
Acid Sphingomyelinase Deficiency Type B Patient-Derived Liver Organoids Reveals Altered Lysosomal Gene Expression and Lipid Homeostasis
 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Generation of Liver Organoids from a Niemann–Pick Type B Patient (ASMD Type B)
2.2. Expression of Differentiation Markers in Liver Organoids
2.3. The NPB Patient Has the Pathogenic p.Arg610del Variant
2.4. Altered Expression of Sphingomyelin Metabolism Genes
2.5. Increased Intracellular Lipid Content in ASMD Type B Organoids
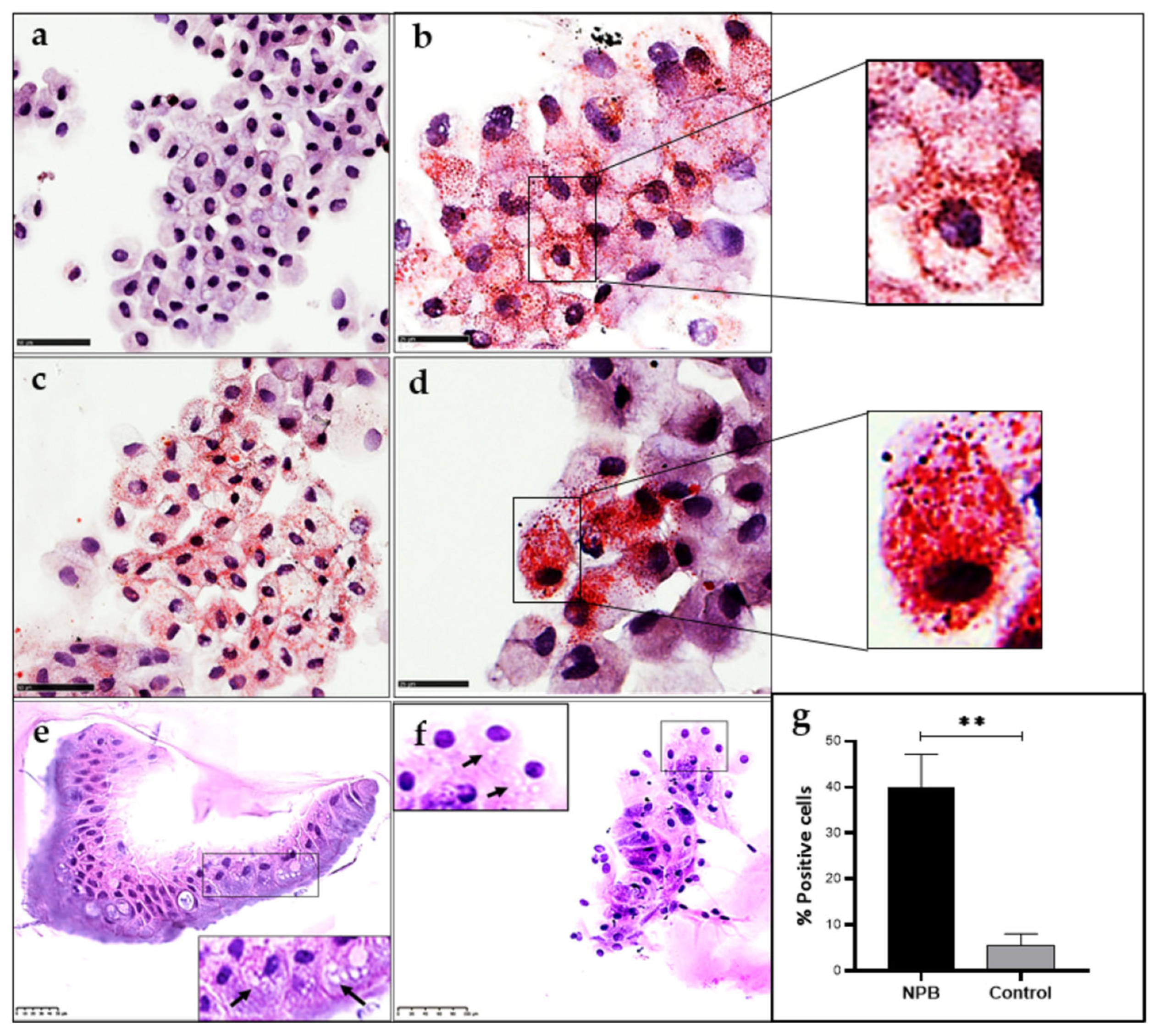
2.6. Identification of Foam Cells in ASMD Type B Liver Organoids
2.7. Alteration of Lipid Homeostasis in the ASMD Type B Liver Organoids
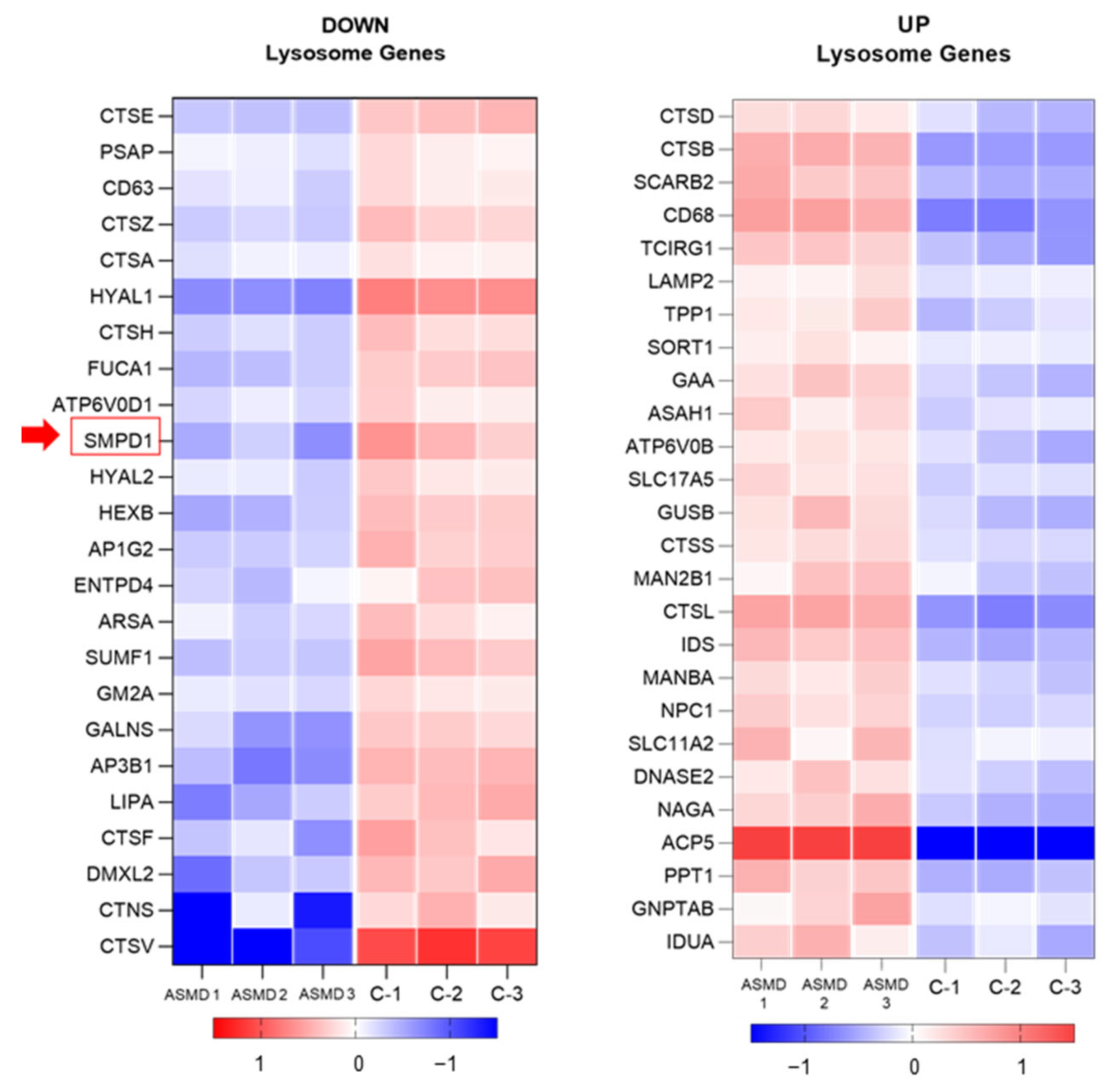
2.8. Transcriptomic Analysis of Liver Organoids (RNA-Seq)
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Genotyping
4.3. Establishment and Culture of Human Liver Organoids
4.4. Validation of Progenitor, Ductal, and Differentiation Cell Markers
4.5. Expression Analysis of Sphingomyelin Metabolism Genes
4.6. Determination of Intracellular Neutral Lipid
4.7. Lipid Extraction, Mass Spectrometry (MS) Data Acquisition and Data Analysis and Post-Processing
4.8. Foam Cell Detection
4.9. Transcriptomic Analysis of Liver Organoids (RNA-Seq)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACD | Acid Ceramidase |
| ACP5 | Acid phosphatase 5, tartrate resistant |
| AdSCs | Adult Stem Cells |
| ALB | Albumin |
| AP1G2 | Adaptor-related protein complex 1 subunit gamma 2 |
| AP3B1 | Adaptor-related protein complex 3 subunit beta 1 |
| APOB | Apolipoprotein B |
| ARSA | Arylsulfatase A |
| ASAH1 | N-acylsphingosine amidohydrolase 1 |
| ASM | Acid Sphingomyelinase |
| ASMD | Acid Sphingomyelinase Deficiency |
| ATP6V0B | ATPase H+ transporting V0 subunit b |
| ATP6V0D1 | ATPase H+ transporting V0 subunit d1 |
| BME2 | Basement Membrane Extract BME Type 2 |
| CD63 | CD63 molecule |
| CD68 | CD68 molecule |
| CE | Cholesterol Esters |
| Cer | Ceramide |
| Chol | Cholesterol |
| CL | Cardiolipin |
| CTNS | Cystinosin, lysosomal cystine transporter |
| CTSA | Cathepsin A |
| CTSB | Cathepsin B |
| CTSD | Cathepsin D |
| CTSE | Cathepsin E |
| CTSF | Cathepsin F |
| CTSH | Cathepsin H |
| CTSL | Cathepsin L |
| CTSS | Cathepsin S |
| CTSV | Cathepsin V |
| CTSZ | Cathepsin Z |
| DAG | Diacylglycerol |
| DAPT | Gamma-Secretase inhibitor |
| DEGs | Differentially Expressed Genes |
| Dif | Differentiation |
| DMXL2 | Dmx like 2 |
| DNASE2 | Deoxyribonuclease 2, lysosomal |
| EBSS | Earle’s equilibration saline solution |
| EGF | Epidermal Growth Factor |
| ENTPD4 | Ectonucleoside triphosphate diphosphohydrolase 4 |
| Exp | Expansion |
| FDR | False Discovery Rate |
| FGF | Fibroblast Growth Factor |
| FUCA1 | Alpha-L-fucosidase 1 |
| GAA | Alpha glucosidase |
| GALNS | Galactosamine (N-acetyl)-6-sulfatase |
| GAPDH | Gliceralhedído-3-fosfato deshidrogenasa |
| GEO | Gene Expression Omnibus |
| GM2A | Ganglioside GM2 activator |
| GNPTAB | N-acetylglucosamine-1-phosphate transferase subunits alpha and beta |
| GUSB | Glucuronidase beta |
| HDL-C | High-Density Lipoprotein Cholesterol |
| HEXB | Hexosaminidase subunit beta |
| HexCer | Hexosylceramide |
| HPS | Hermansky-Pudlak syndrome |
| HYAL1 | Hyaluronidase 1 |
| HYAL2 | Hyaluronidase 2 |
| IDS | Iduronate 2-sulfatase |
| IDUA | Alpha-L-iduronidase |
| IMM | Mitochondrial Inner Membrane |
| KRT19 | Keratin 19 |
| LAMP2 | Lysosomal-associated membrane protein 2 |
| LDL-C | High Low-density Lipoprotein Cholesterol |
| LGR5 | Leucine-rich repeat-containing G protein-coupled receptor 5 |
| LIPA | Lipase A, lysosomal acid type |
| LPA | Lyso-Phosphatidate |
| LPC | Lyso-Phosphatidylcholine |
| LPC O- | Lyso-Phosphatidylcholine (-ether) |
| LPE | Lyso-Phosphatidylethanolamine |
| LPE O- | Lyso-Phosphatidylethanolamine (-ether) |
| LPG | Lyso-phosphatidylglycerol |
| LPI | Lyso-Phosphatidylinositol |
| LPS | Lyso-Phosphatidylserine |
| MAN2B1 | Mannosidase alpha class 2B member 1 |
| MANBA | Mannosidase beta |
| MS | Mass spectrometry |
| NAGA | Alpha-N-acetylgalactosaminidase |
| nCD | Neutral Ceramidase |
| NP | Niemann-Pick |
| NPA | Niemann–Pick type A |
| NPA/B | Niemann–Pick type A/B |
| NPB | Niemann–Pick type B |
| NPC1 | NPC intracellular cholesterol transporter 1 |
| NPD | Niemann-Pick disease |
| nSM | Neutral Sphingomyelinase |
| nSM 2 | Neutral Sphingomyelinase 2 |
| PA | Phosphatidate |
| PAS | Periodic Acid–Schiff |
| PBS | Phosphate Buffered Saline |
| PC | Phosphatidylcholine |
| PC O- | Phosphatidylcholine (-ether) |
| PE | Phosphatidylethanolamine |
| PE O- | Phosphatidylethanolamine (-ether) |
| PFA | Paraformaldehyde |
| PG | Phosphatidylglycerol |
| PI | Phosphatidylinositol |
| PPT1 | Palmitoyl-protein thioesterase 1 |
| PS | Phosphatidylserine |
| PSAP | Prosaposin |
| Rspo1 | R-Spondin 1 |
| SCARB2 | Scavenger receptor class B member 2 |
| SGMS1 | Esfingomielina Sintasa 1 |
| SLC11A2 | Solute carrier family 11 member 2 |
| SLC17A5 | Solute carrier family 17 member 5 |
| SM | Sphingomyelin |
| SMPD1 | Sphingomyelin phosphodiesterase 1 |
| SMPD2 | Sphingomyelin phosphodiesterase 2 |
| SMS1 | Sphingomyelin Synthase 1 |
| SORT1 | Sortilin 1 |
| Sph | Sphingosine |
| STGD | Stargardt’s disease |
| SUMF1 | Sulfatase modifying factor 1 |
| TAG | Triacylglycerol |
| TCIRG1 | T cell immune regulator 1, ATPase H+ transporting V0 subunit a3 |
| TPP1 | Tripeptidyl peptidase 1 |
References
- Schuchman, E.H.; Wasserstein, M.P. Types A and B Niemann-Pick disease. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 237–247. [Google Scholar] [CrossRef] [Green Version]
- Wasserstein, M.P.; Aron, A.; Brodie, S.E.; Simonaro, C.; Desnick, R.J.; McGovern, M.M. Acid sphingomyelinase deficiency: Prevalence and characterization of an intermediate phenotype of Niemann-Pick disease. J. Pediatr. 2006, 149, 554–559. [Google Scholar] [CrossRef] [PubMed]
- Wasserstein, M.P.; Desnick, R.J.; Schuchman, E.H.; Hossain, S.; Wallenstein, S.; Lamm, C.; McGovern, M.M. The natural history of type B Niemann-Pick disease: Results from a 10-year longitudinal study. Pediatrics 2004, 114, e672–e677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGovern, M.M.; Wasserstein, M.P.; Giugliani, R.; Bembi, B.; Vanier, M.T.; Mengel, E.; Brodie, S.E.; Mendelson, D.; Skloot, G.; Desnick, R.J.; et al. A Prospective, Cross-sectional Survey Study of the Natural History of Niemann-Pick Disease Type B. Pediatrics 2008, 122, e341–e349. [Google Scholar] [CrossRef] [Green Version]
- Wasserstein, M.P.; Schuchman, E.H. Acid Sphingomyelinase Deficiency. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- McGovern, M.M.; Aron, A.; Brodie, S.E.; Desnick, R.J.; Wasserstein, M.P. Natural history of Type A Niemann-Pick disease: Possible endpoints for therapeutic trials. Neurology 2006, 66, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Schuchman, E.H.; Levran, O.; Pereira, L.V.; Desnick, R.J. Structural organization and complete nucleotide sequence of the gene encoding human acid sphingomyelinase (SMPD1). Genomics 1992, 12, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Schuchman, E.H.; Miranda, S.R. Niemann-Pick disease: Mutation update, genotype/phenotype correlations, and prospects for genetic testing. Genet. Test. 1997, 1, 13–19. [Google Scholar] [CrossRef]
- Zampieri, S.; Filocamo, M.; Pianta, A.; Lualdi, S.; Gort, L.; Coll, M.J.; Sinnott, R.; Geberhiwot, T.; Bembi, B.; Dardis, A. SMPD1 Mutation Update: Database and Comprehensive Analysis of Published and Novel Variants. Hum. Mutat. 2016, 37, 139–147. [Google Scholar] [CrossRef]
- Simonaro, C.M.; Desnick, R.J.; McGovern, M.M.; Wasserstein, M.P.; Schuchman, E.H. The Demographics and Distribution of Type B Niemann-Pick Disease: Novel Mutations Lead to New Genotype/Phenotype Correlations. Am. J. Hum. Genet. 2002, 71, 1413–1419. [Google Scholar] [CrossRef] [Green Version]
- Pittis, M.; Donnarumma, M.; Montalvo, A.; Dominissini, S.; Kroos, M.; Rosano, C.; Stroppiano, M.; Bianco, M.; Donati, M.; Parenti, G.; et al. Molecular and functional characterization of eight novel GAA mutations in Italian infants with Pompe disease. Hum. Mutat. 2008, 29, E27–E36. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Thurberg, B.L.; Wasserstein, M.P.; Schiano, T.; O’brien, F.; Richards, S.; Cox, G.F.; McGovern, M.M. Liver and Skin Histopathology in Adults with Acid Sphingomyelinase Deficiency (Niemann-Pick Disease Type B). Am. J. Surg. Pathol. 2012, 36, 1234–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz, G.A.; Giugliani, R.; Guffon, N.; Jones, S.A.; Mengel, E.; Scarpa, M.; Witters, P.; Yarramaneni, A.; Li, J.; Armstrong, N.M.; et al. Long-term safety and clinical outcomes of olipudase alfa enzyme replacement therapy in pediatric patients with acid sphingomyelinase deficiency: Two-year results. Orphanet J. Rare Dis. 2022, 17, 437. [Google Scholar] [CrossRef]
- Wasserstein, M.P.; Larkin, A.E.; Glass, R.B.; Schuchman, E.H.; Desnick, R.J.; McGovern, M.M. Growth restriction in children with type B Niemann-Pick disease. J. Pediatr. 2003, 142, 424–428. [Google Scholar] [CrossRef] [PubMed]
- McGovern, M.M.; Wasserstein, M.P.; Aron, A.; Desnick, R.J.; Schuchman, E.H.; Brodie, S.E. Ocular manifestations of Niemann-Pick disease type B. Ophthalmology 2004, 111, 1424–1427. [Google Scholar] [CrossRef] [PubMed]
- McGovern, M.M.; Pohl-Worgall, T.; Deckelbaum, R.J.; Simpson, W.; Mendelson, D.; Desnick, R.J.; Schuchman, E.H.; Wasserstein, M.P. Lipid abnormalities in children with types A and B Niemann Pick disease. J. Pediatr. 2004, 145, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Thurberg, B.L. Autopsy pathology of infantile neurovisceral ASMD (Niemann-Pick Disease type A): Clinicopathologic correlations of a case report. Mol. Genet. Metab. Rep. 2020, 24, 100626. [Google Scholar] [CrossRef]
- Gómez-Mariano, G.; Matamala, N.; Martínez, S.; Justo, I.; Marcacuzco, A.; Jimenez, C.; Monzón, S.; Cuesta, I.; Garfia, C.; Martínez, M.T.; et al. Liver organoids reproduce alpha-1 antitrypsin deficiency-related liver disease. Hepatol. Int. 2020, 14, 127–137. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Maegawa, G.H.; Zhan, X.; Gao, X.; Wang, Y.; Xu, F.; Qiu, W.; Han, L.; Gu, X.; Zhang, H. Clinical, biochemical, and genotype-phenotype correlations of 118 patients with Niemann-Pick disease Types A/B. Hum. Mutat. 2021, 42, 614–625. [Google Scholar] [CrossRef]
- Schuchman, E.H.; Desnick, R.J. Types A and B Niemann-Pick disease. Mol. Genet. Metab. 2017, 120, 27–33. [Google Scholar] [CrossRef] [Green Version]
- Narayan, N.J.; Requena, D.; Lalazar, G.; Ramos-Espiritu, L.; Ng, D.; Levin, S.; Shebl, B.; Wang, R.; Hammond, W.J.; Saltsman, J.A.; et al. Human liver organoids for disease modeling of fibrolamellar carcinoma. Stem Cell Rep. 2022, 17, 1874–1888. [Google Scholar] [CrossRef]
- Sharma, S.; Rawal, P.; Kaur, S.; Puria, R. Liver organoids as a primary human model to study HBV-mediated Hepatocellular carcinoma. A review. Exp. Cell Res. 2023, 428, 113618. [Google Scholar] [CrossRef]
- Levran, O.; Desnick, R.J.; Schuchman, E.H. Niemann-Pick type B disease. Identification of a single codon deletion in the acid sphingomyelinase gene and genotype/phenotype correlations in type A and B patients. J. Clin. Investig. 1991, 88, 806–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Burriel, M.; Peña, L.; Ramos, J.C.; Cabrera, J.C.; Marti, M.; Rodríguez-Quiñones, F.; Chabás, A. The R608del mutation in the acid sphingomyelinase gene (SMPD1) is the most prevalent among patients from Gran Canaria Island with Niemann-Pick disease type B. Clin. Genet. 2003, 63, 235–236. [Google Scholar] [CrossRef]
- Rodrã guez-Pascau, L.; Gort, L.; Schuchman, E.H.; Vilageliu, L.; Grinberg, D.; Chabã¡S, A. Identification and characterization ofSMPD1mutations causing Niemann-Pick types A and B in Spanish patients. Hum. Mutat. 2009, 30, 1117–1122. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Sun, L.; Bracko, O.; Choi, J.W.; Jia, Y.; Nana, A.L.; Brady, O.A.; Hernandez, J.C.C.; Nishimura, N.; Seeley, W.W.; et al. Impaired prosaposin lysosomal trafficking in frontotemporal lobar degeneration due to progranulin mutations. Nat. Commun. 2017, 8, 15277. [Google Scholar] [CrossRef] [Green Version]
- Vaccaro, A.M.; Motta, M.; Tatti, M.; Scarpa, S.; Masuelli, L.; Bhat, M.; Vanier, M.T.; Tylki-Szymanska, A.; Salvioli, R. Saposin C mutations in Gaucher disease patients resulting in lysosomal lipid accumulation, saposin C deficiency, but normal prosaposin processing and sorting. Hum. Mol. Genet. 2010, 19, 2987–2997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cesani, M.; Lorioli, L.; Grossi, S.; Amico, G.; Fumagalli, F.; Spiga, I.; Filocamo, M.; Biffi, A. Mutation Update of ARSA and PSAP Genes Causing Metachromatic Leukodystrophy. Hum. Mutat. 2016, 37, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Stepien, K.M.; Ciara, E.; Jezela-Stanek, A. Fucosidosis-Clinical Manifestation, Long-Term Outcomes, and Genetic Profile-Review and Case Series. Genes 2020, 11, 1383. [Google Scholar] [CrossRef] [PubMed]
- Elmonem, M.A.; Veys, K.R.; Soliman, N.A.; van Dyck, M.; van den Heuvel, L.P.; Levtchenko, E. Cystinosis: A review. Orphanet J. Rare Dis. 2016, 11, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leal, A.F.; Benincore-Flórez, E.; Solano-Galarza, D.; Garzón Jaramillo, R.G.; Echeverri-Peña, O.Y.; Suarez, D.A.; Alméciga-Díaz, C.J.; Espejo-Mojica, A.J. GM2 Gangliosidoses: Clinical Features, Pathophysiological Aspects, and Current Therapies. Int. J. Mol. Sci. 2020, 21, 6213. [Google Scholar] [CrossRef] [PubMed]
- Matte, U.; Yogalingam, G.; Brooks, D.; Leistner, S.; Schwartz, I.; Lima, L.; Norato, D.Y.; Brum, J.M.; Beesley, C.; Winchester, B.; et al. Identification and characterization of 13 new mutations in mucopolysaccharidosis type I patients. Mol. Genet. Metab. 2003, 78, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Hiraiwa, M. Cathepsin A/protective protein: An unusual lysosomal multifunctional protein. Cell. Mol. Life Sci. 1999, 56, 894–907. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, J.; Gu, Y.; Wang, H.; Jiang, M.; Zhao, S.; Qing, H.; Ni, J. Cathepsin H: Molecular characteristics and clues to function and mechanism. Biochem. Pharmacol. 2023, 212, 115585. [Google Scholar] [CrossRef]
- Öörni, K.; Sneck, M.; Brömme, D.; Pentikäinen, M.O.; Lindstedt, K.A.; Mäyränpää, M.; Aitio, H.; Kovanen, P.T. Cysteine Protease Cathepsin F Is Expressed in Human Atherosclerotic Lesions, Is Secreted by Cultured Macrophages, and Modifies Low Density Lipoprotein Particles in Vitro. J. Biol. Chem. 2004, 279, 34776–34784. [Google Scholar] [CrossRef] [Green Version]
- Lecaille, F.; Chazeirat, T.; Saidi, A.; Lalmanach, G. Cathepsin V: Molecular characteristics and significance in health and disease. Mol. Asp. Med. 2022, 88, 101086. [Google Scholar] [CrossRef]
- Takiguchi, K.; Itoh, K.; Shimmoto, M.; Ozand, P.T.; Doi, H.; Sakuraba, H. Structural and functional study of K453E mutant protective protein/cathepsin A causing the late infantile form of galactosialidosis. J. Hum. Genet. 2000, 45, 200–206. [Google Scholar] [CrossRef] [Green Version]
- Moles, A.; Tarrats, N.; Fernández-Checa, J.C.; Marí, M. Cathepsin B overexpression due to acid sphingomyelinase ablation promotes liver fibrosis in Niemann-Pick disease. J. Biol. Chem. 2012, 287, 1178–1188. [Google Scholar] [CrossRef] [Green Version]
- Eskelinen, E.-L.; Illert, A.L.; Tanaka, Y.; Schwarzmann, G.; Blanz, J.; von Figura, K.; Saftig, P.; Styers, M.L.; Salazar, G.; Love, R.; et al. Role of LAMP-2 in Lysosome Biogenesis and Autophagy. Mol. Biol. Cell 2002, 13, 3355–3368. [Google Scholar] [CrossRef] [Green Version]
- Eskelinen, E.-L. Roles of LAMP-1 and LAMP-2 in lysosome biogenesis and autophagy. Mol. Asp. Med. 2006, 27, 495–502. [Google Scholar] [CrossRef]
- Lu, M.; Ammar, D.; Ives, H.; Albrecht, F.; Gluck, S.L. Physical Interaction between Aldolase and Vacuolar H+-ATPase Is Essential for the Assembly and Activity of the Proton Pump. J. Biol. Chem. 2007, 282, 24495–24503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.-M.; Park, J.-H.; He, X.; Levy, B.; Chen, F.; Arai, K.; Adler, D.A.; Disteche, C.M.; Koch, J.; Sandhoff, K.; et al. The Human Acid Ceramidase Gene (ASAH): Structure, Chromosomal Location, Mutation Analysis, and Expression. Genomics 1999, 62, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Ni, X.; Morales, C.R. The lysosomal trafficking of acid sphingomyelinase is mediated by sortilin and mannose 6-phosphate receptor. Traffic 2006, 7, 889–902. [Google Scholar] [CrossRef] [PubMed]
- Pohl, S.; Angermann, A.; Jeschke, A.; Hendrickx, G.; Yorgan, T.A.; Makrypidi-Fraune, G.; Steigert, A.; Kuehn, S.C.; Rolvien, T.; Schweizer, M.; et al. The Lysosomal Protein Arylsulfatase B Is a Key Enzyme Involved in Skeletal Turnover. J. Bone Miner. Res. 2018, 33, 2186–2201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayman, A.R.; Macary, P.; Lehner, P.J.; Cox, T.M. Tartrate-resistant Acid Phosphatase (Acp 5): Identification in Diverse Human Tissues and Dendritic Cells. J. Histochem. Cytochem. 2001, 49, 675–683. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Kar, R.; Dutta, S.; Pati, H.P.; Saxena, R. Niemann-Pick disease, type B with TRAP-positive storage cells and secondary sea blue histiocytosis. Eur. J. Histochem. 2009, 53, e22. [Google Scholar] [CrossRef] [Green Version]
- Brömme, D.; Li, Z.; Barnes, M.; Mehler, E. Human Cathepsin V Functional Expression, Tissue Distribution, Electrostatic Surface Potential, Enzymatic Characterization, and Chromosomal Localization. Biochemistry 1999, 38, 2377–2385. [Google Scholar] [CrossRef]
- Esmaeilzadeh, F.; Bladt, S.; Beukinga, I.; Wijns, W.; van de Borne, P.; Pradier, O.; Argacha, J.-F.; Wauters, A. Pro-thrombotic effect of exercise in a polluted environment: A P-selectin- and CD63-related platelet activation effect. Thromb. Haemost. 2015, 113, 118–124. [Google Scholar] [CrossRef]
- Armstrong, L.W.; Rom, W.N.; Martiniuk, F.T. The Gene for Lysosomal Protein CD63 Is Normal in Patients with Hermansky-Pudlak Syndrome. Lung 1998, 176, 249–256. [Google Scholar] [CrossRef]
- Bisretinoids of the Retina: Photo-Oxidation, Iron-Catalyzed Oxidation, and Disease Consequences—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/34573014/ (accessed on 2 June 2023).
- Lu, Y.W.; Claypool, S.M. Disorders of phospholipid metabolism: An emerging class of mitochondrial disease due to defects in nuclear genes. Front Genet. 2015, 6, 3. [Google Scholar] [CrossRef]
- Torres, S.; Balboa, E.; Zanlungo, S.; Enrich, C.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Lysosomal and Mitochondrial Liaisons in Niemann-Pick Disease. Front. Physiol. 2017, 8, 982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Defective Autophagy, Mitochondrial Clearance and Lipophagy in Niemann-Pick Type B Lymphocytes—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/27798705/ (accessed on 4 July 2023).
- Wu, J.; Zhang, Y.; Wu, Q.; Xie, D.; Dai, W.; Zhang, X.; Yang, Z.; Wang, D. Integrative analyses of myocardial lipidome and proteome implicate mitochondrial dysfunction in lethal ventricular tachyarrhythmia (LVTA) induced by acute myocardial ischemia (AMI). J. Proteom. 2019, 197, 14–22. [Google Scholar] [CrossRef]
- Jiang, Z.; Shen, T.; Huynh, H.; Fang, X.; Han, Z.; Ouyang, K. Cardiolipin Regulates Mitochondrial Ultrastructure and Function in Mammalian Cells. Genes 2022, 13, 1889. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Yang, J.; Cheng, H.; Yang, K.; Abendschein, D.R.; Gross, R.W. Shotgun Lipidomics Identifies Cardiolipin Depletion in Diabetic Myocardium Linking Altered Substrate Utilization with Mitochondrial Dysfunction. Biochemistry 2005, 44, 16684–16694. [Google Scholar] [CrossRef] [PubMed]
- Huch, M.; Gehart, H.; Van Boxtel, R.; Hamer, K.; Blokzijl, F.; Verstegen, M.M.; Ellis, E.; Van Wenum, M.; Fuchs, S.A.; de Ligt, J.; et al. Long-term culture of genome-stable bipotent stem cells from adult human liver. Cell 2015, 160, 299–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surma, M.A.; Gerl, M.J.; Herzog, R.; Helppi, J.; Simons, K.; Klose, C. Mouse lipidomics reveals inherent flexibility of a mammalian lipidome. Sci. Rep. 2021, 11, 19364. [Google Scholar] [CrossRef] [PubMed]
- Ejsing, C.S.; Sampaio, J.L.; Surendranath, V.; Duchoslav, E.; Ekroos, K.; Klemm, R.W.; Simons, K.; Shevchenko, A. Global analysis of the yeast lipidome by quantitative shotgun mass spectrometry. Proc. Natl. Acad. Sci. USA 2009, 106, 2136–2141. [Google Scholar] [CrossRef]
- Liebisch, G.; Binder, M.; Schifferer, R.; Langmann, T.; Schulz, B.; Schmitz, G. High throughput quantification of cholesterol and cholesteryl ester by electrospray ionization tandem mass spectrometry (ESI-MS/MS). Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2006, 1761, 121–128. [Google Scholar] [CrossRef]
- Herzog, R.; Schuhmann, K.; Schwudke, D.; Sampaio, J.L.; Bornstein, S.R.; Schroeder, M.; Shevchenko, A. LipidXplorer: A Software for Consensual Cross-Platform Lipidomics. PLoS ONE 2012, 7, e29851. [Google Scholar] [CrossRef] [Green Version]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomez-Mariano, G.; Perez-Luz, S.; Ramos-Del Saz, S.; Matamala, N.; Hernandez-SanMiguel, E.; Fernandez-Prieto, M.; Gil-Martin, S.; Justo, I.; Marcacuzco, A.; Martinez-Delgado, B. Acid Sphingomyelinase Deficiency Type B Patient-Derived Liver Organoids Reveals Altered Lysosomal Gene Expression and Lipid Homeostasis. Int. J. Mol. Sci. 2023, 24, 12645. https://doi.org/10.3390/ijms241612645
Gomez-Mariano G, Perez-Luz S, Ramos-Del Saz S, Matamala N, Hernandez-SanMiguel E, Fernandez-Prieto M, Gil-Martin S, Justo I, Marcacuzco A, Martinez-Delgado B. Acid Sphingomyelinase Deficiency Type B Patient-Derived Liver Organoids Reveals Altered Lysosomal Gene Expression and Lipid Homeostasis. International Journal of Molecular Sciences. 2023; 24(16):12645. https://doi.org/10.3390/ijms241612645
Chicago/Turabian StyleGomez-Mariano, Gema, Sara Perez-Luz, Sheila Ramos-Del Saz, Nerea Matamala, Esther Hernandez-SanMiguel, Marta Fernandez-Prieto, Sara Gil-Martin, Iago Justo, Alberto Marcacuzco, and Beatriz Martinez-Delgado. 2023. "Acid Sphingomyelinase Deficiency Type B Patient-Derived Liver Organoids Reveals Altered Lysosomal Gene Expression and Lipid Homeostasis" International Journal of Molecular Sciences 24, no. 16: 12645. https://doi.org/10.3390/ijms241612645
APA StyleGomez-Mariano, G., Perez-Luz, S., Ramos-Del Saz, S., Matamala, N., Hernandez-SanMiguel, E., Fernandez-Prieto, M., Gil-Martin, S., Justo, I., Marcacuzco, A., & Martinez-Delgado, B. (2023). Acid Sphingomyelinase Deficiency Type B Patient-Derived Liver Organoids Reveals Altered Lysosomal Gene Expression and Lipid Homeostasis. International Journal of Molecular Sciences, 24(16), 12645. https://doi.org/10.3390/ijms241612645