


Investigation of Inflammation in Lewy Body Dementia: A Systematic Scoping Review

 , , , and
, , , and
Abstract
:1. Introduction
1.1. Lewy Body Dementia
1.2. Inflammation and Lewy Body Disease
2. Objectives
- Review the methods used to investigate inflammation in LBD and how these are applied to conceptualize inflammation.
- Identify alterations in inflammation signals in LBD compared to people without neurodegenerative disease and compared to related neurodegenerative diseases, in particular AD and PD.
3. Methods
- Original peer-reviewed research study.
- People with clinically diagnosed dementia with Lewy bodies (DLB), Parkinson’s disease dementia (PDD), or Lewy body dementia (LBD) without specification of DLB or PDD. For postmortem studies, the use of international neuropathological criteria for diagnosis was sufficient.
- Inflammation is assessed either postmortem or in vivo.
- Studies where LBD participants have a comorbid inflammatory condition that might impact the assessment of inflammation, e.g., encephalitis, multiple sclerosis, inflammatory arthritis, or sepsis.
- People with a Lewy body disease (e.g., Parkinson’s disease) without dementia, including people with mild cognitive impairment (MCI).
- In interventional studies, assessments of inflammation are taken while receiving or after the intervention.
- Non-English language.
- 1.
- (Lewy.mp AND dementia) OR dementia with lewy bodies.mp OR ((Parkinson Disease/OR parkinson’s disease.mp) AND dementia.mp) OR Parkinson’s disease dementia.mp
- 2.
- inflammat* OR neuroinflammat* OR Neurogenic inflammation/OR microglia.mp
4. Results
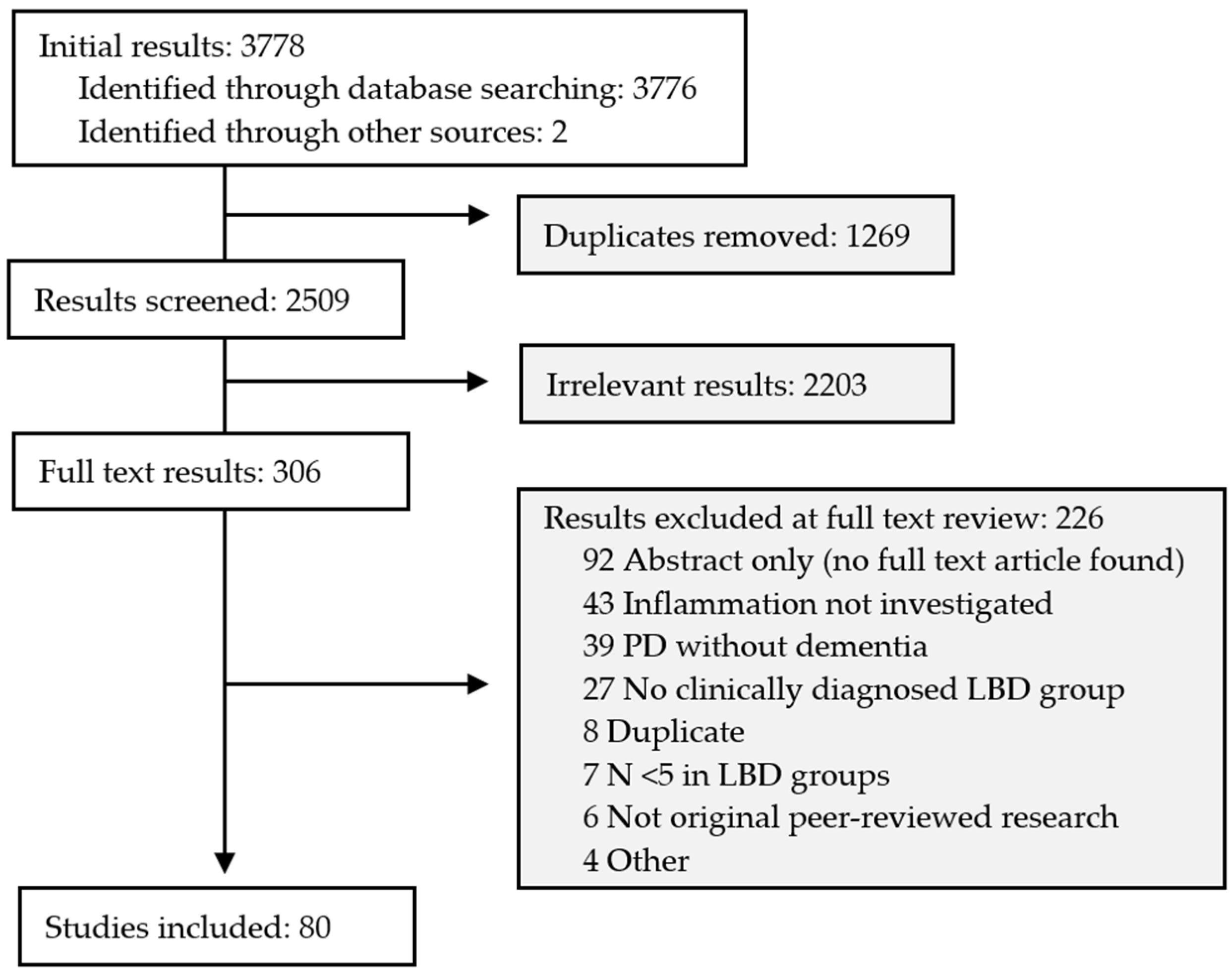
4.1. Search Results
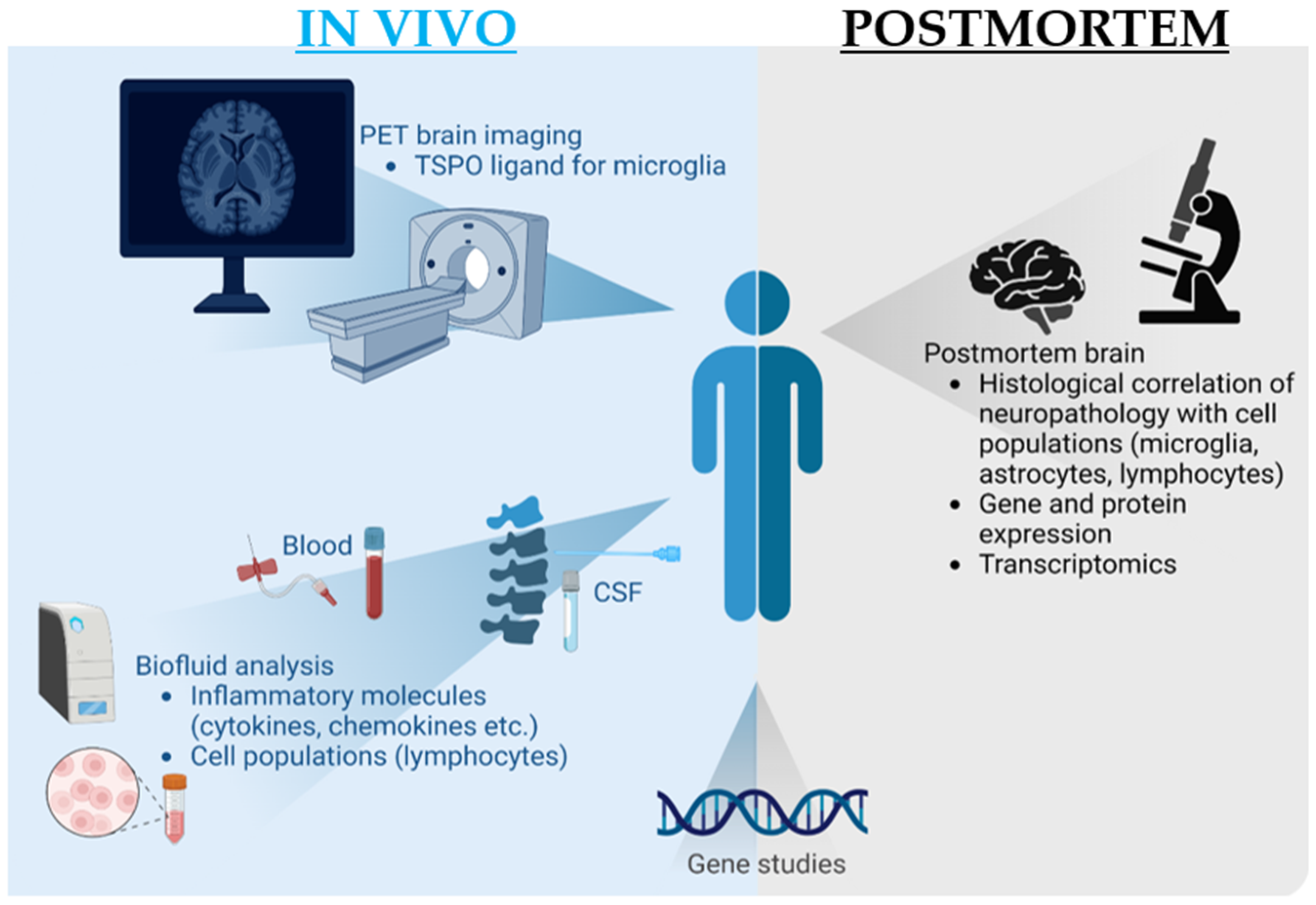
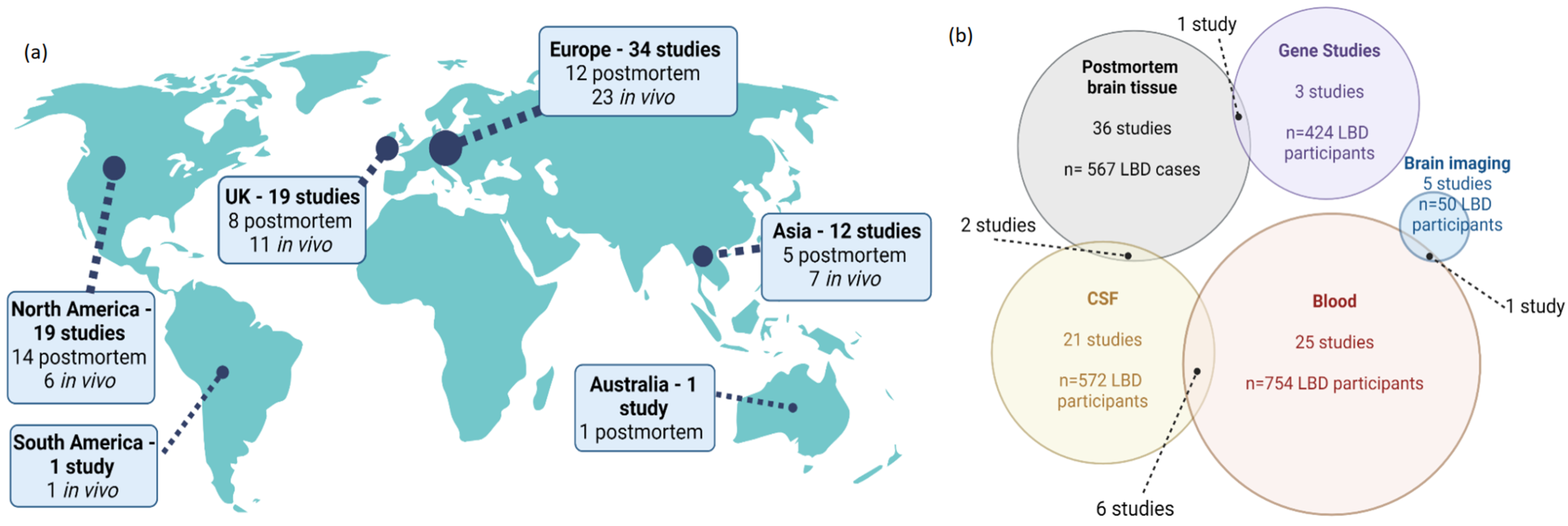
4.2. Characteristics of Included Studies
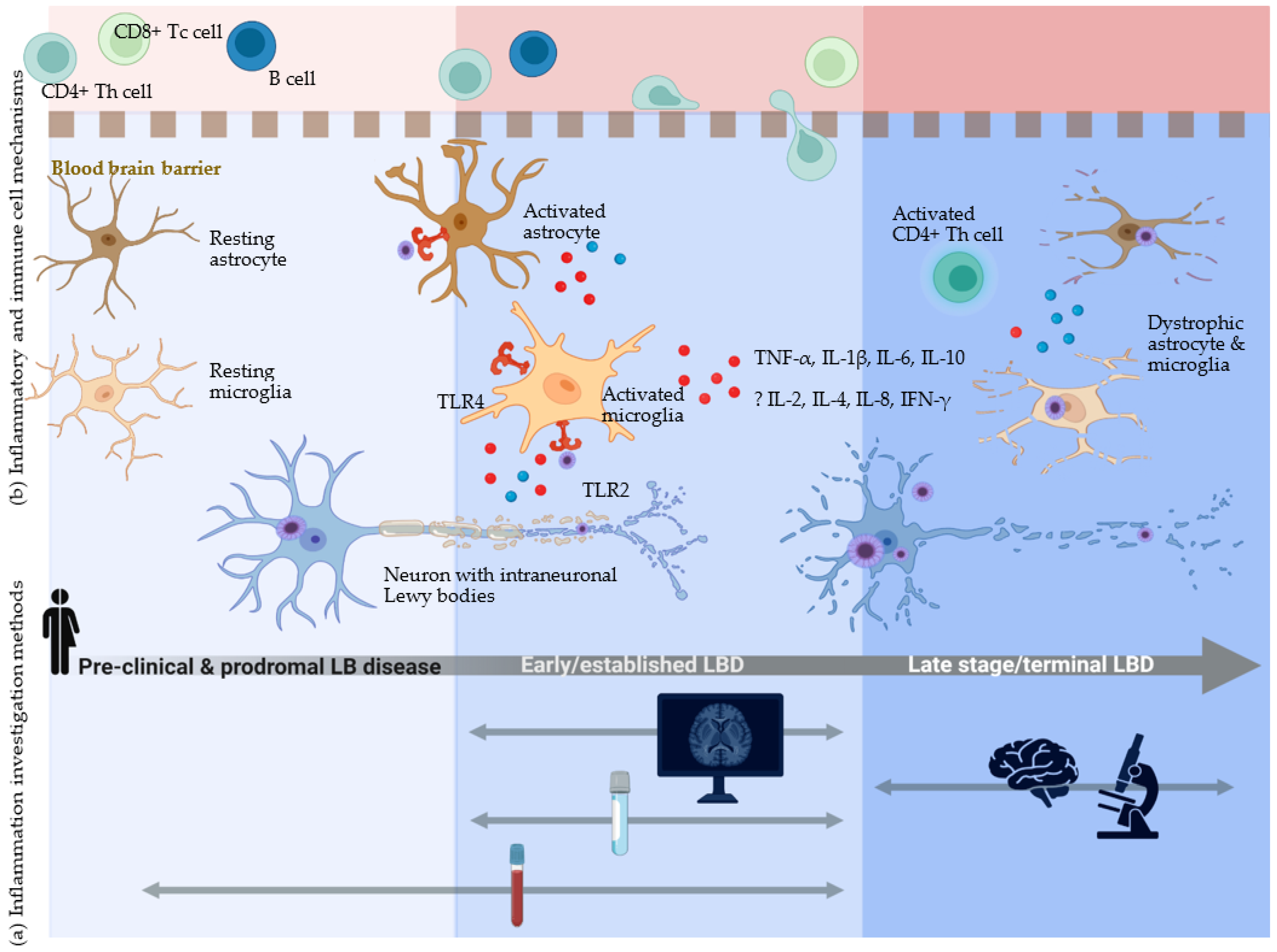
4.3. Investigation of Inflammation in LBD—Insights on Disease Pathobiology from across the Disease Timeline
4.3.1. Microglia—Activation or Dysfunction?
Postmortem Investigation of Microglia
PET Imaging of Microglia In Vivo
4.3.2. Astrocytes—Associated with LBD Pathology at Postmortem but Difficult to Assess in Life
Postmortem Investigation of Astrocytes
Biofluid Assessment of Astrocyte-Associated Inflammation In Vivo
4.3.3. Lymphocytes and the Adaptive Immune Response in LBD-Associated Inflammation
Postmortem Investigation of Lymphocytes
In Vivo Biofluid Assessment of Lymphocytes
4.3.4. Inflammatory Molecules in LBD: Cytokines, Vascular Mediators, and Others
Cytokines and Chemokines—Postmortem and In Vivo Studies
Vascular Inflammatory Mechanisms in LBD
Clinical Acute Phase Inflammatory Markers—CRP and Procalcitonin
Other Inflammatory Molecules Measured Postmortem and In Vivo
4.3.5. Other Approaches to Inflammation Assessment in LBD
Serum Small Extracellular Vesicles
Transcriptomics
Genetic Factors
5. Discussion
5.1. Contribution of Inflammatory and Related Immune System Components to LBD Pathogenesis
5.2. AD Co-Pathology and Inflammation in LBD
5.3. Differences between the Lewy Body Dementias—DLB and PDD
5.4. Limitations of Identified Studies
5.4.1. Limitations of Postmortem Studies
5.4.2. Limitations of In Vivo Studies
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Galvin, J.E.; Tarawneh, R. Distinguishing Lewy body dementias from Alzheimer’s disease. Expert Rev. Neurother. 2007, 7, 1499–1516. [Google Scholar]
- McKeith, I.G.; Boeve, B.F.; Dickson, D.W.; Halliday, G.; Taylor, J.-P.; Weintraub, D.; Kosaka, K. Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology 2017, 89, 88–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emre, M.; Aarsland, D.; Brown, R.; Burn, D.J.; Duyckaerts, C.; Mizuno, Y.; Dubois, B. Clinical diagnostic criteria for dementia associated with Parkinson’s disease. Mov. Disord. 2007, 22, 1689–1707. [Google Scholar] [CrossRef] [PubMed]
- Williams-Gray, C.H.; Mason, S.L.; Evans, J.R.; Foltynie, T.; Brayne, C.; Robbins, T.W.; Barker, R.A. The CamPaIGN study of Parkinson’s disease: 10-year outlook in an incident population-based cohort. J. Neurol. Neurosurg. Psychiatry 2013, 84, 1258–1264. [Google Scholar] [CrossRef] [Green Version]
- Hely, M.A.; Reid, W.G.; Adena, M.A.; Halliday, G.M.; Morris, J.G. The Sydney multicenter study of Parkinson’s disease: The inevitability of dementia at 20 years. Mov. Disord. 2008, 23, 837–844. [Google Scholar] [CrossRef]
- Aarsland, D.; Perry, R.; Brown, A.; Larsen, J.P.; Ballard, C. Neuropathology of dementia in Parkinson’s disease: A prospective, community-based study. Ann. Neurol. 2005, 58, 773–776. [Google Scholar] [CrossRef]
- McKeith, I.G.; Ferman, T.J.; Thomas, A.J.; Blanc, F.; Boeve, B.F.; Fujishiro, H.; Kantarci, K.; Muscio, C.; O’Brien, J.T.; Postuma, R.B.; et al. Research criteria for the diagnosis of prodromal dementia with Lewy bodies. Neurology 2020, 94, 743–755. [Google Scholar] [CrossRef]
- Amor, S.; Puentes, F.; Baker, D.; van der Valk, P. Inflammation in neurodegenerative diseases. Immunology 2010, 129, 154–169. [Google Scholar] [CrossRef]
- Baechle, J.J.; Chen, N.; Makhijani, P.; Winer, S.; Furman, D.; Winer, D.A. Chronic inflammation and the hallmarks of aging. Mol. Metab. 2023, 74, 101755. [Google Scholar] [CrossRef]
- Weiss, F.; Labrador-Garrido, A.; Dzamko, N.; Halliday, G. Immune responses in the Parkinson’s disease brain. Neurobiol. Dis. 2022, 168, 105700. [Google Scholar] [CrossRef]
- Surendranathan, A.; O’Brien, J.; Rowe, J. Neuroinflammation in lewy body dementia. Am. J. Neurodegener. Dis. 2015, 4 (Suppl. 1), 150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benarroch, E.E. Microglia: Multiple roles in surveillance, circuit shaping, and response to injury. Neurology 2013, 81, 1079–1088. [Google Scholar] [CrossRef] [PubMed]
- Kreisl, W.C.; Kim, M.J.; Coughlin, J.M.; Henter, I.D.; Owen, D.R.; Innis, R.B. PET imaging of neuroinflammation in neurological disorders. Lancet Neurol. 2020, 19, 940–950. [Google Scholar] [CrossRef]
- Dupont, A.-C.; Largeau, B.; Santiago Ribeiro, M.J.; Guilloteau, D.; Tronel, C.; Arlicot, N. Translocator Protein-18 kDa (TSPO) Positron Emission Tomography (PET) Imaging and Its Clinical Impact in Neurodegenerative Diseases. Int. J. Mol. Sci. 2017, 18, 785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acioglu, C.; Li, L.; Elkabes, S. Contribution of astrocytes to neuropathology of neurodegenerative diseases. Brain Res. 2021, 1758, 147291. [Google Scholar] [CrossRef]
- Myers, A.J.; Brahimi, A.; Jenkins, I.J.; Koob, A.O. The Synucleins and the Astrocyte. Biology 2023, 12, 155. [Google Scholar] [CrossRef]
- Abdelhak, A.; Foschi, M.; Abu-Rumeileh, S.; Yue, J.K.; D’Anna, L.; Huss, A.; Tumani, H. Blood GFAP as an emerging biomarker in brain and spinal cord disorders. Nat. Rev. Neurol. 2022, 18, 158–172. [Google Scholar] [CrossRef]
- Luckheeram, R.V.; Zhou, R.; Verma, A.D.; Xia, B. CD4⁺T cells: Differentiation and functions. Clin. Dev. Immunol. 2012, 2012, 925135. [Google Scholar] [CrossRef] [Green Version]
- McGeer, P.L.; Itagaki, S.; Akiyama, H.; McGeer, E.G. Rate of cell death in parkinsonism indicates active neuropathological process. Ann. Neurol. 1988, 24, 574–576. [Google Scholar] [CrossRef]
- Sulzer, D.; Alcalay, R.N.; Garretti, F.; Cote, L.; Kanter, E.; Agin-Liebes, J.; Sette, A. T cells from patients with Parkinson’s disease recognize α-synuclein peptides. Nature 2017, 546, 656–661. [Google Scholar] [CrossRef] [Green Version]
- Baird, J.K.; Bourdette, D.; Meshul, C.K.; Quinn, J.F. The key role of T cells in Parkinson’s disease pathogenesis and therapy. Park. Relat. Disord. 2019, 60, 25–31. [Google Scholar] [CrossRef]
- Porrini, V.; Pilotto, A.; Vezzoli, M.; Lanzillotta, A.; Gennari, M.M.; Bonacina, S.; Pizzi, M. NF-κB/c-Rel DNA-binding is reduced in substantia nigra and peripheral blood mononuclear cells of Parkinson’s disease patients. Neurobiol. Dis. 2023, 180, 106067. [Google Scholar] [CrossRef]
- Khalil, M.; Teunissen, C.E.; Otto, M.; Piehl, F.; Sormani, M.P.; Gattringer, T.; Kuhle, J. Neurofilaments as biomarkers in neurological disorders. Nat. Rev. Neurol. 2018, 14, 577–589. [Google Scholar] [CrossRef]
- Qin, X.-Y.; Zhang, S.-P.; Cao, C.; Loh, Y.P.; Cheng, Y. Aberrations in Peripheral Inflammatory Cytokine Levels in Parkinson Disease: A Systematic Review and Meta-analysis. JAMA Neurol. 2016, 73, 1316–1324. [Google Scholar] [CrossRef]
- Williams-Gray, C.H.; Wijeyekoon, R.; Yarnall, A.J.; Lawson, R.A.; Breen, D.P.; Evans, J.R.; Cummins, G.A.; Duncan, G.W.; Khoo, T.K.; Burn, D.J.; et al. Serum immune markers and disease progression in an incident Parkinson’s disease cohort (ICICLE-PD). Mov. Disord. 2016, 31, 995–1003. [Google Scholar] [CrossRef] [Green Version]
- Rayaprolu, S.; Mullen, B.; Baker, M.; Lynch, T.; Finger, E.; Seeley, W.W.; Hatanpaa, K.J.; Lomen-Hoerth, C.; Kertesz, A.; Bigio, E.H.; et al. TREM2 in neurodegeneration: Evidence for association of the p.R47H variant with frontotemporal dementia and Parkinson’s disease. Mol. Neurodegener. 2013, 8, 19. [Google Scholar] [CrossRef] [Green Version]
- Ohara, T.; Hata, J.; Tanaka, M.; Honda, T.; Yamakage, H.; Yoshida, D.; Inoue, T.; Hirakawa, Y.; Kusakabe, T.; Shibata, M.; et al. Serum Soluble Triggering Receptor Expressed on Myeloid Cells 2 as a Biomarker for Incident Dementia: The Hisayama Study. Ann. Neurol. 2019, 85, 47–58. [Google Scholar] [CrossRef]
- Chowdhury, A.; Rajkumar, A.P. Systematic review of gene expression studies in people with Lewy body dementia. Acta Neuropsychiatrica. 2020, 32, 281–292. [Google Scholar] [CrossRef]
- Munn, Z.; Peters, M.D.J.; Stern, C.; Tufanaru, C.; McArthur, A.; Aromataris, E. Systematic review or scoping review? Guidance for authors when choosing between a systematic or scoping review approach. BMC Med. Res. Methodol. 2018, 18, 143. [Google Scholar] [CrossRef]
- Tricco, A.C.; Lillie, E.; Zarin, W.; O’Brien, K.K.; Colquhoun, H.; Levac, D.; Moher, D.; Peters, M.D.; Horsley, T.; Weeks, L.; et al. PRISMA Extension for Scoping Reviews (PRISMA-ScR): Checklist and Explanation. Ann. Intern. Med. 2018, 169, 467–473. [Google Scholar] [CrossRef] [Green Version]
- Thomas, A.J.; Hamilton, C.A.; Donaghy, P.C.; Martin-Ruiz, C.; Morris, C.M.; Barnett, N.; O’Brien, J.T. Prospective longitudinal evaluation of cytokines in mild cognitive impairment due to AD and Lewy body disease. Int. J. Geriatr. Psychiatry 2020, 35, 1250–1259. [Google Scholar] [CrossRef] [PubMed]
- Laguna, A.; Xicoy, H.; Tolosa, E.; Serradell, M.; Vilas, D.; Gaig, C.; Fernández, M.; Yanes, O.; Santamaria, J.; Amigó, N.; et al. Serum metabolic biomarkers for synucleinopathy conversion in isolated REM sleep behavior disorder. NPJ Park. Dis. 2021, 7, 40. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Huang, Y. Effects of butylphthalide soft capsules on cognitive function, ability of daily living, and related factors in patients with parkinson’s disease dementia. Int. J. Clin. Exp. 2020, 13, 7758–7765. [Google Scholar]
- McGeer, P.L.; Itagaki, S.; Boyes, B.E.; McGeer, E.G. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 1988, 38, 1285–1291. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, I.R. Activated microglia in dementia with Lewy bodies. Neurology 2000, 55, 132–134. [Google Scholar] [CrossRef]
- Imamura, K.; Hishikawa, N.; Ono, K.; Suzuki, H.; Sawada, M.; Nagatsu, T.; Yoshida, M.; Hashizume, Y. Cytokine production of activated microglia and decrease in neurotrophic factors of neurons in the hippocampus of Lewy body disease brains. Acta Neuropathol. 2005, 109, 141–150. [Google Scholar] [CrossRef]
- Iseki, E.; Marui, W.; Akiyama, H.; Ueda, K.; Kosaka, K. Degeneration process of Lewy bodies in the brains of patients with dementia with Lewy bodies using alpha-synuclein-immunohistochemistry. Neurosci. Lett. 2000, 286, 69–73. [Google Scholar] [CrossRef]
- Togo, T.; Iseki, E.; Marui, W.; Akiyama, H.; Ueda, K.; Kosaka, K. Glial involvement in the degeneration process of Lewy body-bearing neurons and the degradation process of Lewy bodies in brains of dementia with Lewy bodies. J. Neurol. Sci. 2001, 184, 71–75. [Google Scholar] [CrossRef]
- Shepherd, C.E.; Thiel, E.; McCann, H.; Harding, A.J.; Halliday, G.M. Cortical inflammation in Alzheimer disease but not dementia with Lewy bodies. Arch. Neurol. 2000, 57, 817–822. [Google Scholar] [CrossRef] [Green Version]
- Rozemuller, A.J.; Eikelenboom, P.; Theeuwes, J.W.; Jansen Steur, E.N.; de Vos, R.A. Activated microglial cells and complement factors are unrelated to cortical Lewy bodies. Acta Neuropathol. 2000, 100, 701–708. [Google Scholar] [CrossRef]
- Loeffler, D.A.; Camp, D.M.; Conant, S.B. Complement activation in the Parkinson’s disease substantia nigra: An immunocytochemical study. J. Neuroinflamm. 2006, 3, 29. [Google Scholar] [CrossRef] [Green Version]
- Katsuse, O.; Iseki, E.; Kosaka, K. Immunohistochemical study of the expression of cytokines and nitric oxide synthases in brains of patients with dementia with Lewy bodies. Neuropathology 2003, 23, 9–15. [Google Scholar] [CrossRef]
- Bachstetter, A.D.; Van Eldik, L.J.; Schmitt, F.A.; Neltner, J.H.; Ighodaro, E.T.; Webster, S.J.; Patel, E.; Abner, E.L.; Kryscio, R.J.; Nelson, P.T. Disease-related microglia heterogeneity in the hippocampus of Alzheimer’s disease, dementia with Lewy bodies, and hippocampal sclerosis of aging. Acta Neuropathol. Commun. 2015, 3, 32. [Google Scholar] [CrossRef] [Green Version]
- Streit, W.J.; Xue, Q.-S. Microglia in dementia with Lewy bodies. Brain Behav. Immun. 2016, 55, 191–201. [Google Scholar] [CrossRef]
- Kohl, Z.; Feldewerth, J.; Hornauer, P.; Munch, M.; Winkler, J.; Schlachetzki, J.C.M.; Masliah, E. Distinct pattern of microgliosis in the olfactory bulb of neurodegenerative proteinopathies. Neural Plast. 2017, 2017, 3851262. [Google Scholar] [CrossRef] [Green Version]
- Amin, J.; Holmes, C.; Dorey, R.B.; Tommasino, E.; Casal, Y.R.; Williams, D.M.; Dupuy, C.; Nicoll, J.A.R.; Boche, D. Neuroinflammation in dementia with Lewy bodies: A human post-mortem study. Transl. Psychiatry 2020, 10, 267. [Google Scholar] [CrossRef]
- Fixemer, S.; Ameli, C.; Hammer, G.; Salamanca, L.; Huarte, O.U.; Schwartz, C.; Gérardy, J.J.; Mechawar, N.; Skupin, A.; Mittelbronn, M.; et al. Microglia phenotypes are associated with subregional patterns of concomitant tau, amyloid-beta and alpha-synuclein pathologies in the hippocampus of patients with Alzheimer’s disease and dementia with Lewy bodies. Acta Neuropathol. Commun. 2022, 10, 36. [Google Scholar] [CrossRef]
- Terreros-Roncal, J.; Moreno-Jimenez, E.P.; Flor-Garcia, M.; Rodriguez-Moreno, C.B.; Trinchero, M.F.; Cafini, F.; Rábano, A.; Llorens-Martín, M. Impact of neurodegenerative diseases on human adult hippocampal neurogenesis. Science 2021, 374, 1106–1113. [Google Scholar] [CrossRef]
- Iannaccone, S.; Cerami, C.; Alessio, M.; Garibotto, V.; Panzacchi, A.; Olivieri, S.; Gelsomino, G.; Moresco, R.M.; Perani, D. In vivo microglia activation in very early dementia with Lewy bodies, comparison with Parkinson’s disease. Park. Relat. Disord. 2013, 19, 47–52. [Google Scholar] [CrossRef] [Green Version]
- Fan, Z.; Aman, Y.; Ahmed, I.; Chetelat, G.; Landeau, B.; Chaudhuri, K.R.; Brooks, D.J.; Edison, P. Influence of microglial activation on neuronal function in Alzheimer’s and Parkinson’s disease dementia. Alzheimers Dement. 2015, 11, 608. [Google Scholar] [CrossRef]
- Edison, P.; Ahmed, I.; Fan, Z.; Hinz, R.; Gelosa, G.; Chaudhuri, K.R.; Walker, Z.; Turkheimer, F.E.; Brooks, D.J. Microglia, amyloid, and glucose metabolism in Parkinson’s disease with and without dementia. Neuropsychopharmacology 2013, 38, 938–949. [Google Scholar] [CrossRef] [Green Version]
- Femminella, G.D.; Ninan, S.; Atkinson, R.; Fan, Z.; Brooks, D.J.; Edison, P. Does Microglial Activation Influence Hippocampal Volume and Neuronal Function in Alzheimer’s Disease and Parkinson’s Disease Dementia? J. Alzheimers Dis. 2016, 51, 1275–1289. [Google Scholar] [CrossRef]
- Surendranathan, A.; Su, L.; Mak, E.; Passamonti, L.; Hong, Y.T.; Arnold, R.; Rodríguez, P.V.; Bevan-Jones, W.R.; Brain, S.A.E.; Fryer, T.D.; et al. Early microglial activation and peripheral inflammation in dementia with Lewy bodies. Brain 2018, 141, 3415–3427. [Google Scholar] [CrossRef] [Green Version]
- Nicastro, N.; Surendranathan, A.; Mak, E.; Rowe, J.B.; O’Brien, J.T. (11) C-PK11195 PET imaging and white matter changes in Parkinson’s disease dementia. Ann. Clin. Transl. Neurol. 2019, 6, 2133–2136. [Google Scholar] [CrossRef] [Green Version]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Esparcia, P.; Lopez-Gonzalez, I.; Grau-Rivera, O.; Garcia-Garrido, M.F.; Konetti, A.; Llorens, F.; Zafar, S.; Carmona, M.; Del Rio, J.A.; Zerr, I.; et al. Dementia with Lewy Bodies: Molecular Pathology in the Frontal Cortex in Typical and Rapidly Progressive Forms. Front. Neurol. 2017, 8, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.; Beilina, A.; Smith, N.; Li, Y.; Kim, M.; Kumaran, R.; Kaganovich, A.; Mamais, A.; Adame, A.; Iba, M.; et al. LRRK2 mediates microglial neurotoxicity via NFATc2 in rodent models of synucleinopathies. Sci. Transl. Med. 2020, 12, eaay0399. [Google Scholar] [CrossRef]
- Low, C.Y.B.; Lee, J.H.; Lim, F.T.W.; Lee, C.; Ballard, C.; Francis, P.T.; Lai, M.K.P.; Tan, M.G.K. Isoform-specific upregulation of FynT kinase expression is associated with tauopathy and glial activation in Alzheimer’s disease and Lewy body dementias. Brain Pathol. 2021, 31, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Llorens, F.; Thune, K.; Tahir, W.; Kanata, E.; Diaz-Lucena, D.; Xanthopoulos, K.; Kovatsi, E.; Pleschka, C.; Garcia-Esparcia, P.; Schmitz, M.; et al. YKL-40 in the brain and cerebrospinal fluid of neurodegenerative dementias. Mol. Neurodegener. 2017, 12, 83. [Google Scholar] [CrossRef] [Green Version]
- Erskine, D.; Ding, J.; Thomas, A.J.; Kaganovich, A.; Khundakar, A.A.; Hanson, P.S.; Taylor, J.-P.; McKeith, I.G.; Attems, J.; Cookson, M.R.; et al. Molecular changes in the absence of severe pathology in the pulvinar in dementia with Lewy bodies. Mov. Disord. 2018, 33, 982–991. [Google Scholar] [CrossRef]
- Wennstrom, M.; Surova, Y.; Hall, S.; Nilsson, C.; Minthon, L.; Hansson, O.; Nielsen, H.M. The Inflammatory Marker YKL-40 Is Elevated in Cerebrospinal Fluid from Patients with Alzheimer’s but Not Parkinson’s Disease or Dementia with Lewy Bodies. PLoS ONE 2015, 10, e0135458. [Google Scholar] [CrossRef]
- Janelidze, S.; Hertze, J.; Zetterberg, H.; Waldö, M.L.; Santillo, A.; Blennow, K.; Hansson, O. Cerebrospinal fluid neurogranin and YKL-40 as biomarkers of Alzheimer’s disease. Ann. Clin. Transl. Neurol. 2016, 3, 12–20. [Google Scholar] [CrossRef] [Green Version]
- Paterson, R.W.; Slattery, C.F.; Poole, T.; Nicholas, J.M.; Magdalinou, N.K.; Toombs, J.; Chapman, M.D.; Lunn, M.P.; Heslegrave, A.J.; Foiani, M.S.; et al. Cerebrospinal fluid in the differential diagnosis of Alzheimer’s disease: Clinical utility of an extended panel of biomarkers in a specialist cognitive clinic. Alzheimers Res. Ther. 2018, 10, 32. [Google Scholar] [CrossRef]
- Morenas-Rodríguez, E.; Alcolea, D.; Suárez-Calvet, M.; Muñoz-Llahuna, L.; Vilaplana, E.; Sala, I.; Subirana, A.; Querol-Vilaseca, M.; Carmona-Iragui, M.; Illán-Gala, I.; et al. Different pattern of CSF glial markers between dementia with Lewy bodies and Alzheimer’s disease. Sci. Rep. 2019, 9, 7803. [Google Scholar] [CrossRef] [Green Version]
- Villar-Pique, A.; Schmitz, M.; Hermann, P.; Goebel, S.; Bunck, T.; Varges, D.; Ferrer, I.; Riggert, J.; Llorens, F.; Zerr, I. Plasma YKL-40 in the spectrum of neurodegenerative dementia. J. Neuroinflamm. 2019, 16, 145. [Google Scholar] [CrossRef]
- Del Campo, M.; Galimberti, D.; Elias, N.; Boonkamp, L.; Pijnenburg, Y.A.; van Swieten, J.C.; Watts, K.; Paciotti, S.; Beccari, T.; Hu, W.; et al. Novel CSF biomarkers to discriminate FTLD and its pathological subtypes. Ann. Clin. Transl. Neurol. 2018, 5, 1163–1175. [Google Scholar] [CrossRef]
- Schulz, I.; Kruse, N.; Gera, R.G.; Kremer, T.; Cedarbaum, J.; Barbour, R.; Zago, W.; Schade, S.; Otte, B.; Bartl, M.; et al. Systematic Assessment of 10 Biomarker Candidates Focusing on α-Synuclein-Related Disorders. Mov. Disord. 2021, 36, 2874–2887. [Google Scholar] [CrossRef]
- Castellani, R.J.; Nugent, S.L.; Morrison, A.L.; Zhu, X.; Lee, H.-G.; Harris, P.L.R.; Bajić, V.; Sharma, H.S.; Chen, S.G.; Oettgen, P.; et al. CD3 in Lewy pathology: Does the abnormal recall of neurodevelopmental processes underlie Parkinson’s disease. J. Neural Transm. 2011, 118, 23–26. [Google Scholar] [CrossRef] [Green Version]
- Iba, M.; Kim, C.; Sallin, M.; Kwon, S.; Verma, A.; Overk, C.; Rissman, R.A.; Sen, R.; Sen, J.M.; Masliah, E. Neuroinflammation is associated with infiltration of T cells in Lewy body disease and alpha-synuclein transgenic models. J. Neuroinflamm. 2020, 17, 214. [Google Scholar] [CrossRef]
- Kouli, A.; Camacho, M.; Allinson, K.; Williams-Gray, C.H. Neuroinflammation and protein pathology in Parkinson’s disease dementia. Acta Neuropathol. Commun. 2020, 8, 211. [Google Scholar] [CrossRef]
- Gate, D.; Tapp, E.; Leventhal, O.; Shahid, M.; Nonninger, T.J.; Yang, A.C.; Strempfl, K.; Unger, M.S.; Fehlmann, T.; Oh, H.; et al. CD4(+) T cells contribute to neurodegeneration in Lewy body dementia. Science 2021, 374, 868–874. [Google Scholar] [CrossRef]
- Amin, J.; Boche, D.; Clough, Z.; Teeling, J.; Williams, A.; Gao, Y.; Chudley, L.; Lau, L.; Smith, F.; Harris, S.; et al. Peripheral immunophenotype in dementia with Lewy bodies and Alzheimer’s disease: An observational clinical study. J. Neurol. Neurosurg. Psychiatry 2020, 91, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Lanuti, P.; Ciccocioppo, F.; Bonanni, L.; Marchisio, M.; Lachmann, R.; Tabet, N.; Pierdomenico, L.; Santavenere, E.; Catinella, V.; Iacone, A.; et al. Amyloid-specific T-cells differentiate Alzheimer’s disease from Lewy body dementia. Neurobiol. Aging 2012, 33, 2599–2611. [Google Scholar] [CrossRef] [PubMed]
- Wennstrom, M.; Hall, S.; Hansson, O. Levels of CSF IL-6 are decreased and correlate with Mmse and CSF levels of alpha-synuclein in DLB patients. Neurodegener. Dis. 2015, 15 (Suppl. 1), 1227. [Google Scholar]
- Hu, W.T.; Howell, J.C.; Ozturk, T.; Gangishetti, U.; Kollhoff, A.L.; Hatcher-Martin, J.M.; Anderson, A.M.; Tyor, W.R. CSF Cytokines in Aging, Multiple Sclerosis, and Dementia. Front. Immunol. 2019, 10, 480. [Google Scholar] [CrossRef] [Green Version]
- Hall, S.; Janelidze, S.; Surova, Y.; Widner, H.; Zetterberg, H.; Hansson, O. Cerebrospinal fluid concentrations of inflammatory markers in Parkinson’s disease and atypical parkinsonian disorders. Sci. Rep. 2018, 8, 13276. [Google Scholar] [CrossRef] [Green Version]
- Chua, X.Y.; Chong, J.R.; Cheng, A.L.; Lee, J.H.; Ballard, C.; Aarsland, D.; Francis, P.T.; Lai, M.K.P. Elevation of inactive cleaved annexin A1 in the neocortex is associated with amyloid, inflammatory and apoptotic markers in neurodegenerative dementias. Neurochem. Int. 2022, 152, 105251. [Google Scholar] [CrossRef]
- Bjorkqvist, M.; Ohlsson, M.; Minthon, L.; Hansson, O. Evaluation of a previously suggested plasma biomarker panel to identify Alzheimer’s disease. PLoS ONE 2012, 7, e29868. [Google Scholar] [CrossRef]
- Lue, L.F.; Schmitz, C.T.; Snyder, N.L.; Chen, K.W.; Walker, D.G.; Davis, K.J.; Belden, C.; Caviness, J.N.; Driver-Dunckley, E.; Adler, C.H.; et al. Converging mediators from immune and trophic pathways to identify Parkinson disease dementia. Neurol. Neuroimmunol. NeuroInflammation. 2016, 3, e193. [Google Scholar] [CrossRef] [Green Version]
- Clough, Z.; Jeyapaul, P.; Zotova, E.; Holmes, C. Proinflammatory cytokines and the clinical features of dementia with lewy bodies. Alzheimer Dis. Assoc. Disord. 2015, 29, 97–99. [Google Scholar] [CrossRef]
- Usenko, T.S.; Nikolaev, M.A.; Miliukhina, I.V.; Bezrukova, A.I.; Senkevich, K.A.; Gomzyakova, N.A.; Beltceva, Y.A.; Zalutskaya, N.M.; Gracheva, E.V.; Timofeeva, A.A.; et al. Plasma cytokine profile in synucleinophaties with dementia. J. Clin. Neurosci. 2020, 78, 323–326. [Google Scholar] [CrossRef]
- Costantini, E.; Carrarini, C.; Borrelli, P.; De Rosa, M.; Calisi, D.; Consoli, S.; D’Ardes, D.; Cipollone, F.; Di Nicola, M.; Onofrj, M.; et al. Different peripheral expression patterns of the nicotinic acetylcholine receptor in dementia with Lewy bodies and Alzheimer’s disease. Immun. Ageing. 2023, 20, 3. [Google Scholar] [CrossRef]
- King, E.; O’Brien, J.T.; Donaghy, P.; Morris, C.; Barnett, N.; Olsen, K.; Martin-Ruiz, C.; Taylor, J.-P.; Thomas, A.J. 1. Peripheral inflammation in prodromal Alzheimer’s and Lewy body dementias. J. Neurol. Neurosurg. Psychiatry 2018, 89, 339–345. [Google Scholar] [CrossRef]
- Nielsen, H.M.; Janciauskiene, S.M.; Londos, E.; Minthon, L. Soluble adhesion molecules and angiotensin-converting enzyme in dementia. Neurobiol. Dis. 2007, 26, 27–35. [Google Scholar] [CrossRef]
- Nielsen, H.M.; Minthon, L.; Londos, E.; Blennow, K.; Miranda, E.; Perez, J.; Crowther, D.C.; Lomas, D.A.; Janciauskiene, S.M. Plasma and CSF serpins in Alzheimer disease and dementia with Lewy bodies. Neurology 2007, 69, 1569–1579. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, H.M.; Palmqvist, S.; Minthon, L.; Londos, E.; Wennstrom, M. Gender-dependent levels of hyaluronic acid in cerebrospinal fluid of patients with neurodegenerative dementia. Curr. Alzheimer Res. 2012, 9, 257–266. [Google Scholar] [CrossRef] [Green Version]
- Wennstrom, M.; Nielsen, H.M.; Orhan, F.; Londos, E.; Minthon, L.; Erhardt, S. Kynurenic Acid levels in cerebrospinal fluid from patients with Alzheimer’s disease or dementia with lewy bodies. Int. J. Tryptophan Res. 2014, 7, 1–7. [Google Scholar] [CrossRef]
- Walker, D.G.; Lue, L.F.; Tang, T.M.; Adler, C.H.; Caviness, J.N.; Sabbagh, M.N.; Serrano, G.E.; Sue, L.I.; Beach, T.G. Changes in CD200 and intercellular adhesion molecule-1 (ICAM-1) levels in brains of Lewy body disorder cases are associated with amounts of Alzheimer’s pathology not alpha-synuclein pathology. Neurobiol. Aging 2017, 54, 175–186. [Google Scholar] [CrossRef]
- Rajkumar, A.P.; Aarsland, D.; Bidkhori, G.; Shoaie, S.; Clarke, E.; Williams, G.; Ballard, C.; Francis, P.; Aarsland, D. Postmortem Cortical Transcriptomics of Lewy Body Dementia Reveal Mitochondrial Dysfunction and Lack of Neuroinflammation. Am. J. Geriatr. Psychiatry 2020, 28, 75–86. [Google Scholar] [CrossRef]
- Song, I.-U.; Kim, Y.-D.; Cho, H.-J.; Chung, S.-W. Is neuroinflammation involved in the development of dementia in patients with Parkinson’s disease? Intern. Med. 2013, 52, 1787–1792. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.-M.; Kim, B.C.; Kang, K.W.; Choi, K.-H.; Nam, T.-S.; Kim, J.-T.; Lee, S.-H.; Park, M.-S.; Kim, M.-K.; Cho, K.-H. Relationship between serum high-sensitivity C-reactive protein levels and cognitive function in patients with Parkinson’s disease. Neurol. Asia 2016, 21, 349–356. [Google Scholar]
- Liu, H.; Deng, B.; Zhou, H.; Wu, Z.; Chen, Y.; Weng, G.; Zhu, S.; Xu, J.; Wang, H.; Zhou, Z.; et al. QEEG indices are associated with inflammatory and metabolic risk factors in Parkinson’s disease dementia: An observational study. eClinicalMedicine 2022, 52, 101615. [Google Scholar] [CrossRef] [PubMed]
- Ernst, A.; Morgenthaler, N.G.; Buerger, K.; Dodel, R.; Noelker, C.; Sommer, N.; Schwarz, M.; Koehrle, J.; Bergmann, A.; Hampel, H. Procalcitonin is elevated in the cerebrospinal fluid of patients with dementia and acute neuroinflammation. J. Neuroimmunol. 2007, 189, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Ren, Q.; Ma, M.; Yang, J.; Nonaka, R.; Yamaguchi, A.; Ishikawa, K.-I.; Kobayashi, L.; Murayama, S.; Hwang, S.H.; Saiki, S.; et al. Soluble epoxide hydrolase plays a key role in the pathogenesis of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2018, 115, E5815–E5823. [Google Scholar] [CrossRef] [Green Version]
- Saldana, M.; Pujols, L.; Mullol, J.; Roca-Ferrer, J.; Cardozo, A.; Aguilar, E.; Bonastre, M.; Marin, C. Relevance of COX-2 gene expression in dementia with Lewy bodies associated with Alzheimer pathology. Mov. Disord. 2008, 23, 804–810. [Google Scholar] [CrossRef]
- De Wit, N.M.; De Vries, H.E.; Den Hoedt, S.; Mulder, M.T.; Martinez-Martinez, P.; Rozemuller, A.J. Astrocytic ceramide as possible indicator of neuroinflammation. J. Neuroinflamm. 2019, 16, 48. [Google Scholar] [CrossRef] [Green Version]
- Maetzler, W.; Berg, D.; Schalamberidze, N.; Melms, A.; Schott, K.; Mueller, J.C.; Liaw, L.; Gasser, T.; Nitsch, C. Osteopontin is elevated in Parkinson’s disease and its absence leads to reduced neurodegeneration in the MPTP model. Neurobiol. Dis. 2007, 25, 473–482. [Google Scholar] [CrossRef]
- Maetzler, W.; Michelis, J.; Tomiuk, J.; Melms, A.; Becker, C.; Gasser, T.; Schulte, C.; Berg, D. A single-nucleotide polymorphism of the osteopontin gene may contribute to a susceptibility to Lewy body disease. J. Neural Transm. 2009, 116, 599–605. [Google Scholar] [CrossRef]
- Oizumi, H.; Sugimura, Y.; Totsune, T.; Kawasaki, I.; Ohshiro, S.; Baba, T.; Kimpara, T.; Sakuma, H.; Hasegawa, T.; Kawahata, I.; et al. Plasma sphingolipid abnormalities in neurodegenerative diseases. PLoS ONE 2022, 17, e0279315. [Google Scholar] [CrossRef]
- Wang, Q. Vascular, inflammatory and metabolic risk factors in relation to dementia in parkinson’s disease patients with type 2 diabetes mellitus. Mov. Disord. 2020, 35 (Suppl. 1), S190. [Google Scholar] [CrossRef]
- Wang, Y.; Hancock, A.M.; Bradner, J.; Chung, K.A.; Quinn, J.F.; Peskind, E.R.; Galasko, D.; Jankovic, J.; Zabetian, C.P.; Kim, H.M.; et al. Complement 3 and factor h in human cerebrospinal fluid in Parkinson’s disease, Alzheimer’s disease, and multiple-system atrophy. Am. J. Pathol. 2011, 178, 1509–1516. [Google Scholar] [CrossRef]
- Janssen, J.C.; Godbolt, A.K.; Ioannidis, P.; Thompson, E.J.; Rossor, M.N. The prevalence of oligoclonal bands in the CSF of patients with primary neurodegenerative dementia. J. Neurol. 2004, 251, 184–188. [Google Scholar] [CrossRef]
- Jesse, S.; Brettschneider, J.; Sussmuth, S.D.; Landwehrmeyer, B.G.; Von Arnim, C.A.F.; Ludolph, A.C.; Tumani, H.; Otto, M. Summary of cerebrospinal fluid routine parameters in neurodegenerative diseases. J. Neurol. 2011, 258, 1034–1041. [Google Scholar] [CrossRef] [Green Version]
- Rajkumar, A.P.; Hye, A.; Lange, J.; Manesh, Y.R.; Ballard, C.; Fladby, T.; Aarsland, D. Next-Generation RNA-Sequencing of Serum Small Extracellular Vesicles Discovers Potential Diagnostic Biomarkers for Dementia With Lewy Bodies. Am. J. Geriatr. Psychiatry Off. J. Am. Assoc. Geriatr. Psychiatry 2021, 29, 573–584. [Google Scholar] [CrossRef]
- Santpere, G.; Garcia-Esparcia, P.; Andres-Benito, P.; Lorente-Galdos, B.; Navarro, A.; Ferrer, I. Transcriptional network analysis in frontal cortex in Lewy body diseases with focus on dementia with Lewy bodies. Brain Pathol. 2018, 28, 315–333. [Google Scholar] [CrossRef] [Green Version]
- Donaghy, P.C.; Cockell, S.J.; Martin-Ruiz, C.; Coxhead, J.; Kane, J.; Erskine, D.; Koss, D.; Taylor, J.-P.; Morris, C.M.; O’Brien, J.T.; et al. Blood mRNA Expression in Alzheimer’s Disease and Dementia With Lewy Bodies. Am. J. Geriatr. Psychiatry 2022, 30, 964–975. [Google Scholar] [CrossRef]
- Chahine, L.M.; Qiang, J.; Ashbridge, E.; Minger, J.; Horn, S.; Colcher, A.; Hurtig, H.I.; Lee, V.M.-Y.; Van Deerlin, V.M.; Leverenz, J.B.; et al. Clinical and biochemical differences in patients having parkinson disease with vs without GBA mutations. JAMA Neurol. 2013, 70, 852–858. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Guo, J.; Wang, Y.; Li, K.; Kang, J.; Wei, Y.; Sun, Q.; Xu, Q.; Xu, C.; Yan, X.; et al. Lack of association between IL-10 and IL-18 gene promoter polymorphisms and Parkinson’s disease with cognitive impairment in a Chinese population. Sci. Rep. 2016, 6, 19021. [Google Scholar] [CrossRef]
- Conway, O.J.; Carrasquillo, M.M.; Wang, X.; Bredenberg, J.M.; Reddy, J.S.; Strickland, S.L.; Younkin, C.S.; Burgess, J.D.; Allen, M.; Lincoln, S.J.; et al. ABI3 and PLCG2 missense variants as risk factors for neurodegenerative diseases in Caucasians and African Americans. Mol. Neurodegener. 2018, 13, 53. [Google Scholar] [CrossRef] [Green Version]
- Amin, J.; Erskine, D.; Donaghy, P.C.; Surendranathan, A.; Swann, P.; Kunicki, A.P.; Boche, D.; Holmes, C.; McKeith, I.G.; O’Brien, J.T.; et al. Inflammation in dementia with Lewy bodies. Neurobiol. Dis. 2022, 168, 105698. [Google Scholar] [CrossRef]
- Lee, H.J.; Bae, E.J.; Lee, S.J. Extracellular alpha-synuclein—A novel and crucial factor in Lewy body diseases. Nat. Rev. Neurol. 2014, 10, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Lee, H.J.; Masliah, E.; Lee, S.J. Non-cell-autonomous Neurotoxicity of alpha-synuclein Through Microglial Toll-like Receptor 2. Exp. Neurobiol. 2016, 25, 113–119. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.; Ho, D.H.; Suk, J.E.; You, S.; Michael, S.; Kang, J.; Lee, S.J.; Masliah, E.; Hwang, D.; Lee, H.-J.; et al. Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun. 2013, 4, 1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Erviti, L.; Couch, Y.; Richardson, J.; Cooper, J.M.; Wood, M.J. Alpha-synuclein release by neurons activates the inflammatory response in a microglial cell line. Neurosci. Res. 2011, 69, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Fellner, L.; Irschick, R.; Schanda, K.; Reindl, M.; Klimaschewski, L.; Poewe, W.; Wenning, G.K.; Stefanova, N. Toll-like receptor 4 is required for α-synuclein dependent activation of microglia and astroglia. Glia 2013, 61, 349–360. [Google Scholar] [CrossRef] [Green Version]
- Choi, I.; Seegobin, S.P.; Liang, D.; Yue, Z. Synucleinphagy: A microglial “community cleanup program” for neuroprotection. Autophagy 2020, 16, 1718–1720. [Google Scholar] [CrossRef]
- Kim, C.; Spencer, B.; Rockenstein, E.; Yamakado, H.; Mante, M.; Adame, A.; Fields, J.A.; Masliah, D.; Iba, M.; Lee, H.-J.; et al. Immunotherapy targeting toll-like receptor 2 alleviates neurodegeneration in models of synucleinopathy by modulating alpha-synuclein transmission and neuroinflammation. Mol. Neurodegener. 2018, 13, 43. [Google Scholar] [CrossRef]
- Kwon, S.; Beilina, A.; Kumaran, R.; Kaganovich, A.; Mamais, A.; Iba, M.; Singleton, A.B.; Cookson, M.R.; Masliah, E.; Kim, C.; et al. LRRK2-mediated microglial activation via NFATc2: A novel mechanism of neurotoxic inflammation in synucleinopathies. J. Immunol. 2020, 204 (Suppl. 1), 64.7. [Google Scholar] [CrossRef]
- Duffy, M.F.; Collier, T.J.; Patterson, J.R.; Kemp, C.J.; Luk, K.C.; Tansey, M.G.; Paumier, K.L.; Kanaan, N.M.; Fischer, D.L.; Polinski, N.K.; et al. Lewy body-like alpha-synuclein inclusions trigger reactive microgliosis prior to nigral degeneration. J. Neuroinflamm. 2018, 15, 129. [Google Scholar] [CrossRef]
- Schonhoff, A.M.; Figge, D.A.; Williams, G.P.; Jurkuvenaite, A.; Gallups, N.J.; Childers, G.M.; Webster, J.M.; Standaert, D.G.; Goldman, J.E.; Harms, A.S. Border-associated macrophages mediate the neuroinflammatory response in an alpha-synuclein model of Parkinson disease. Nat. Commun. 2023, 14, 3754. [Google Scholar] [CrossRef]
- Xu, E.; Boddu, R.; Abdelmotilib, H.A.; Sokratian, A.; Kelly, K.; Liu, Z.; Bryant, N.; Chandra, S.; Carlisle, S.M.; Lefkowitz, E.J.; et al. Pathological α-synuclein recruits LRRK2 expressing pro-inflammatory monocytes to the brain. Mol. Neurodegener. 2022, 17, 7. [Google Scholar] [CrossRef]
- Bradburn, S.; Murgatroyd, C.; Ray, N. Neuroinflammation in mild cognitive impairment and Alzheimer’s disease: A meta-analysis. Ageing Res. Rev. 2019, 50, 1–8. [Google Scholar] [CrossRef]
- Montoliu-Gaya, L.; Alcolea, D.; Ashton, N.J.; Pegueroles, J.; Levin, J.; Bosch, B.; Lantero-Rodriguez, J.; Carmona-Iragui, M.; Wagemann, O.; Balasa, M.; et al. Plasma and cerebrospinal fluid glial fibrillary acidic protein levels in adults with Down syndrome: A longitudinal cohort study. eBioMedicine 2023, 90, 104547. [Google Scholar] [CrossRef]
- Guo, Y.; Shen, X.-N.; Wang, H.-F.; Chen, S.-D.; Zhang, Y.-R.; Chen, S.-F.; Cui, M.; Cheng, W.; Dong, Q.; Ma, T.; et al. The dynamics of plasma biomarkers across the Alzheimer’s continuum. Alzheimers Res. Ther. 2023, 15, 31. [Google Scholar] [CrossRef]
- Silva-Spínola, A.; Lima, M.; Leitão, M.J.; Bernardes, C.; Durães, J.; Duro, D.; Tábuas-Pereira, M.; Santana, I.; Baldeiras, I. Blood biomarkers in mild cognitive impairment patients: Relationship between analytes and progression to Alzheimer disease dementia. Eur. J. Neurol. 2023, 30, 1565–1573. [Google Scholar] [CrossRef]
- Ishiki, A.; Kamada, M.; Kawamura, Y.; Terao, C.; Shimoda, F.; Tomita, N.; Arai, H.; Furukawa, K. Glial fibrillar acidic protein in the cerebrospinal fluid of Alzheimer’s disease, dementia with Lewy bodies, and frontotemporal lobar degeneration. J. Neurochem. 2016, 136, 258–261. [Google Scholar] [CrossRef]
- Chouliaras, L.; Thomas, A.; Malpetti, M.; Donaghy, P.; Kane, J.; Mak, E.; Savulich, G.; Prats-Sedano, M.A.; Heslegrave, A.J.; Zetterberg, H. Differential levels of plasma biomarkers of neurodegeneration in Lewy body dementia, Alzheimer’s disease, frontotemporal dementia and progressive supranuclear palsy. J. Neurol. Neurosurg. Psychiatry 2022, 93, 651–658. [Google Scholar] [CrossRef]
- Oeckl, P.; Halbgebauer, S.; Anderl-Straub, S.; Steinacker, P.; Huss, A.M.; Neugebauer, H.; von Arnim, C.A.F.; Diehl-Schmid, J.; Grimmer, T.; Kornhuber, J.; et al. Glial Fibrillary Acidic Protein in Serum is Increased in Alzheimer’s Disease and Correlates with Cognitive Impairment. J. Alzheimers Dis. 2019, 67, 481–488. [Google Scholar] [CrossRef]
- Baiardi, S.; Quadalti, C.; Mammana, A.; Dellavalle, S.; Zenesini, C.; Sambati, L.; Pantieri, R.; Polischi, B.; Romano, L.; Suffritti, M.; et al. Diagnostic value of plasma p-tau181, NfL, and GFAP in a clinical setting cohort of prevalent neurodegenerative dementias. Alzheimers Res. Ther. 2022, 14, 153. [Google Scholar] [CrossRef]
- Bolsewig, K.; Hok, A.H.Y.S.; Sepe, F.N.; Boonkamp, L.; Jacobs, D.; Bellomo, G.; Paoletti, F.P.; Vanmechelen, E.; Teunissen, C.E.; Parnetti, L.; et al. A Combination of Neurofilament Light, Glial Fibrillary Acidic Protein, and Neuronal Pentraxin-2 Discriminates Between Frontotemporal Dementia and Other Dementias. J. Alzheimers Dis. 2022, 90, 363–380. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, G.P.; Schonhoff, A.M.; Jurkuvenaite, A.; Gallups, N.J.; Standaert, D.G.; Harms, A.S. CD4 T cells mediate brain inflammation and neurodegeneration in a mouse model of Parkinson’s disease. Brain 2021, 144, 2047–2059. [Google Scholar] [CrossRef]
- Rostami, J.; Fotaki, G.; Sirois, J.; Mzezewa, R.; Bergström, J.; Essand, M.; Healy, L.; Erlandsson, A. Astrocytes have the capacity to act as antigen-presenting cells in the Parkinson’s disease brain. J. Neuroinflamm. 2020, 17, 119. [Google Scholar] [CrossRef] [PubMed]
- Webers, A.; Heneka, M.T.; Gleeson, P.A. The role of innate immune responses and neuroinflammation in amyloid accumulation and progression of Alzheimer’s disease. Immunol. Cell Biol. 2020, 98, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; Khoury, J.E.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Parbo, P.; Madsen, L.S.; Ismail, R.; Zetterberg, H.; Blennow, K.; Eskildsen, S.F.; Vorup-Jensen, T.; Brooks, D.J. Low plasma neurofilament light levels associated with raised cortical microglial activation suggest inflammation acts to protect prodromal Alzheimer’s disease. Alzheimers Res. Ther. 2020, 12, 3. [Google Scholar] [CrossRef] [Green Version]
- Rakic, S.; Hung, Y.M.A.; Smith, M.; So, D.; Tayler, H.M.; Varney, W.; Wild, J.; Harris, S.; Holmes, C.; Love, S.; et al. Systemic infection modifies the neuroinflammatory response in late stage Alzheimer’s disease. Acta Neuropathol. Commun. 2018, 6, 88. [Google Scholar] [CrossRef] [Green Version]
- Hamelin, L.; Lagarde, J.; Dorothée, G.; Leroy, C.; Labit, M.; Comley, R.A.; de Souza, L.C.; Corne, H.; Dauphinot, L.; Bertoux, M.; et al. Early and protective microglial activation in Alzheimer’s disease: A prospective study using 18F-DPA-714 PET imaging. Brain 2016, 139 Pt 4, 1252–1264. [Google Scholar] [CrossRef] [Green Version]
- Elahi, F.M.; Miller, B.L. A clinicopathological approach to the diagnosis of dementia. Nat. Rev. Neurol. 2017, 13, 457–476. [Google Scholar] [CrossRef] [Green Version]
- Jellinger, K.A.; Korczyn, A.D. Are dementia with Lewy bodies and Parkinson’s disease dementia the same disease? BMC Med. 2018, 16, 34. [Google Scholar] [CrossRef] [Green Version]
- Englund, E.; Badri, M.; Oskooi, M.; Brunnstrom, H. The concordance of clinical and neuropathological diagnosis of dementia. Clin. Neuropathol. 2014, 33, 204. [Google Scholar]
- Bousiges, O.; Blanc, F. Biomarkers of Dementia with Lewy Bodies: Differential Diagnostic with Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 6371. [Google Scholar] [CrossRef]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Thijssen, E.H.; Rabinovici, G.D. Rapid Progress Toward Reliable Blood Tests for Alzheimer Disease. JAMA Neurol. 2021, 78, 143–145. [Google Scholar] [CrossRef]
- Chaudhry, A.; Houlden, H.; Rizig, M. Novel fluid biomarkers to differentiate frontotemporal dementia and dementia with Lewy bodies from Alzheimer’s disease: A systematic review. J. Neurol. Sci. 2020, 415, 116886. [Google Scholar] [CrossRef]
- Manne, S.; Kondru, N.; Hepker, M.; Jin, H.; Anantharam, V.; Kanthasamy, A.; Kanthasamy, A.G. Ultrasensitive Detection of Aggregated alpha-Synuclein in Glial Cells, Human Cerebrospinal Fluid, and Brain Tissue Using the RT-QuIC Assay: New High-Throughput Neuroimmune Biomarker Assay for Parkinsonian Disorders. J. Neuroimmune Pharmacol. 2019, 14, 423–435. [Google Scholar] [CrossRef]
- Coysh, T.; Mead, S. The Future of Seed Amplification Assays and Clinical Trials. Front. Aging Neurosci. 2022, 14, 872629. [Google Scholar] [CrossRef]
- Mammana, A.; Baiardi, S.; Quadalti, C.; Rossi, M.; Donadio, V.; Capellari, S.; Liguori, R.; Parchi, P. RT-QuIC Detection of Pathological α-Synuclein in Skin Punches of Patients with Lewy Body Disease. Mov. Disord. 2021, 36, 2173–2177. [Google Scholar] [CrossRef]
- Hall, S.; Orrù, C.D.; Serrano, G.E.; Galasko, D.; Hughson, A.G.; Groveman, B.R.; Adler, C.H.; Beach, T.G.; Caughey, B. Hansson Performance of αSynuclein RT-QuIC in relation to neuropathological staging of Lewy body disease. Acta Neuropathol. Commun. 2022, 10, 90. [Google Scholar] [CrossRef]
- Iranzo, A.; Fairfoul, G.; Ayudhaya, A.C.N.; Serradell, M.; Gelpi, E.; Vilaseca, I.; Sanchez-Valle, R.; Gaig, C.; Santamaria, J.; Tolosa, E.; et al. Detection of α-synuclein in CSF by RT-QuIC in patients with isolated rapid-eye-movement sleep behaviour disorder: A longitudinal observational study. Lancet Neurol. 2021, 20, 203–212. [Google Scholar] [CrossRef]
- Okuzumi, A.; Hatano, T.; Matsumoto, G.; Nojiri, S.; Ueno, S.-I.; Imamichi-Tatano, Y.; Kimura, H.; Kakuta, S.; Kondo, A.; Fukhara, T.; et al. Propagative α-synuclein seeds as serum biomarkers for synucleinopathies. Nat. Med. 2023, 29, 1448–1455. [Google Scholar] [CrossRef] [PubMed]
- Chin, K.S.; Yassi, N.; Churilov, L.; Masters, C.L.; Watson, R. Prevalence and clinical associations of tau in Lewy body dementias: A systematic review and meta-analysis. Parkinsonism Relat. Disord. 2020, 80, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Hijazi, Z.; Yassi, N.; O’Brien, J.T.; Watson, R. The influence of cerebrovascular disease in dementia with Lewy bodies and Parkinson’s disease dementia. Eur. J. Neurol. 2022, 29, 1254–1265. [Google Scholar] [CrossRef]
- Iba, M.; McDevitt, R.A.; Kim, C.; Roy, R.; Sarantopoulou, D.; Tommer, E.; Siegars, B.; Sallin, M.; Kwon, S.; Sen, J.M.; et al. Aging exacerbates the brain inflammatory micro-environment contributing to alpha-synuclein pathology and functional deficits in a mouse model of DLB/PD. Mol. Neurodegener. 2022, 17, 60. [Google Scholar] [CrossRef] [PubMed]
- Rota, E.; Bellone, G.; Rocca, P.; Bergamasco, B.; Emanuelli, G.; Ferrero, P. Increased intrathecal TGF-beta1, but not IL-12, IFN-gamma and IL-10 levels in Alzheimer’s disease patients. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 2006, 27, 33–39. [Google Scholar]
- Lourenco, M.V.; Ribeiro, F.C.; Santos, L.E.; Beckman, D.; Melo, H.M.; Sudo, F.K.; Drummond, C.; Assunção, N.; Vanderborght, B.; Tovar-Moll, F.; et al. Cerebrospinal fluid neurotransmitters, cytokines, and chemokines in Alzheimer’s and Lewy body diseases. J. Alzheimes Dis. 2021, 82, 1067–1074. [Google Scholar] [CrossRef]
- Petrovsky, N.; McNair, P.; Harrison, L.C. Diurnal rhythms of pro-inflammatory cytokines: Regulation by plasma cortisol and therapeutic implications. Cytokine 1998, 10, 307–312. [Google Scholar] [CrossRef]
- Zhou, X.; Fragala, M.S.; McElhaney, J.E.; Kuchel, G.A. Conceptual and methodological issues relevant to cytokine and inflammatory marker measurements in clinical research. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 541–547. [Google Scholar] [CrossRef] [Green Version]
- Casaletto, K.B.; Elahi, F.M.; Fitch, R.; Walters, S.; Fox, E.; Staffaroni, A.M.; Bettcher, B.M.; Zetterberg, H.; Karydas, A.; Rojas, J.C.; et al. A comparison of biofluid cytokine markers across platform technologies: Correspondence or divergence? Cytokine 2018, 111, 481–489. [Google Scholar] [CrossRef]
- Chong, J.R.; Chai, Y.L.; Lee, J.H.; Howlett, D.; Attems, J.; Ballard, C.G.; Aarsland, D.; Francis, P.T.; Chen, C.P.; Lai, M.K.P. Increased Transforming Growth Factor beta 2 in the Neocortex of Alzheimer’s Disease and Dementia with Lewy Bodies is Correlated with Disease Severity and Soluble A beta(42) Load. J. Alzheimers Dis. 2017, 56, 157–166. [Google Scholar] [CrossRef]
- Wilhelmsson, U.; Andersson, D.; de Pablo, Y.; Pekny, R.; Ståhlberg, A.; Mulder, J.; Mitsios, N.; Hortobágyi, T.; Pekny, M.; Pekna, M. Injury Leads to the Appearance of Cells with Characteristics of Both Microglia and Astrocytes in Mouse and Human Brain. Cereb. Cortex 2017, 27, 3360–3377. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Sun, J.; Perrin, R.J.; Mach, R.H.; Bales, K.R.; Morris, J.C.; Benzinger, T.L.S.; Holtzman, D.M. Translocator protein in late stage Alzheimer’s disease and Dementia with Lewy bodies brains. Ann. Clin. Transl. Neurol. 2019, 6, 1423–1434. [Google Scholar] [CrossRef]
- Li, H.; Knight, W.C.; Yang, P.; Guo, Y.; Perlmutter, J.S.; Morris, J.C.; Bateman, R.J.; Benzinger, T.L.S.; Xu, J. Microglia Implicated in Tauopathy in the Striatum of Neurodegenerative Disease Patients from Genotype to Phenotype. Int. J. Mol. Sci. 2020, 21, 6047. [Google Scholar] [CrossRef]
- Xu, J.; Li, H.; Knight, W.C. Striatal oxidative damages and neuroinflammation correlate with progression and survival of Lewy body and Alzheimer diseases. Neural Regen. Res. 2022, 17, 867–874. [Google Scholar] [CrossRef]
- Tu, H.; Zhang, Z.W.; Qiu, L.; Lin, Y.; Jiang, M.; Chia, S.-Y.; Wei, Y.; Ng, A.S.L.; Reynolds, R.; Tan, E.-K.; et al. Increased expression of pathological markers in Parkinson’s disease dementia post-mortem brains compared to dementia with Lewy bodies. BMC Neurosci. 2022, 23, 3. [Google Scholar] [CrossRef]
- Nicastro, N.; Mak, E.; Williams, G.B.; Surendranathan, A.; Bevan-Jones, W.R.; Passamonti, L.; Rodrìguez, P.V.; Su, L.; Arnold, R.; Fryer, T.D.; et al. Correlation of microglial activation with white matter changes in dementia with Lewy bodies. Neuroimage-Clin. 2020, 25, 102200. [Google Scholar] [CrossRef]
- Gómez-Tortosa, E.; Gonzalo, I.; Fanjul, S.; Sainz, M.J.; Cantarero, S.; Cemillán, C.; Yébenes, J.G.; del Ser, T. Cerebrospinal Fluid Markers in Dementia With Lewy Bodies Compared With Alzheimer Disease. Arch. Neurol. 2003, 60, 1218–1222. [Google Scholar] [CrossRef] [Green Version]
- Morgenthaler, N.G.; Struck, J.; Fischer-Schulz, C.; Bergmann, A. Sensitive immunoluminometric assay for the detection of procalcitonin. Clin. Chem. 2002, 48, 788–790. [Google Scholar] [CrossRef]
- Wennström, M.; Hall, S.; Nägga, K.; Londos, E.; Minthon, L.; Hansson, O. Cerebrospinal fluid levels of IL-6 are decreased and correlate with cognitive status in DLB patients. Alzheimers Res. Ther. 2015, 7, 63. [Google Scholar] [CrossRef] [Green Version]
- Janelidze, S.; Lindqvist, D.; Francardo, V.; Hall, S.; Zetterberg, H.; Blennow, K.; Adler, C.H.; Beach, T.G.; Serrano, G.E.; van Westen, D.; et al. Increased CSF biomarkers of angiogenesis in Parkinson disease. Neurology 2015, 85, 1834–1842. [Google Scholar] [CrossRef] [Green Version]
- Lindqvist, D.; Hall, S.; Surova, Y.; Nielsen, H.M.; Janelidze, S.; Brundin, L.; Hansson, O. Cerebrospinal fluid inflammatory markers in Parkinson’s disease--associations with depression, fatigue, and cognitive impairment. Brain Behav. Immun. 2013, 33, 183–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayton, S.; Hall, S.; Janelidze, S.; Kalinowski, P.; Palmqvist, S.; Belaidi, A.A.; Roberts, B.; Roberts, A.; Stomrud, E.; Bush, A.I.; et al. The Neuroinflammatory Acute Phase Response in Parkinsonian-Related Disorders. Mov. Disord. 2022, 37, 993–1003. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.T.; Ramesh, T.; Toh, X.R.; Nguyen, L.N. Emerging roles of lysophospholipids in health and disease. Prog. Lipid Res. 2020, 80, 101068. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Glia | Tissue Markers |
|---|---|
| Microglia |
|
| Astrocytes |
|
| Lymphocytes | Tissue markers |
| T cells |
|
| B cells |
|
| Signaling molecules | Molecules |
| Cytokines and chemokines | Pro-inflammatory:
Anti-inflammatory/regulatory:
|
| Investigation Method | Number of Studies | DLB, n | PDD, n | LBD NOS, n | Total LBD, n | AD, n | PD, n | Controls, n |
|---|---|---|---|---|---|---|---|---|
| Postmortem | 36 | 334 | 151 | 82 | 567 | 242 | 183 | 587 |
| Living subjects | 44 | 608 | 493 | 130 | 1231 | 1641 | 1623 | 2614 |
| Imaging | 5 | 25 | 25 | 0 | 50 | 18 | 6 | 61 |
| CSF | 21 | 313 | 163 | 96 | 572 | 1292 | 801 | 1671 |
| Blood | 25 | 390 | 330 | 34 | 754 | 672 | 1045 | 1097 |
| Gene studies | 3 | 316 | 108 | 0 | 424 | 2770 | 955 | 3834 |
| Total * | 80 | 1236 | 698 | 172 | 2106 | 4542 | 2661 | 6801 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loveland, P.M.; Yu, J.J.; Churilov, L.; Yassi, N.; Watson, R. Investigation of Inflammation in Lewy Body Dementia: A Systematic Scoping Review. Int. J. Mol. Sci. 2023, 24, 12116. https://doi.org/10.3390/ijms241512116
Loveland PM, Yu JJ, Churilov L, Yassi N, Watson R. Investigation of Inflammation in Lewy Body Dementia: A Systematic Scoping Review. International Journal of Molecular Sciences. 2023; 24(15):12116. https://doi.org/10.3390/ijms241512116
Chicago/Turabian StyleLoveland, Paula M., Jenny J. Yu, Leonid Churilov, Nawaf Yassi, and Rosie Watson. 2023. "Investigation of Inflammation in Lewy Body Dementia: A Systematic Scoping Review" International Journal of Molecular Sciences 24, no. 15: 12116. https://doi.org/10.3390/ijms241512116
APA StyleLoveland, P. M., Yu, J. J., Churilov, L., Yassi, N., & Watson, R. (2023). Investigation of Inflammation in Lewy Body Dementia: A Systematic Scoping Review. International Journal of Molecular Sciences, 24(15), 12116. https://doi.org/10.3390/ijms241512116