinger, K.; Bernstein, B.E.; Jaenisch, R. In Vitro Reprogramming of Fibroblasts into a Pluripotent ES-Cell-like State. Nature 2007, 448, 318–324. [Google Scholar] [CrossRef]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced Pluripotent Stem Cell Lines Derived from Human Somatic Cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, S. Pluripotent Stem Cell-Based Cell Therapy—Promise and Challenges. Cell Stem Cell 2020, 27, 523–531. [Google Scholar] [CrossRef]
- Andrews, P.W.; Ben-David, U.; Benvenisty, N.; Coffey, P.; Eggan, K.; Knowles, B.B.; Nagy, A.; Pera, M.; Reubinoff, B.; Rugg-Gunn, P.J.; et al. Assessing the Safety of Human Pluripotent Stem Cells and Their Derivatives for Clinical Applications. Stem Cell Rep. 2017, 9, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Santaguida, S.; Amon, A. Short- and Long-Term Effects of Chromosome Mis-Segregation and Aneuploidy. Nat. Rev. Mol. Cell Biol. 2015, 16, 473–485. [Google Scholar] [CrossRef] [Green Version]
- The International Stem Cell Initiative. Screening Ethnically Diverse Human Embryonic Stem Cells Identifies a Chromosome 20 Minimal Amplicon Conferring Growth Advantage. Nat. Biotechnol. 2011, 29, 1132–1144. [Google Scholar] [CrossRef] [Green Version]
- Taapken, S.M.; Nisler, B.S.; Newton, M.A.; Sampsell-Barron, T.L.; Leonhard, K.A.; McIntire, E.M.; Montgomery, K.D. Karyotypic Abnormalities in Human Induced Pluripotent Stem Cells and Embryonic Stem Cells. Nat. Biotechnol. 2011, 29, 313–314. [Google Scholar] [CrossRef]
- Mayshar, Y.; Ben-David, U.; Lavon, N.; Biancotti, J.-C.; Yakir, B.; Clark, A.T.; Plath, K.; Lowry, W.E.; Benvenisty, N. Identification and Classification of Chromosomal Aberrations in Human Induced Pluripotent Stem Cells. Cell Stem Cell 2010, 7, 521–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Draper, J.S.; Smith, K.; Gokhale, P.; Moore, H.D.; Maltby, E.; Johnson, J.; Meisner, L.; Zwaka, T.P.; Thomson, J.A.; Andrews, P.W. Recurrent Gain of Chromosomes 17q and 12 in Cultured Human Embryonic Stem Cells. Nat. Biotechnol. 2004, 22, 53–54. [Google Scholar] [CrossRef] [PubMed]
- Martins-Taylor, K.; Nisler, B.S.; Taapken, S.M.; Compton, T.; Crandall, L.; Montgomery, K.D.; Lalande, M.; Xu, R.-H. Recurrent Copy Number Variations in Human Induced Pluripotent Stem Cells. Nat. Biotechnol. 2011, 29, 488–491. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.E.C.; Harrison, N.J.; Maltby, E.; Smith, K.; Moore, H.D.; Shaw, P.J.; Heath, P.R.; Holden, H.; Andrews, P.W. Adaptation to Culture of Human Embryonic Stem Cells and Oncogenesis in Vivo. Nat. Biotechnol. 2007, 25, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Laurent, L.C.; Ulitsky, I.; Slavin, I.; Tran, H.; Schork, A.; Morey, R.; Lynch, C.; Harness, J.V.; Lee, S.; Barrero, M.J.; et al. Dynamic Changes in the Copy Number of Pluripotency and Cell Proliferation Genes in Human ESCs and IPSCs during Reprogramming and Time in Culture. Cell Stem Cell 2011, 8, 106–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, C.; Cavazza, T.; Schuh, M. Aneuploidy in Human Eggs: Contributions of the Meiotic Spindle. Biochem. Soc. Trans. 2021, 49, 107–118. [Google Scholar] [CrossRef]
- Chunduri, N.K.; Storchová, Z. The Diverse Consequences of Aneuploidy. Nat. Cell Biol. 2019, 21, 54–62. [Google Scholar] [CrossRef]
- Weaver, B.A.; Cleveland, D.W. Does Aneuploidy Cause Cancer? Curr. Opin. Cell Biol. 2006, 18, 658–667. [Google Scholar] [CrossRef]
- Weaver, B.A.; Cleveland, D.W. The Aneuploidy Paradox in Cell Growth and Tumorigenesis. Cancer Cell 2008, 14, 431–433. [Google Scholar] [CrossRef] [Green Version]
- Holland, A.J.; Cleveland, D.W. Losing Balance: The Origin and Impact of Aneuploidy in Cancer: ‘Exploring Aneuploidy: The Significance of Chromosomal Imbalance’ Review Series. EMBO Rep. 2012, 13, 501–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, K.; Zambelli, F.; Mertzanidou, A.; Smolders, I.; Geens, M.; Nguyen, H.T.; Barbé, L.; Sermon, K.; Spits, C. Higher-Density Culture in Human Embryonic Stem Cells Results in DNA Damage and Genome Instability. Stem Cell Rep. 2016, 6, 330–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, D.J.; Resio, B.; Pellman, D. Causes and Consequences of Aneuploidy in Cancer. Nat. Rev. Genet. 2012, 13, 189–203. [Google Scholar] [CrossRef]
- Compton, D.A. Mechanisms of Aneuploidy. Curr. Opin. Cell Biol. 2011, 23, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Musacchio, A.; Salmon, E.D. The Spindle-Assembly Checkpoint in Space and Time. Nat. Rev. Mol. Cell Biol. 2007, 8, 379–393. [Google Scholar] [CrossRef]
- Mirkovic, M.; Oliveira, R.A. Centromeric Cohesin: Molecular Glue and Much More. Prog. Mol. Subcell. Biol. 2017, 56, 485–513. [Google Scholar] [CrossRef]
- Piskadlo, E.; Oliveira, R.A. A Topology-Centric View on Mitotic Chromosome Architecture. IJMS 2017, 18, 2751. [Google Scholar] [CrossRef] [Green Version]
- Soto, M.; Raaijmakers, J.A.; Medema, R.H. Consequences of Genomic Diversification Induced by Segregation Errors. Trends Genet. 2019, 35, 279–291. [Google Scholar] [CrossRef]
- Becker, K.A.; Ghule, P.N.; Therrien, J.A.; Lian, J.B.; Stein, J.L.; van Wijnen, A.J.; Stein, G.S. Self-Renewal of Human Embryonic Stem Cells Is Supported by a Shortened G1 Cell Cycle Phase. J. Cell. Physiol. 2006, 209, 883–893. [Google Scholar] [CrossRef]
- Kobayashi, H.; Kikyo, N. Epigenetic Regulation of Open Chromatin in Pluripotent Stem Cells. Transl. Res. 2015, 165, 18–27. [Google Scholar] [CrossRef] [Green Version]
- Lamm, N.; Ben-David, U.; Golan-Lev, T.; Storchová, Z.; Benvenisty, N.; Kerem, B. Genomic Instability in Human Pluripotent Stem Cells Arises from Replicative Stress and Chromosome Condensation Defects. Cell Stem Cell 2016, 18, 253–261. [Google Scholar] [CrossRef] [Green Version]
- Halliwell, J.A.; Frith, T.J.R.; Laing, O.; Price, C.J.; Bower, O.J.; Stavish, D.; Gokhale, P.J.; Hewitt, Z.; El-Khamisy, S.F.; Barbaric, I.; et al. Nucleosides Rescue Replication-Mediated Genome Instability of Human Pluripotent Stem Cells. Stem Cell Rep. 2020, 14, 1009–1017. [Google Scholar] [CrossRef]
- Simara, P.; Tesarova, L.; Rehakova, D.; Matula, P.; Stejskal, S.; Hampl, A.; Koutna, I. DNA Double-Strand Breaks in Human Induced Pluripotent Stem Cell Reprogramming and Long-Term in Vitro Culturing. Stem Cell Res. Ther. 2017, 8, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallabhaneni, H.; Lynch, P.J.; Chen, G.; Park, K.; Liu, Y.; Goehe, R.; Mallon, B.S.; Boehm, M.; Hursh, D.A. High Basal Levels of ΓH2AX in Human Induced Pluripotent Stem Cells Are Linked to Replication-Associated DNA Damage and Repair. Stem Cells 2018, 36, 1501–1513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kislova, A.V.; Zheglo, D.; Pozhitnova, V.O.; Sviridov, P.S.; Gadzhieva, E.P.; Voronina, E.S. Replication Stress Causes Delayed Mitotic Entry and Chromosome 12 Fragility at the ANKS1B Large Neuronal Gene in Human Induced Pluripotent Stem Cells. preprint 2023. [Google Scholar] [CrossRef]
- Mankouri, H.W.; Huttner, D.; Hickson, I.D. How Unfinished Business from S-Phase Affects Mitosis and Beyond. EMBO J. 2013, 32, 2661–2671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz, S.; Lopez-Contreras, A.J.; Gabut, M.; Marion, R.M.; Gutierrez-Martinez, P.; Bua, S.; Ramirez, O.; Olalde, I.; Rodrigo-Perez, S.; Li, H.; et al. Limiting Replication Stress during Somatic Cell Reprogramming Reduces Genomic Instability in Induced Pluripotent Stem Cells. Nat. Commun. 2015, 6, 8036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desmarais, J.A.; Hoffmann, M.J.; Bingham, G.; Gagou, M.E.; Meuth, M.; Andrews, P.W. Human Embryonic Stem Cells Fail to Activate CHK1 and Commit to Apoptosis in Response to DNA Replication Stress. Stem Cells 2012, 30, 1385–1393. [Google Scholar] [CrossRef]
- Vessoni, A.T.; Zhang, T.; Quinet, A.; Jeong, H.-C.; Munroe, M.; Wood, M.; Tedone, E.; Vindigni, A.; Shay, J.W.; Greenberg, R.A.; et al. Telomere Erosion in Human Pluripotent Stem Cells Leads to ATR-Mediated Mitotic Catastrophe. J. Cell Biol. 2021, 220, e202011014. [Google Scholar] [CrossRef]
- Delgado-Olguin, P.; Recillas-Targa, F. Chromatin Structure of Pluripotent Stem Cells and Induced Pluripotent Stem Cells. Brief. Funct. Genom. 2011, 10, 37–49. [Google Scholar] [CrossRef]
- Li, D.; Shu, X.; Zhu, P.; Pei, D. Chromatin Accessibility Dynamics during Cell Fate Reprogramming. EMBO Rep. 2021, 22, e51644. [Google Scholar] [CrossRef]
- Wilkins, B.J.; Rall, N.A.; Ostwal, Y.; Kruitwagen, T.; Hiragami-Hamada, K.; Winkler, M.; Barral, Y.; Fischle, W.; Neumann, H. A Cascade of Histone Modifications Induces Chromatin Condensation in Mitosis. Science 2014, 343, 77–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weir, J.R.; Faesen, A.C.; Klare, K.; Petrovic, A.; Basilico, F.; Fischböck, J.; Pentakota, S.; Keller, J.; Pesenti, M.E.; Pan, D.; et al. Insights from Biochemical Reconstitution into the Architecture of Human Kinetochores. Nature 2016, 537, 249–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Black, B.E.; Jansen, L.E.T.; Foltz, D.R.; Cleveland, D.W. Centromere Identity, Function, and Epigenetic Propagation across Cell Divisions. Cold Spring Harb. Symp. Quant. Biol. 2010, 75, 403–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milagre, I.; Pereira, C.; Oliveira, R.A.; Jansen, L.E.T. Reprogramming of Human Cells to Pluripotency Induces CENP-A Chromatin Depletion. Open Biol. 2020, 10, 200227. [Google Scholar] [CrossRef]
- Bodor, D.L.; Valente, L.P.; Mata, J.F.; Black, B.E.; Jansen, L.E.T. Assembly in G1 Phase and Long-Term Stability Are Unique Intrinsic Features of CENP-A Nucleosomes. MBoC 2013, 24, 923–932. [Google Scholar] [CrossRef] [Green Version]
- Jansen, L.E.T.; Black, B.E.; Foltz, D.R.; Cleveland, D.W. Propagation of Centromeric Chromatin Requires Exit from Mitosis. J. Cell Biol. 2007, 176, 795–805. [Google Scholar] [CrossRef]
- Fachinetti, D.; Han, J.S.; McMahon, M.A.; Ly, P.; Abdullah, A.; Wong, A.J.; Cleveland, D.W. DNA Sequence-Specific Binding of CENP-B Enhances the Fidelity of Human Centromere Function. Dev. Cell 2015, 33, 314–327. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Hirst, A.J.; Duan, F.; Qiu, H.; Huang, R.; Ji, Y.; Bai, L.; Zhang, F.; Robinson, D.; Jones, M.; et al. Anti-Apoptotic Mutations Desensitize Human Pluripotent Stem Cells to Mitotic Stress and Enable Aneuploid Cell Survival. Stem Cell Rep. 2019, 12, 557–571. [Google Scholar] [CrossRef] [Green Version]
- Deng, C.; Ya, A.; Compton, D.A.; Godek, K.M. A Pluripotent Developmental State Confers a Low Fidelity of Chromosome Segregation. Stem Cell Rep. 2023, 18, 475–488. [Google Scholar] [CrossRef]
- Gregan, J.; Polakova, S.; Zhang, L.; Tolić-Nørrelykke, I.M.; Cimini, D. Merotelic Kinetochore Attachment: Causes and Effects. Trends Cell Biol. 2011, 21, 374–381. [Google Scholar] [CrossRef] [Green Version]
- Kitagawa, M.; Lee, S.H. The Chromosomal Passenger Complex (CPC) as a Key Orchestrator of Orderly Mitotic Exit and Cytokinesis. Front. Cell Dev. Biol. 2015, 3, 14. [Google Scholar] [CrossRef] [Green Version]
- Vukušić, K.; Tolić, I.M. Polar Chromosomes—Challenges of a Risky Path. Cells 2022, 11, 1531. [Google Scholar] [CrossRef]
- Lyu, R.; Wu, X.; Ma, N.; Wang, D.; Sun, S.; Luo, Y.; Zhou, J.; Lu, X.; Liu, M.; Li, D. The Specialized Mitotic Behavior of Human Embryonic Stem Cells. Cell Tissue Res. 2022, 387, 85–93. [Google Scholar] [CrossRef]
- Francois, L.; Boskovic, P.; Knerr, J.; He, W.; Sigismondo, G.; Schwan, C.; More, T.H.; Schlotter, M.; Krijgsveld, J.; Hiller, K.; et al. BCAT1 Redox Function Maintains Mitotic Fidelity. Cell Rep. 2022, 41, 111524. [Google Scholar] [CrossRef] [PubMed]
- Dou, Z.; Prifti, D.; Gui, P.; Liu, X.; Elowe, S.; Yao, X. Recent Progress on the Localization of the Spindle Assembly Checkpoint Machinery to Kinetochores. Cells 2019, 8, 278. [Google Scholar] [CrossRef] [Green Version]
- Mantel, C.; Guo, Y.; Lee, M.R.; Kim, M.-K.; Han, M.-K.; Shibayama, H.; Fukuda, S.; Yoder, M.C.; Pelus, L.M.; Kim, K.-S.; et al. Checkpoint-Apoptosis Uncoupling in Human and Mouse Embryonic Stem Cells: A Source of Karyotpic Instability. Blood 2007, 109, 4518–4527. [Google Scholar] [CrossRef] [Green Version]
- Padgett, J.; Santos, S.D.M. From Clocks to Dominoes: Lessons on Cell Cycle Remodelling from Embryonic Stem Cells. FEBS Lett. 2020, 594, 2031–2045. [Google Scholar] [CrossRef] [PubMed]
- Barnum, K.J.; O’Connell, M.J. Cell Cycle Regulation by Checkpoints. In Cell Cycle Control; Noguchi, E., Gadaleta, M.C., Eds.; Methods in Molecular Biology; Springer New York: New York, NY, USA, 2014; Volume 1170, pp. 29–40. ISBN 978-1-4939-0887-5. [Google Scholar]
- Damelin, M.; Sun, Y.E.; Sodja, V.B.; Bestor, T.H. Decatenation Checkpoint Deficiency in Stem and Progenitor Cells. Cancer Cell 2005, 8, 479–484. [Google Scholar] [CrossRef] [Green Version]
- Rieder, C.L.; Maiato, H. Stuck in Division or Passing Through. Dev. Cell 2004, 7, 637–651. [Google Scholar] [CrossRef] [Green Version]
- Becker, K.A.; Stein, J.L.; Lian, J.B.; van Wijnen, A.J.; Stein, G.S. Human Embryonic Stem Cells Are Pre-Mitotically Committed to Self-Renewal and Acquire a Lengthened G1 Phase upon Lineage Programming. J. Cell Physiol. 2010, 222, 103–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filipczyk, A.A.; Laslett, A.L.; Mummery, C.; Pera, M.F. Differentiation Is Coupled to Changes in the Cell Cycle Regulatory Apparatus of Human Embryonic Stem Cells. Stem Cell Res. 2007, 1, 45–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pauklin, S.; Vallier, L. The Cell-Cycle State of Stem Cells Determines Cell Fate Propensity. Cell 2013, 155, 135–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzales, K.A.U.; Liang, H.; Lim, Y.-S.; Chan, Y.-S.; Yeo, J.-C.; Tan, C.-P.; Gao, B.; Le, B.; Tan, Z.-Y.; Low, K.-Y.; et al. Deterministic Restriction on Pluripotent State Dissolution by Cell-Cycle Pathways. Cell 2015, 162, 564–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagana, A.; Dorn, J.F.; De Rop, V.; Ladouceur, A.-M.; Maddox, A.S.; Maddox, P.S. A Small GTPase Molecular Switch Regulates Epigenetic Centromere Maintenance by Stabilizing Newly Incorporated CENP-A. Nat. Cell Biol. 2010, 12, 1186–1193. [Google Scholar] [CrossRef]
- Nashun, B.; Hill, P.W.; Hajkova, P. Reprogramming of Cell Fate: Epigenetic Memory and the Erasure of Memories Past. EMBO J. 2015, 34, 1296–1308. [Google Scholar] [CrossRef]
- Milagre, I.; Stubbs, T.M.; King, M.R.; Spindel, J.; Santos, F.; Krueger, F.; Bachman, M.; Segonds-Pichon, A.; Balasubramanian, S.; Andrews, S.R.; et al. Gender Differences in Global but Not Targeted Demethylation in IPSC Reprogramming. Cell Rep. 2017, 18, 1079–1089. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.-S.; Shin, J.-Y.; Tonge, P.D.; Puri, M.C.; Lee, S.; Park, H.; Lee, W.-C.; Hussein, S.M.I.; Bleazard, T.; Yun, J.-Y.; et al. An Epigenomic Roadmap to Induced Pluripotency Reveals DNA Methylation as a Reprogramming Modulator. Nat. Commun. 2014, 5, 5619. [Google Scholar] [CrossRef] [Green Version]
- Hanna, J.H.; Saha, K.; Jaenisch, R. Pluripotency and Cellular Reprogramming: Facts, Hypotheses, Unresolved Issues. Cell 2010, 143, 508–525. [Google Scholar] [CrossRef] [Green Version]
- Cacchiarelli, D.; Trapnell, C.; Ziller, M.J.; Soumillon, M.; Cesana, M.; Karnik, R.; Donaghey, J.; Smith, Z.D.; Ratanasirintrawoot, S.; Zhang, X.; et al. Integrative Analyses of Human Reprogramming Reveal Dynamic Nature of Induced Pluripotency. Cell 2015, 162, 412–424. [Google Scholar] [CrossRef] [Green Version]
- Koche, R.P.; Smith, Z.D.; Adli, M.; Gu, H.; Ku, M.; Gnirke, A.; Bernstein, B.E.; Meissner, A. Reprogramming Factor Expression Initiates Widespread Targeted Chromatin Remodeling. Cell Stem Cell 2011, 8, 96–105. [Google Scholar] [CrossRef] [Green Version]
- van Nuland, R.; Gozani, O. Histone H4 Lysine 20 (H4K20) Methylation, Expanding the Signaling Potential of the Proteome One Methyl Moiety at a Time. Mol. Cell Proteom. 2016, 15, 755–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andonegui-Elguera, M.A.; Cáceres-Gutiérrez, R.E.; López-Saavedra, A.; Cisneros-Soberanis, F.; Justo-Garrido, M.; Díaz-Chávez, J.; Herrera, L.A. The Roles of Histone Post-Translational Modifications in the Formation and Function of a Mitotic Chromosome. IJMS 2022, 23, 8704. [Google Scholar] [CrossRef]
- Bergmann, J.H.; Rodríguez, M.G.; Martins, N.M.C.; Kimura, H.; Kelly, D.A.; Masumoto, H.; Larionov, V.; Jansen, L.E.T.; Earnshaw, W.C. Epigenetic Engineering Shows H3K4me2 Is Required for HJURP Targeting and CENP-A Assembly on a Synthetic Human Kinetochore: H3K4me2 and Kinetochore Maintenance. EMBO J. 2011, 30, 328–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina, O.; Vargiu, G.; Abad, M.A.; Zhiteneva, A.; Jeyaprakash, A.A.; Masumoto, H.; Kouprina, N.; Larionov, V.; Earnshaw, W.C. Epigenetic Engineering Reveals a Balance between Histone Modifications and Transcription in Kinetochore Maintenance. Nat. Commun. 2016, 7, 13334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Tanasa, B.; Tyurina, O.V.; Zhou, T.Y.; Gassmann, R.; Liu, W.T.; Ohgi, K.A.; Benner, C.; Garcia-Bassets, I.; Aggarwal, A.K.; et al. PHF8 Mediates Histone H4 Lysine 20 Demethylation Events Involved in Cell Cycle Progression. Nature 2010, 466, 508–512. [Google Scholar] [CrossRef] [Green Version]
- Schalch, T.; Steiner, F.A. Structure of Centromere Chromatin: From Nucleosome to Chromosomal Architecture. Chromosoma 2017, 126, 443–455. [Google Scholar] [CrossRef] [Green Version]
- Abe, Y.; Sako, K.; Takagaki, K.; Hirayama, Y.; Uchida, K.S.K.; Herman, J.A.; DeLuca, J.G.; Hirota, T. HP1-Assisted Aurora B Kinase Activity Prevents Chromosome Segregation Errors. Dev. Cell 2016, 36, 487–497. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Higgins, J.M.G. Histone Modifications and Mitosis: Countermarks, Landmarks, and Bookmarks. Trends Cell Biol. 2013, 23, 175–184. [Google Scholar] [CrossRef]
- Soufi, A.; Dalton, S. Cycling through Developmental Decisions: How Cell Cycle Dynamics Control Pluripotency, Differentiation and Reprogramming. Development 2016, 143, 4301–4311. [Google Scholar] [CrossRef] [Green Version]
- Kagey, M.H.; Newman, J.J.; Bilodeau, S.; Zhan, Y.; Orlando, D.A.; van Berkum, N.L.; Ebmeier, C.C.; Goossens, J.; Rahl, P.B.; Levine, S.S.; et al. Mediator and Cohesin Connect Gene Expression and Chromatin Architecture. Nature 2010, 467, 430–435. [Google Scholar] [CrossRef] [Green Version]
- Blum, B.; Bar-Nur, O.; Golan-Lev, T.; Benvenisty, N. The Anti-Apoptotic Gene Survivin Contributes to Teratoma Formation by Human Embryonic Stem Cells. Nat. Biotechnol. 2009, 27, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Payer, B.; Lee, J.T. X Chromosome Dosage Compensation: How Mammals Keep the Balance. Annu. Rev. Genet. 2008, 42, 733–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Tsai, H.-J.; Gordon, M.R.; Li, R. Cellular Stress Associated with Aneuploidy. Dev. Cell 2018, 44, 420–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schukken, K.M.; Sheltzer, J.M. Extensive Protein Dosage Compensation in Aneuploid Human Cancers. Genome Res. 2022, 32, 1254–1270. [Google Scholar] [CrossRef] [PubMed]
- Senger, G.; Santaguida, S.; Schaefer, M.H. Regulation of Protein Complex Partners as a Compensatory Mechanism in Aneuploid Tumors. eLife 2022, 11, e75526. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.; Zhao, X.; Katsnelson, L.; Camacho-Hernandez, E.M.; Mermerian, A.; Mays, J.C.; Lippman, S.M.; Rosales-Alvarez, R.E.; Moya, R.; Shwetar, J.; et al. Proteogenomic Analysis of Cancer Aneuploidy and Normal Tissues Reveals Divergent Modes of Gene Regulation across Cellular Pathways. eLife 2022, 11, e75227. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.R.; Prabhu, V.R.; Hunter, K.E.; Glazier, C.M.; Whittaker, C.A.; Housman, D.E.; Amon, A. Aneuploidy Affects Proliferation and Spontaneous Immortalization in Mammalian Cells. Science 2008, 322, 703–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholson, J.M.; Macedo, J.C.; Mattingly, A.J.; Wangsa, D.; Camps, J.; Lima, V.; Gomes, A.M.; Dória, S.; Ried, T.; Logarinho, E.; et al. Chromosome Mis-Segregation and Cytokinesis Failure in Trisomic Human Cells. eLife 2015, 4, e05068. [Google Scholar] [CrossRef]
- Shahbazi, M.N.; Wang, T.; Tao, X.; Weatherbee, B.A.T.; Sun, L.; Zhan, Y.; Keller, L.; Smith, G.D.; Pellicer, A.; Scott, R.T.; et al. Developmental Potential of Aneuploid Human Embryos Cultured beyond Implantation. Nat. Commun. 2020, 11, 3987. [Google Scholar] [CrossRef]
- Yang, S.; Lin, G.; Tan, Y.-Q.; Zhou, D.; Deng, L.-Y.; Cheng, D.-H.; Luo, S.-W.; Liu, T.-C.; Zhou, X.-Y.; Sun, Z.; et al. Tumor Progression of Culture-Adapted Human Embryonic Stem Cells during Long-Term Culture. Genes Chromosomes Cancer 2008, 47, 665–679. [Google Scholar] [CrossRef]
- Ben-David, U.; Arad, G.; Weissbein, U.; Mandefro, B.; Maimon, A.; Golan-Lev, T.; Narwani, K.; Clark, A.T.; Andrews, P.W.; Benvenisty, N.; et al. Aneuploidy Induces Profound Changes in Gene Expression, Proliferation and Tumorigenicity of Human Pluripotent Stem Cells. Nat. Commun. 2014, 5, 4825. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Cheng, L.; Jia, Y.; Liu, G.; Li, C.; Song, S.; Bradley, A.; Huang, Y. Aneuploid Embryonic Stem Cells Exhibit Impaired Differentiation and Increased Neoplastic Potential. EMBO J. 2016, 35, 2285–2300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haim-Abadi, G.; Golan-Lev, T.; Koren, A.; Benvenisty, N. Generation, Genomic Characterization, and Differentiation of Triploid Human Embryonic Stem Cells. Stem Cell Rep. 2023, 18, 1049–1060. [Google Scholar] [CrossRef]
- Mirkovic, M.; Guilgur, L.G.; Tavares, A.; Passagem-Santos, D.; Oliveira, R.A. Induced Aneuploidy in Neural Stem Cells Triggers a Delayed Stress Response and Impairs Adult Life Span in Flies. PLoS Biol. 2019, 17, e3000016. [Google Scholar] [CrossRef] [Green Version]
- Merkle, F.T.; Ghosh, S.; Kamitaki, N.; Mitchell, J.; Avior, Y.; Mello, C.; Kashin, S.; Mekhoubad, S.; Ilic, D.; Charlton, M.; et al. Human Pluripotent Stem Cells Recurrently Acquire and Expand Dominant Negative P53 Mutations. Nature 2017, 545, 229–233. [Google Scholar] [CrossRef] [Green Version]
- Cohen-Sharir, Y.; McFarland, J.M.; Abdusamad, M.; Marquis, C.; Bernhard, S.V.; Kazachkova, M.; Tang, H.; Ippolito, M.R.; Laue, K.; Zerbib, J.; et al. Aneuploidy Renders Cancer Cells Vulnerable to Mitotic Checkpoint Inhibition. Nature 2021, 590, 486–491. [Google Scholar] [CrossRef]
- Barroso-Vilares, M.; Macedo, J.C.; Reis, M.; Warren, J.D.; Compton, D.; Logarinho, E. Small-molecule Inhibition of Aging-associated Chromosomal Instability Delays Cellular Senescence. EMBO Rep. 2020, 21, e49248. [Google Scholar] [CrossRef] [PubMed]
- Orr, B.; Talje, L.; Liu, Z.; Kwok, B.H.; Compton, D.A. Adaptive Resistance to an Inhibitor of Chromosomal Instability in Human Cancer Cells. Cell Rep. 2016, 17, 1755–1763. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Main Findings | Human ESCs | Human iPSCs |
|---|---|---|
| PSCs have recurrent chromosomal aberrations, including whole chromosome aneuploidies | [10,11,12,13,15,16] | [10,11,12,14,16] |
| PSCs have erroneous mitosis | [23,24,25,26] | [26,27] |
| PSCs undergo high replication stress during S phase | [23,27,28,29] | [23,27,28,29,30] |
| Aneuploid PSCs have problems in chromosome condensation | [23] | [23] |
| PSCs have weaker centromeres | [24] | [24] |
| PSCs have high number of erroneous kinetochore–microtubule attachments | [26,31] | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Milagre, I.; Pereira, C.; Oliveira, R.A. Compromised Mitotic Fidelity in Human Pluripotent Stem Cells. Int. J. Mol. Sci. 2023, 24, 11933. https://doi.org/10.3390/ijms241511933
Milagre I, Pereira C, Oliveira RA. Compromised Mitotic Fidelity in Human Pluripotent Stem Cells. International Journal of Molecular Sciences. 2023; 24(15):11933. https://doi.org/10.3390/ijms241511933
Chicago/Turabian StyleMilagre, Inês, Carolina Pereira, and Raquel A. Oliveira. 2023. "Compromised Mitotic Fidelity in Human Pluripotent Stem Cells" International Journal of Molecular Sciences 24, no. 15: 11933. https://doi.org/10.3390/ijms241511933
APA StyleMilagre, I., Pereira, C., & Oliveira, R. A. (2023). Compromised Mitotic Fidelity in Human Pluripotent Stem Cells. International Journal of Molecular Sciences, 24(15), 11933. https://doi.org/10.3390/ijms241511933