

Recent Advances in the Development of Pyrazole Derivatives as Anticancer Agents

 and
and
Abstract
:1. Introduction
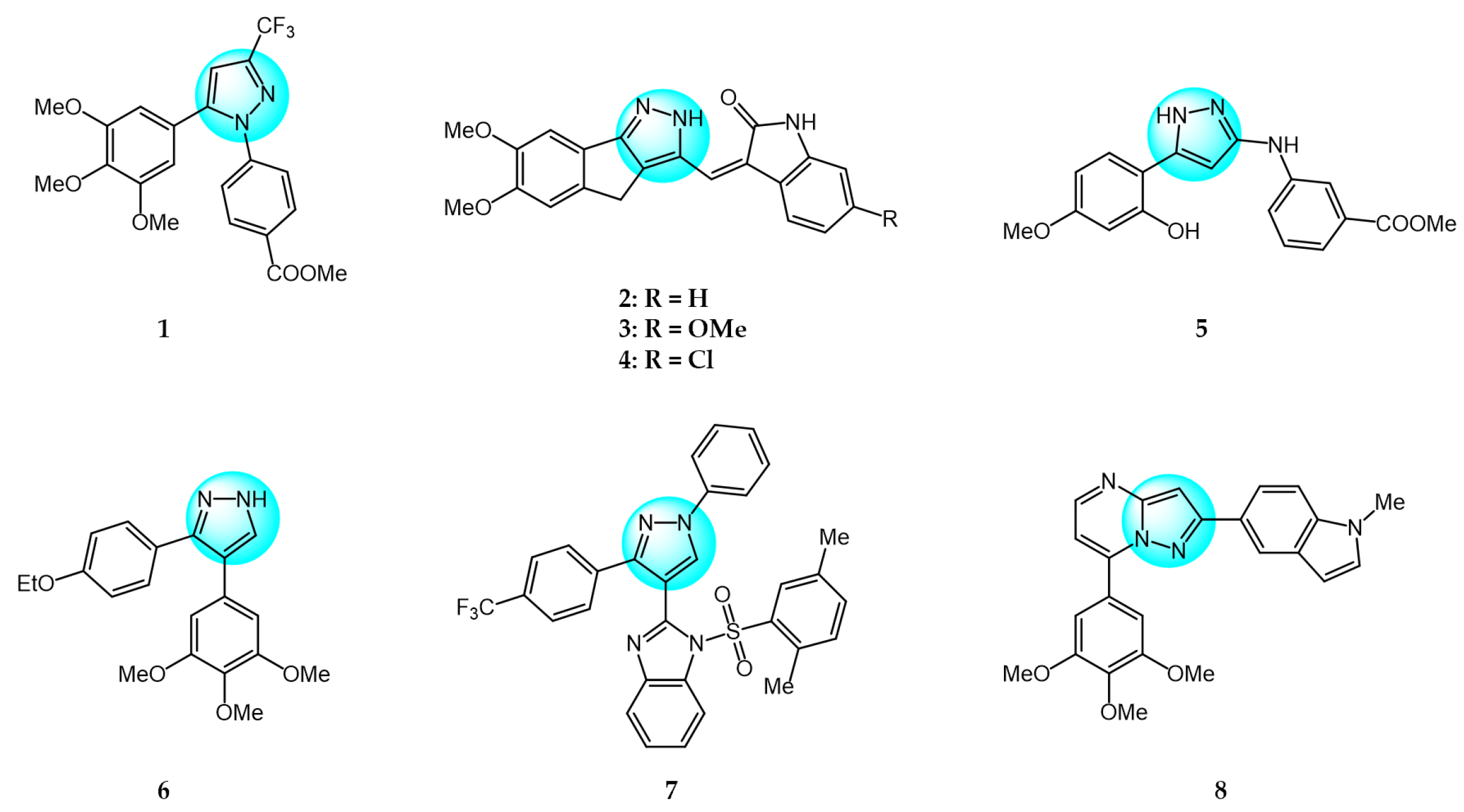
2. Tubulin Polymerization Inhibitors
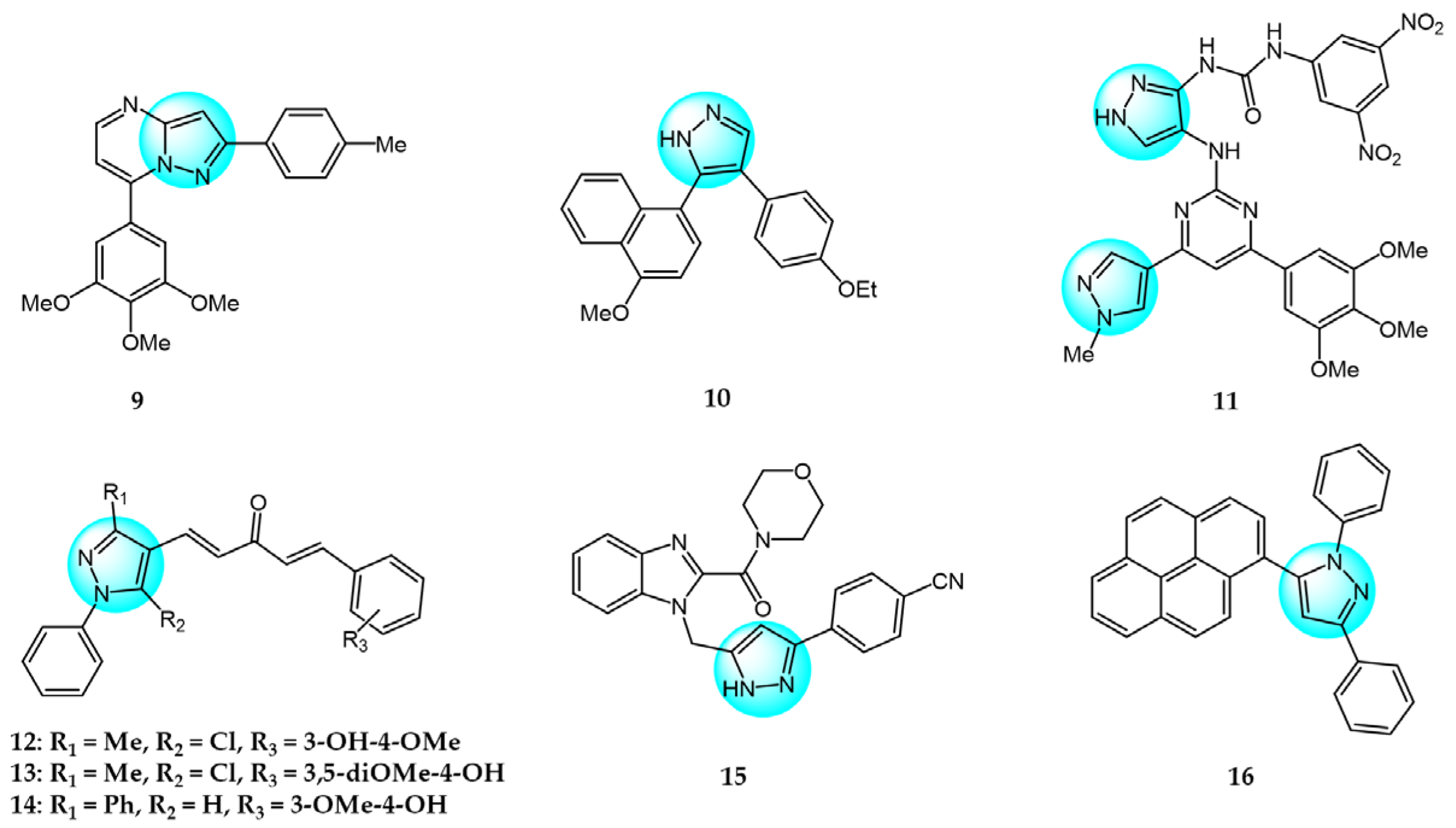
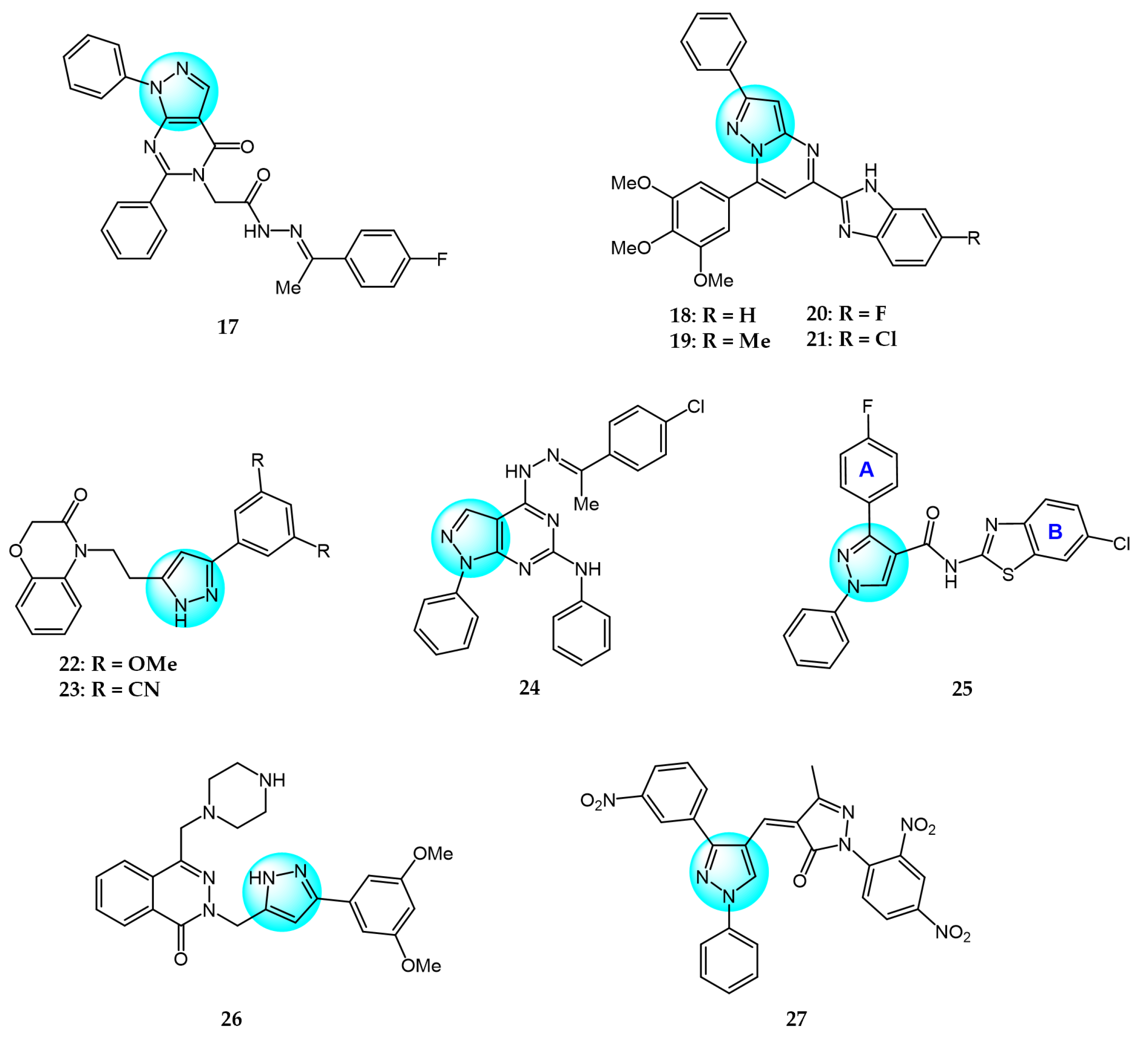
3. Kinase Inhibitors
4. Multitargeted Kinase Inhibitors
5. Inhibitors of Other Targets
5.1. DNA Binding Agents
5.2. Topoisomerase Inhibitors
5.3. Eg5 Inhibitors
5.4. MDM2 Inhibitors
5.5. COX-2 Inhibitors
5.6. hCA IX Isoenzyme Inhibitors
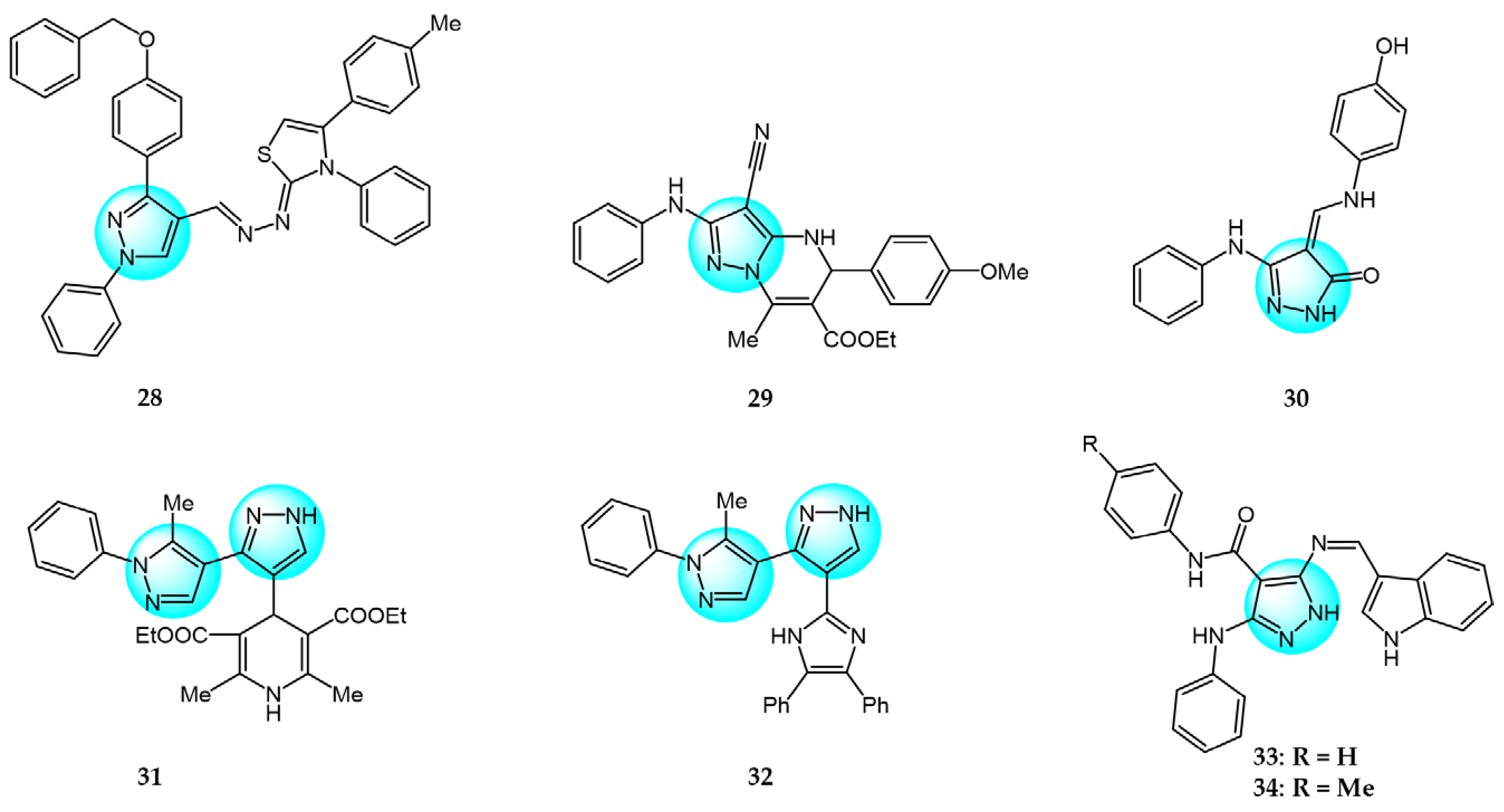
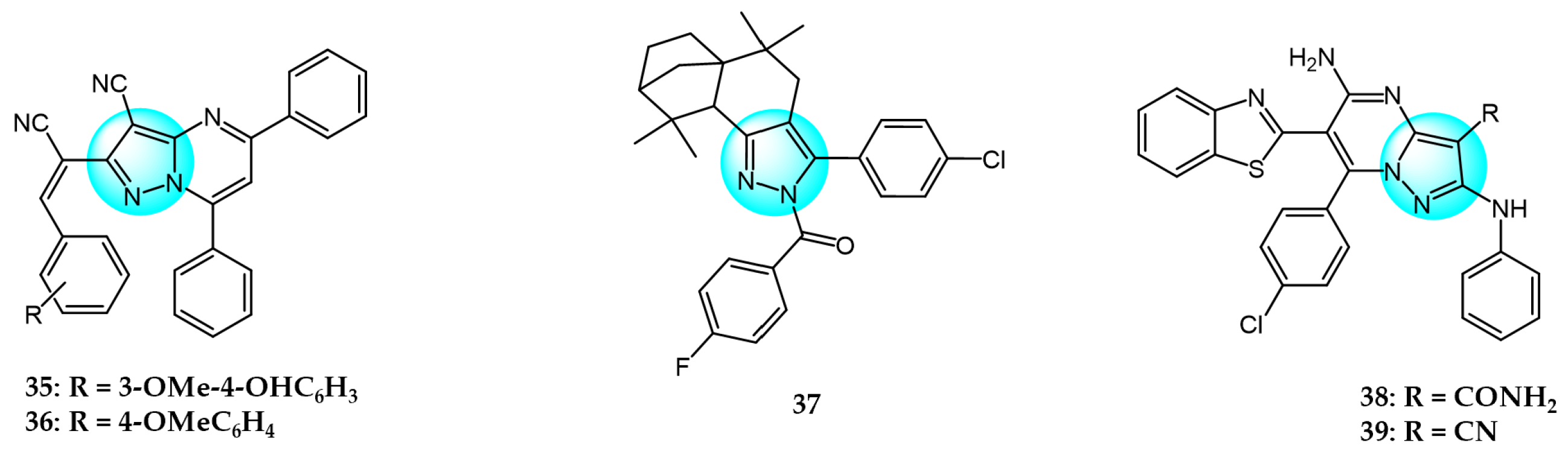
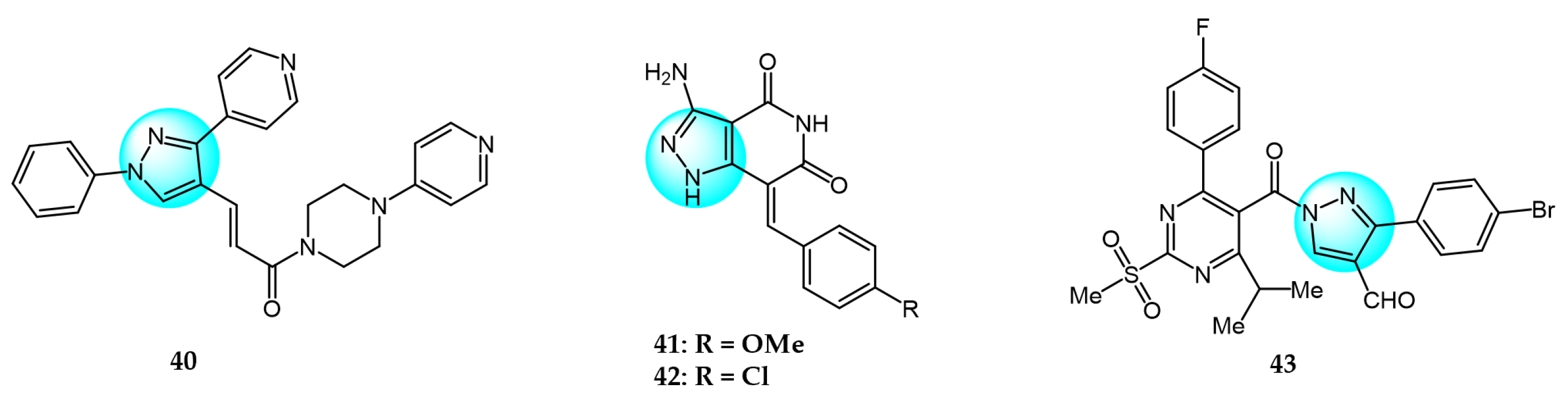
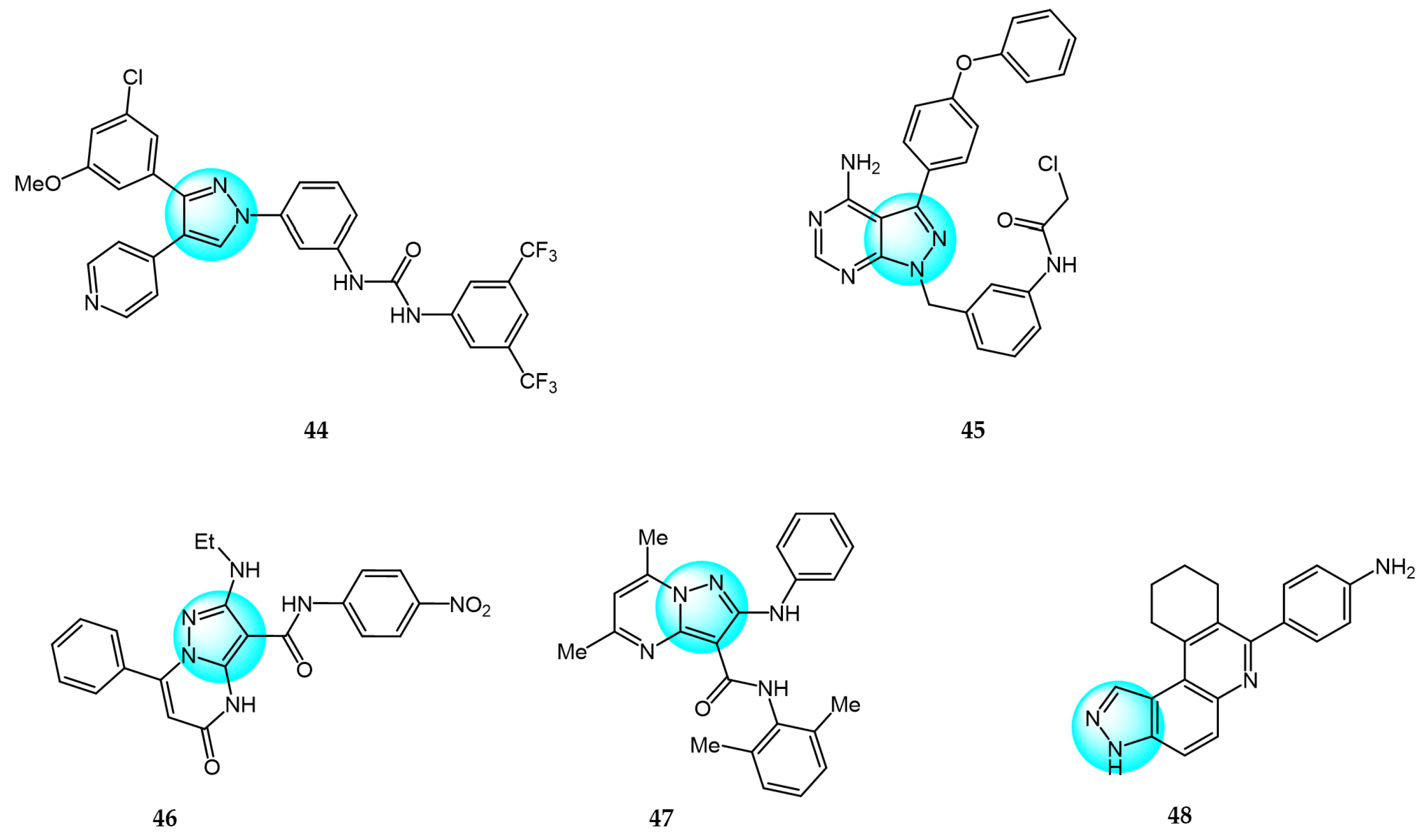
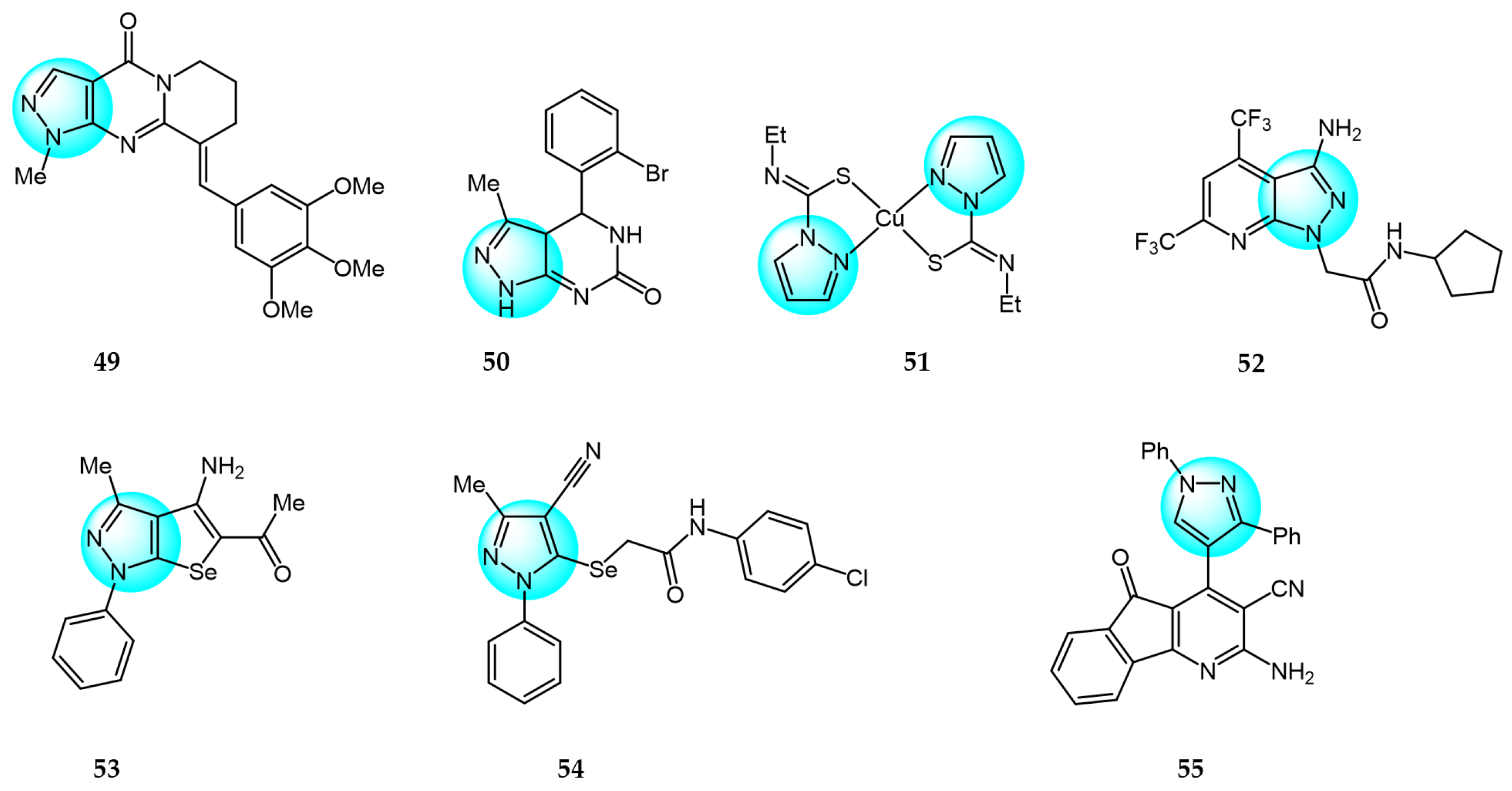
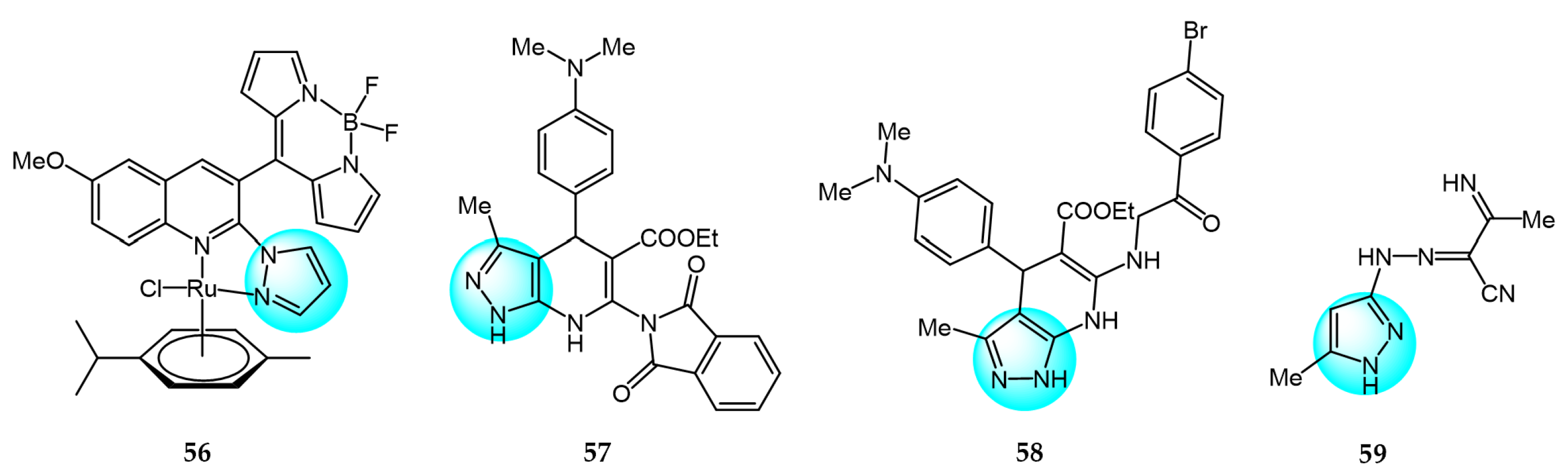
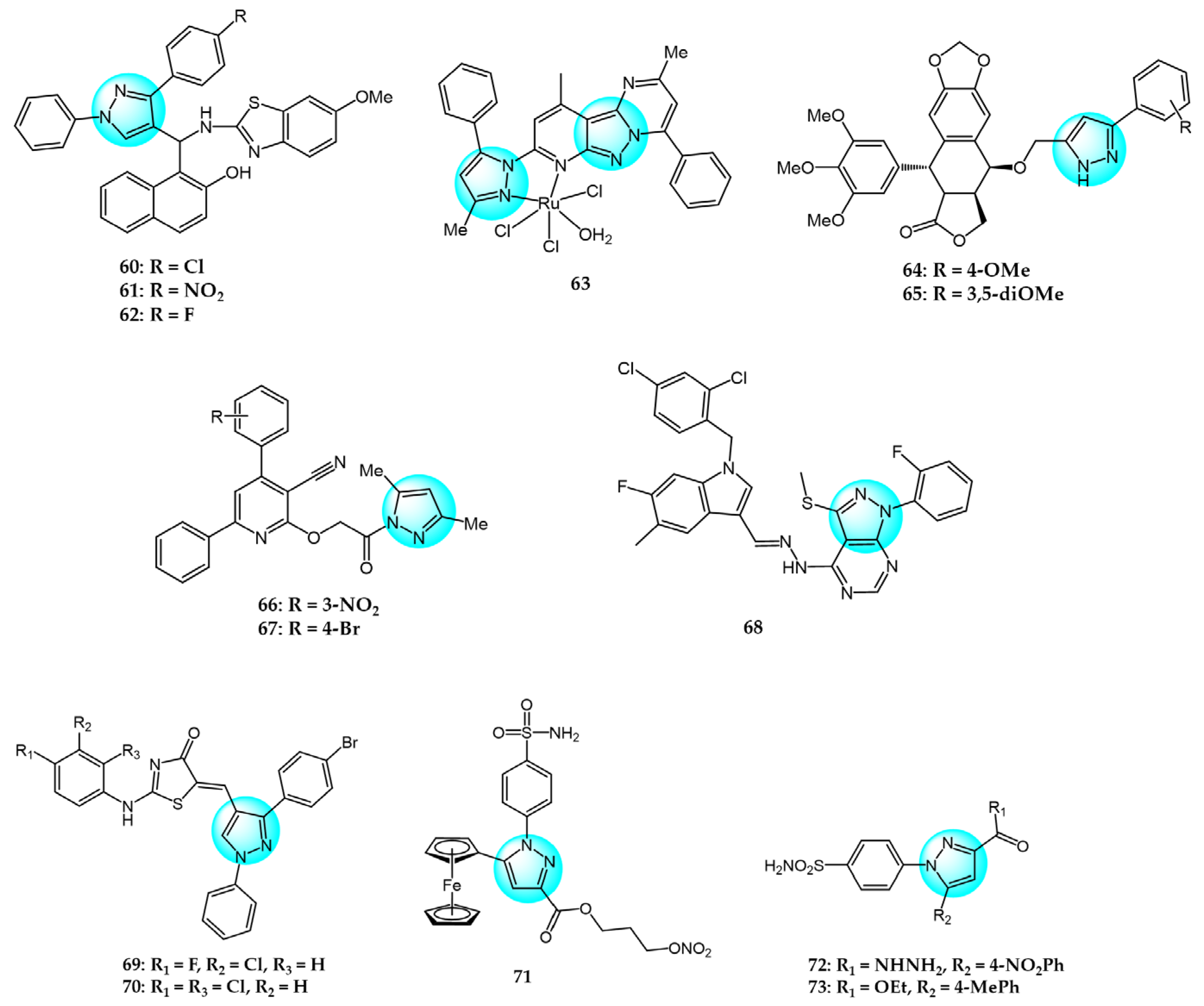
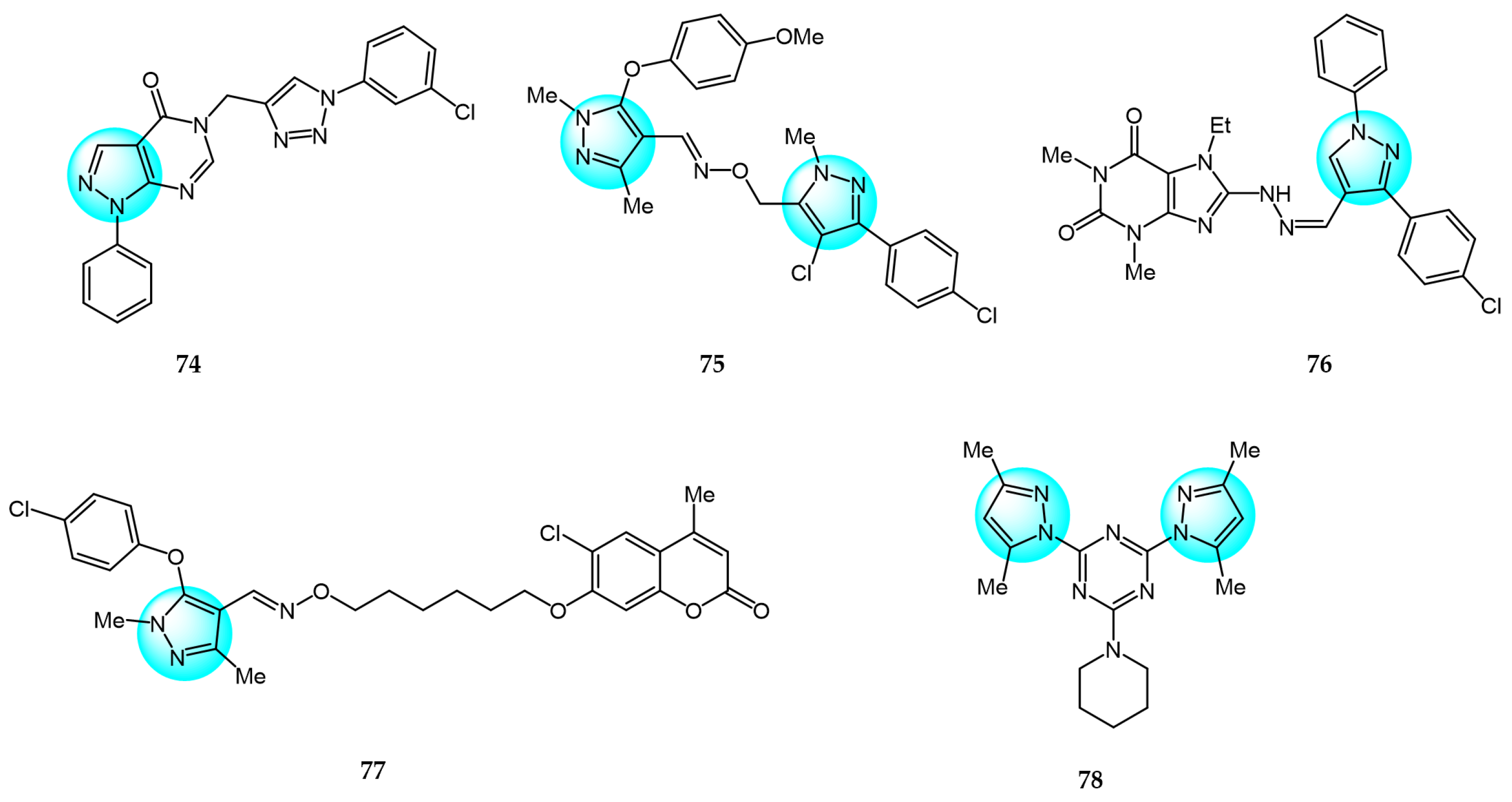
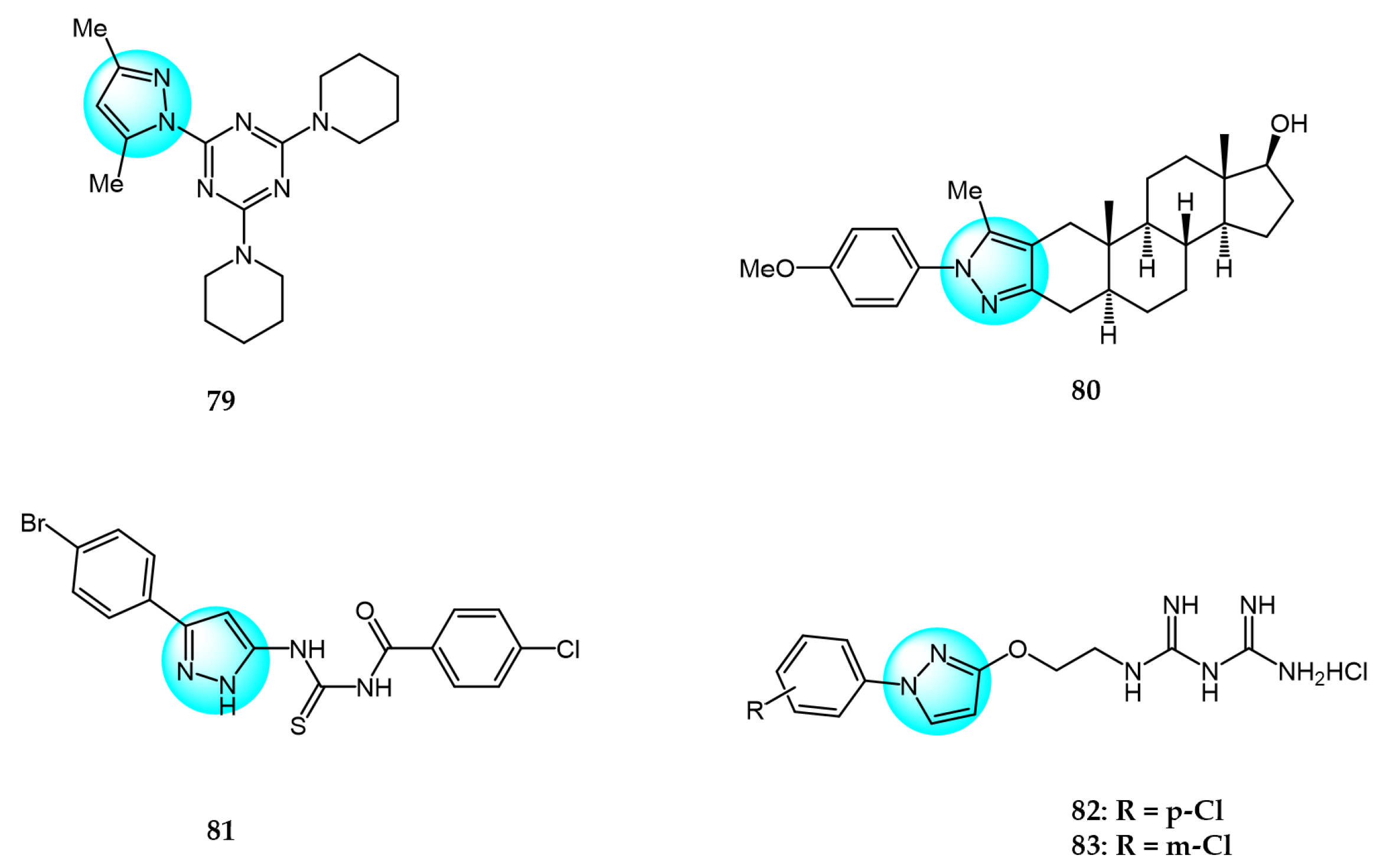
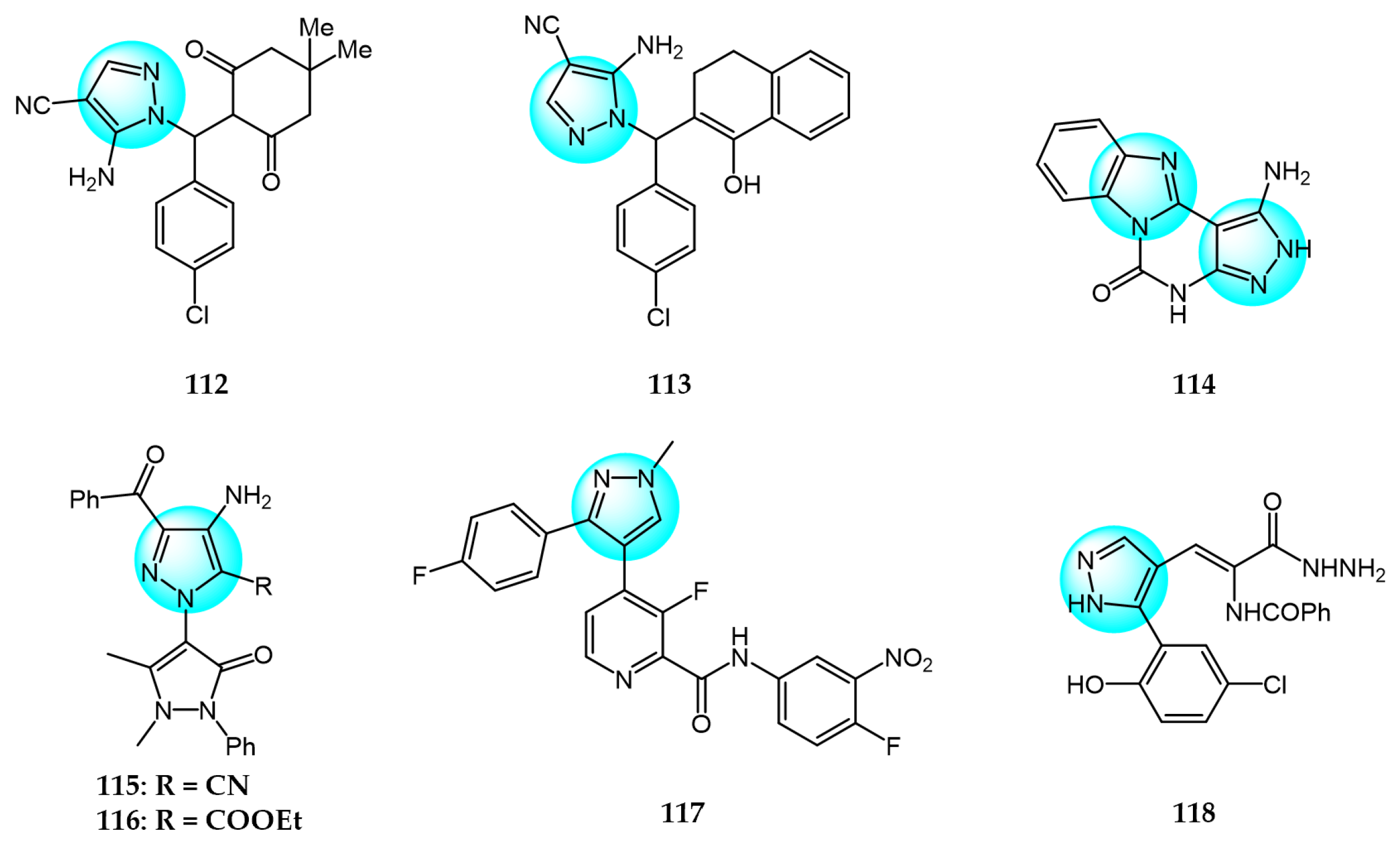
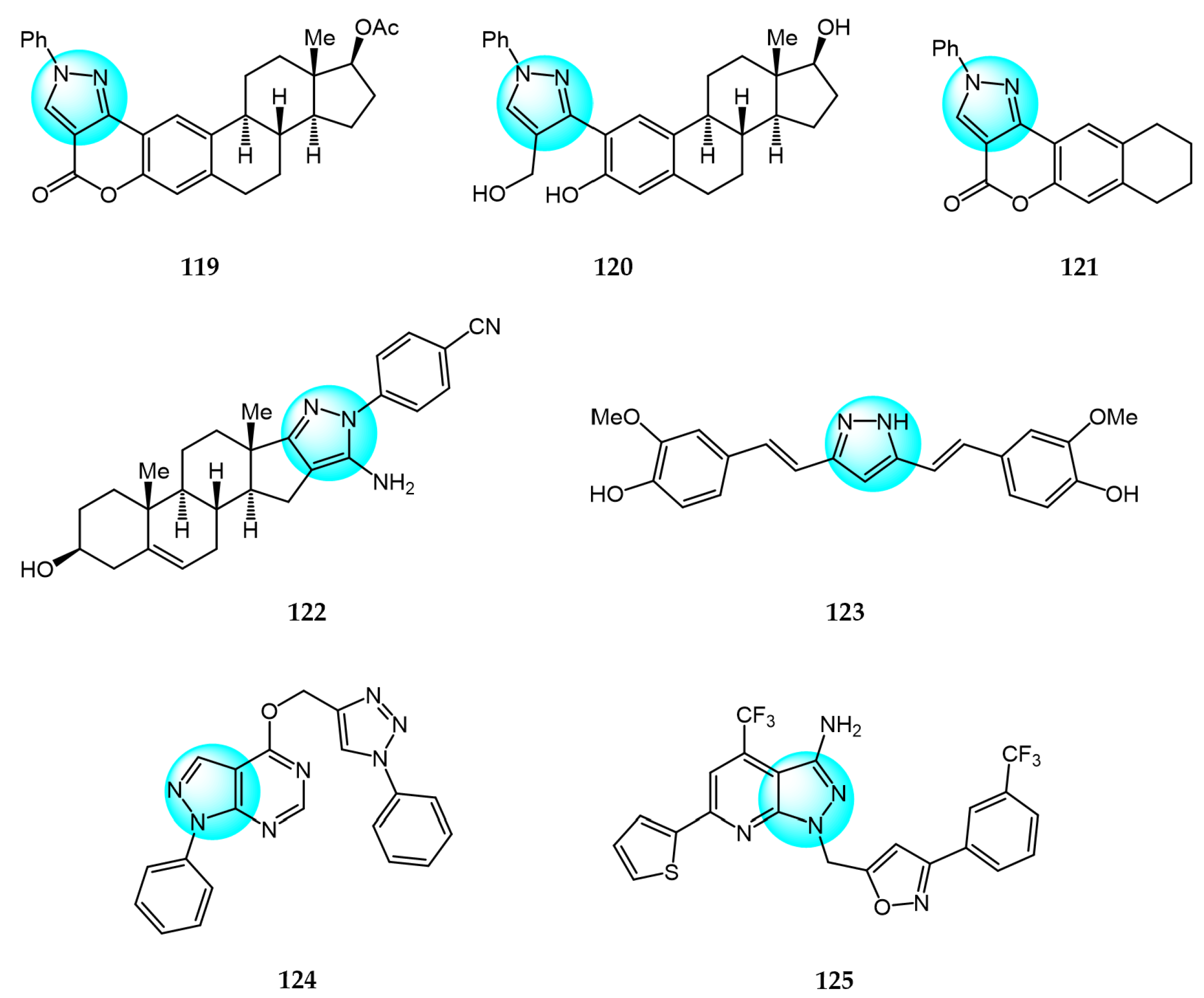
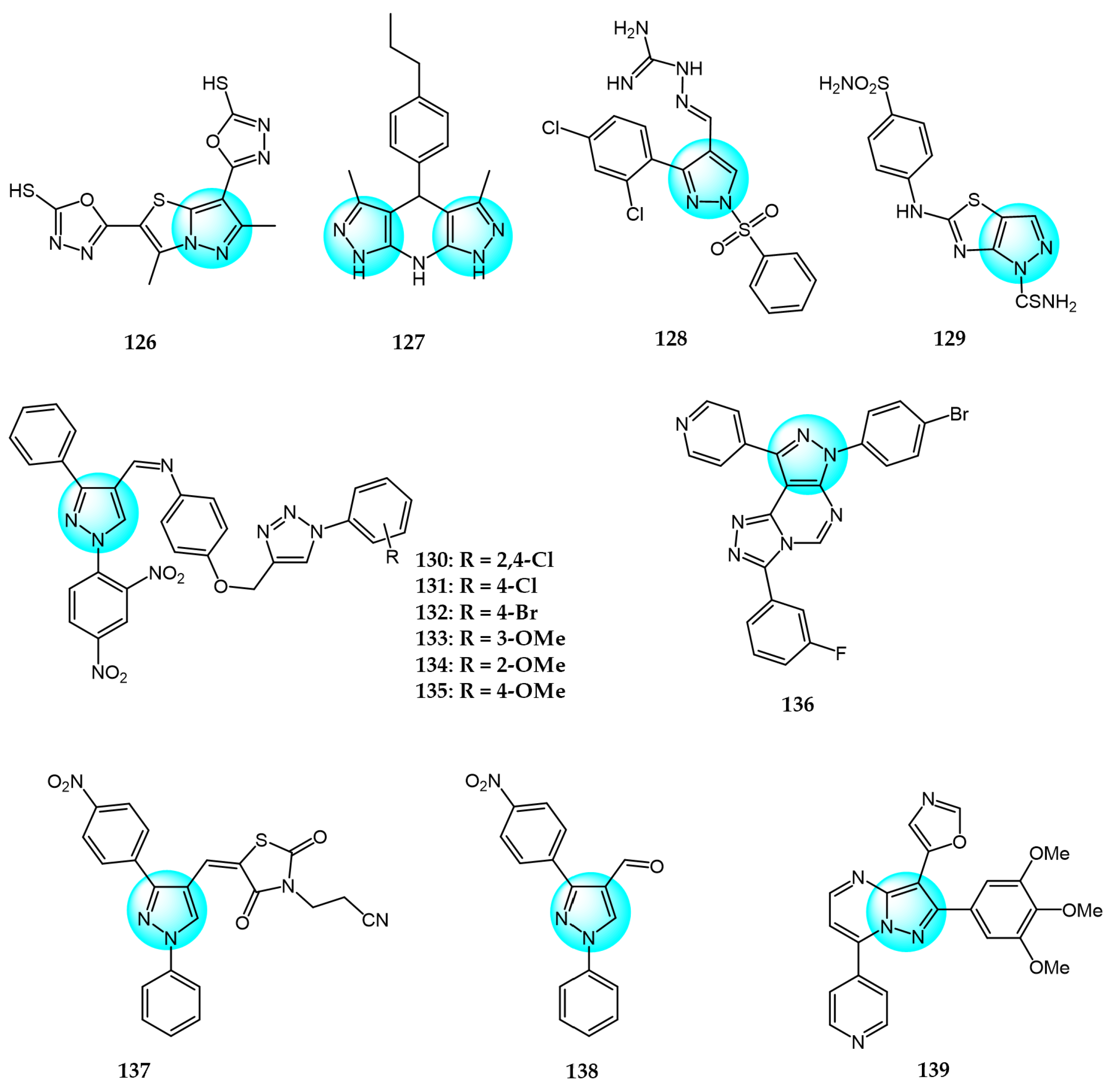
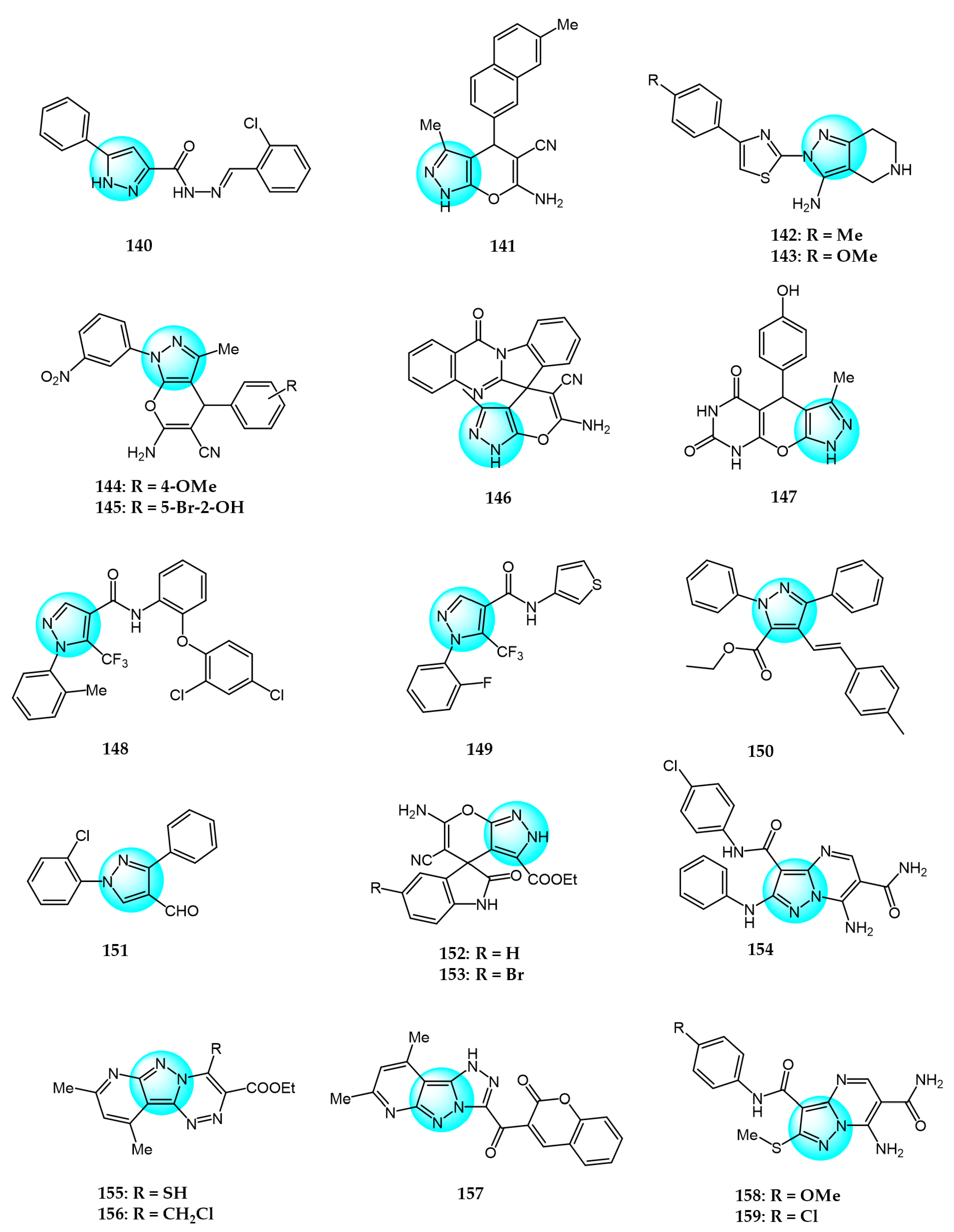
6. Pyrazole Derivatives with Undefined Mechanisms
7. Future Perspective
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hulvat, M.C. Cancer incidence and trends. Surg. Clin. 2020, 100, 469–481. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA-Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.J.; Lei, K.F.; Han, F. Tumor microenvironment: Recent advances in various cancer treatments. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 3855–3864. [Google Scholar]
- Ali, I.; Lone, M.N.; Al-Othman, Z.A.; Al-Warthan, A.; Sanagi, M.M. Heterocyclic scaffolds: Centrality in anticancer drug development. Curr. Drug Targets 2015, 16, 711–734. [Google Scholar] [CrossRef]
- Bekhit, A.A.; Nasralla, S.N.; El-Agroudy, E.J.; Hamouda, N.; El-Fattah, A.A.; Bekhit, S.A.; Amagase, K.; Ibrahim, T.M. Investigation of the anti-inflammatory and analgesic activities of promising pyrazole derivative. Eur. J. Pharm. Sci. 2022, 168, 106080. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.A. Antibacterial Pyrazoles: Tackling resistant bacteria. Future Med. Chem. 2022, 14, 343–362. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Qu, K.; Zhang, W.; Wang, X. Pharmacological activity of pyrazole derivatives as an anticonvulsant for benefit against Epilepsy. Neuroimmunomodulation 2021, 28, 90–98. [Google Scholar] [CrossRef]
- Sau, M.C.; Rajesh, Y.; Mandal, M.; Bhattacharjee, M. Copper catalyzed regioselective N-alkynylation of pyrazoles and evaluation of the anticancer activity of ethynyl-pyrazoles. ChemistrySelect 2018, 3, 3511–3515. [Google Scholar] [CrossRef]
- Kumar, G.; Siva Krishna, V.; Sriram, D.; Jachak, S.M. Pyrazole-coumarin and pyrazole-quinoline chalcones as potential antitubercular agents. Arch. Pharm. 2020, 353, 2000077. [Google Scholar] [CrossRef]
- Rao, P.; Knaus, E.E. Evolution of nonsteroidal anti-inflammatory drugs (NSAIDs): Cyclooxygenase (COX) inhibition and beyond. J. Pharm. Pharm. Sci. 2008, 11, 81s–110s. [Google Scholar] [CrossRef] [Green Version]
- Krasselt, M.; Baerwald, C. Celecoxib for the treatment of musculoskeletal arthritis. Expert. Opin. Pharmacother. 2019, 20, 1689–1702. [Google Scholar] [CrossRef]
- Shaw, A.T.; Yasothan, U.; Kirkpatrick, P. Crizotinib. Nat. Rev. Drug Discov. 2011, 10, 897–898. [Google Scholar] [CrossRef] [PubMed]
- Li, G.C.; Cheng, Y.F.; Han, C.; Song, C.; Huang, N.; Du, Y.F. Pyrazole-containing pharmaceuticals: Target, pharmacological activity, and their SAR studies. RSC Med. Chem. 2022, 13, 1300–1321. [Google Scholar] [CrossRef] [PubMed]
- Derosa, G.; Maffioli, P. Anti-obesity drugs: A review about their effects and their safety. Expert Opin. Drug Saf. 2012, 11, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Greig, S.L.; Garnock-Jones, K.P. Apixaban: A review in venous thromboembolism. Drugs 2016, 76, 1493–1504. [Google Scholar] [CrossRef] [PubMed]
- Luttinger, D.; Hlasta, D.J. Chapter 3 antidepressant agents. Annu. Rep. Med. Chem. 1987, 22, 21–30. [Google Scholar]
- Ozbeyli, D.; Gokalp, A.G.; Koral, T.; Ocal, O.Y.; Dogan, B.; Akakin, D.; Yuksel, M.; Kasimay, O. Protective effect of exercise and sildenafil on acute stress and cognitive function. Physiol. Behav. 2015, 151, 230–237. [Google Scholar] [CrossRef]
- Spitz, I.M.; Novis, B.H.; Ebert, R.; Trestian, S.; LeRoith, D.; Creutzfeldt, W. Betazole Induced GIP secretion is not mediated by gastric HCl. Metabolism 1982, 31, 380–382. [Google Scholar] [CrossRef]
- McMartin, K.; Jacobsen, D.; Hovda, K.E. Antidotes for poisoning by alcohols that form toxic metabolites. Br. J. Clin. Pharmacol. 2016, 81, 505–515. [Google Scholar] [CrossRef] [Green Version]
- Foster, A.C.; Kemp, J.A. Glutamate- and GABA-based CNS therapeutics. Curr. Opin. Pharmacol. 2006, 6, 7–17. [Google Scholar] [CrossRef]
- Mets, M.A.; Volkerts, E.R.; Olivier, B.; Verster, J.C. Effect of hypnotic drugs on body balance and standing steadiness. Sleep. Med. Rev. 2010, 14, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Fiebich, B.L.; Hofer, T.J.; Lieb, K.; Huell, M.; Butcher, R.D.; Schumann, G.; Schulze-Osthoff, K.; Bauer, J. The non-steroidal anti-inflammatory drug tepoxalin inhibits interleukin-6 and alpha(1)-anti-chymotrypsin synthesis in astrocytes by preventing degradation of I kappa B-alpha. Neuropharmacology 1999, 38, 1325–1333. [Google Scholar] [CrossRef] [PubMed]
- King, J.N.; Arnaud, J.P.; Goldenthal, E.I.; Gruet, P.; Jung, M.; Seewald, W.; Lees, P. Robenacoxib in the dog: Target species safety in relation to extent and duration of inhibition of COX-1 and COX-2. J. Vet. Pharmacol. Ther. 2011, 34, 298–311. [Google Scholar] [CrossRef]
- Huang, H.S.; Wang, R.; Chen, W.J.; Chen, J.Z.; Gong, S.S.; Sun, Q. The first chemical synthesis of pyrazofurin 5′-triphosphate. Tetrahedron Lett. 2018, 59, 3423–3427. [Google Scholar] [CrossRef]
- Vemuri, V.K.; Janero, D.R.; Makriyannis, A. Pharmacotherapeutic targeting of the endocannabinoid signaling system: Drugs for obesity and the metabolic syndrome. Physiol. Behav. 2008, 93, 671–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matiadis, D.; Sagnou, M. Pyrazoline hybrids as promising anticancer agents: An up-to-date overview. Int. J. Mol. Sci. 2020, 21, 5507. [Google Scholar] [CrossRef]
- Asati, V.; Anant, A.; Patel, P.; Kaur, K.; Gupta, G.D. Pyrazolopyrimidines as anticancer agents: A review on structural and target-based approaches. Eur. J. Med. Chem. 2021, 225, 113781. [Google Scholar] [CrossRef]
- Zhao, Z.; Dai, X.; Li, C.; Wang, X.; Tian, J.; Feng, Y.; Xie, J.; Ma, C.; Nie, Z.; Fan, P.; et al. Pyrazolone structural motif in medicinal chemistry: Retrospect and prospect. Eur. J. Med. Chem. 2020, 186, 111893. [Google Scholar] [CrossRef]
- Zhu, T.; Wang, S.H.; Li, D.; Wang, S.Y.; Liu, X.; Song, J.; Wang, Y.T.; Zhang, S.Y. Progress of tubulin polymerization activity detection methods. Bioorg. Med. Chem. Lett. 2021, 37, 127698. [Google Scholar] [CrossRef]
- Chen, P.; Zhuang, Y.X.; Diao, P.C.; Yang, F.; Wu, S.Y.; Lv, L.; You, W.W.; Zhao, P.L. Synthesis, biological evaluation, and molecular docking investigation of 3-amidoindoles as potent tubulin polymerization inhibitors. Eur. J. Med. Chem. 2019, 162, 525–533. [Google Scholar] [CrossRef]
- Siddiqui-Jain, A.; Hoj, J.P.; Cescon, D.W.; Hansen, M.D. Pharmacology and in vivo efficacy of pyridine-pyrimidine amides that inhibit microtubule polymerization. Bioorg. Med. Chem. Lett. 2018, 28, 934–941. [Google Scholar] [CrossRef]
- Steinmetz, M.O.; Prota, A.E. Microtubule-targeting agents: Strategies to hijack the cytoskeleton. Trends Cell Biol. 2018, 28, 776–792. [Google Scholar] [CrossRef] [PubMed]
- Hura, N.; Naaz, A.; Prassanawar, S.S.; Guchhait, S.K.; Panda, D. Drug-clinical agent molecular hybrid: Synthesis of diaryl(trifluoromethyl)pyrazoles as tubulin targeting anticancer agents. ACS Omega 2018, 3, 1955–1969. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Garikapati, K.R.; Shaik, A.B.; Makani, V.K.K.; Rahim, A.; Shareef, M.A.; Reddy, V.G.; Pal-Bhadra, M.; Kamal, A.; Kumar, C.G. Design, synthesis and biological evaluation of 1, 4-dihydro indeno[1,2-c] pyrazole linked oxindole analogues as potential anticancer agents targeting tubulin and inducing p53 dependent apoptosis. Eur. J. Med. Chem. 2018, 144, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.J.; Tang, L.Q.; Zhang, C.M.; Liu, Z.P. Synthesis of novel pyrazole derivatives and their tumor cell growth inhibitory activity. Molecules 2019, 24, 279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romagnoli, R.; Oliva, P.; Salvador, M.K.; Camacho, M.E.; Padroni, C.; Brancale, A.; Ferla, S.; Hamel, E.; Ronca, R.; Grillo, E.; et al. Design, synthesis and biological evaluation of novel vicinal diaryl-substituted 1H-pyrazole analogues of combretastatin A-4 as highly potent tubulin polymerization inhibitors. Eur. J. Med. Chem. 2019, 181, 111577. [Google Scholar] [CrossRef]
- Wang, Y.T.; Shi, T.Q.; Zhu, H.L.; Liu, C.H. Synthesis, biological evaluation and molecular docking of benzimidazole grafted benzsulfamide-containing pyrazole ring derivatives as novel tubulin polymerization inhibitors. Bioorg. Med. Chem. 2019, 27, 502–515. [Google Scholar] [CrossRef]
- Li, G.; Wang, Y.; Li, L.; Ren, Y.; Deng, X.; Liu, J.; Wang, W.; Luo, M.; Liu, S.; Chen, J. Design, synthesis, and bioevaluation of pyrazolo[1,5-a]pyrimidine derivatives as tubulin polymerization inhibitors targeting the colchicine binding site with potent anticancer activities. Eur. J. Med. Chem. 2020, 202, 112519. [Google Scholar] [CrossRef]
- Wang, G.; Liu, W.; Peng, Z.; Huang, Y.; Gong, Z.; Li, Y. Design, synthesis, molecular modeling, and biological evaluation of pyrazole-naphthalene derivatives as potential anticancer agents on MCF-7 breast cancer cells by inhibiting tubulin polymerization. Bioorg. Chem. 2020, 103, 104141. [Google Scholar] [CrossRef]
- Cherukumalli, P.K.R.; Tadiboina, B.R.; Gulipalli, K.C.; Bodige, S.; Badavath, V.N.; Sridhar, G.; Gangarapu, K. Design and synthesis of novel urea derivatives of pyrimidine-pyrazoles as anticancer agents. J. Mol. Struct. 2022, 1251, 131937. [Google Scholar] [CrossRef]
- Doan, N.Q.H.; Nguyen, N.T.K.; Duong, V.B.; Nguyen, H.T.T.; Vong, L.B.; Duong, D.N.; Nguyen, N.T.; Nguyen, T.L.T.; Do, T.T.H.; Truong, T.N. Synthesis, biological evaluation, and molecular modeling studies of 1-aryl-1H-pyrazole-fused Curcumin analogues as anticancer agents. ACS Omega 2022, 7, 33963–33984. [Google Scholar] [CrossRef]
- Sagam, R.R.; Nukala, S.K.; Nagavath, R.; Sirassu, N.; Mohammod, M.; Manchal, R.; Thirukovela, N.S. Synthesis of new morpholine-benzimidazole-pyrazole hybrids as tubulin polymerization inhibiting anticancer agents. J. Mol. Struct. 2022, 1268, 133692. [Google Scholar] [CrossRef]
- Sar, D.; Srivastava, I.; Misra, S.K.; Ostadhossein, F.; Fathi, P.; Pan, D. Copper-catalyzed syntheses of pyrene-pyrazole pharmacophores and structure activity studies for tubulin polymerization. ACS Omega 2018, 3, 6378–6387. [Google Scholar] [CrossRef] [PubMed]
- Nitulescu, G.M.; Stancov, G.; Seremet, O.C.; Nitulescu, G.; Mihai, D.P.; Duta-Bratu, C.G.; Barbuceanu, S.F.; Olaru, O.T. The importance of the pyrazole scaffold in the design of protein kinases inhibitors as targeted anticancer therapies. Molecules 2023, 28, 5359. [Google Scholar] [CrossRef]
- Gaber, A.A.; El-Morsy, A.M.; Sherbiny, F.F.; Bayoumi, A.H.; El-Gamal, K.M.; El-Adl, K.; Al-Karmalawy, A.A.; Ezz Eldin, R.R.; Saleh, M.A.; Abulkhair, H.S. Pharmacophore-linked pyrazolo[3,4-d]pyrimidines as EGFR-TK inhibitors: Synthesis, anticancer evaluation, pharmacokinetics, and in silico mechanistic studies. Arch. Pharm. 2021, e2100258. [Google Scholar] [CrossRef] [PubMed]
- Bagul, C.; Rao, G.K.; Veena, I.; Kulkarni, R.; Tamboli, J.R.; Akunuri, R.; Shaik, S.P.; Pal-Bhadra, M.; Kamal, A. Benzimidazole-linked pyrazolo[1,5-a]pyrimidine conjugates: Synthesis and detail evaluation as potential anticancer agents. Mol. Divers. 2023, 27, 1185–1202. [Google Scholar] [CrossRef] [PubMed]
- Benarjee, V.; Saritha, B.; Hari Gangadhar, K.; Sailaja, B.B.V. Synthesis of some new 1,4-benzoxazine-pyrazoles in water as EGFR targeting anticancer agents. J. Mol. Struct. 2022, 1265, 133188. [Google Scholar] [CrossRef]
- Gaber, A.A.; Sobhy, M.; Turky, A.; Abdulwahab, H.G.; Al-Karmalawy, A.A.; Elhendawy, M.A.; Radwan, M.M.; Elkaeed, E.B.; Ibrahim, I.M.; Elzahabi, H.S.A.; et al. Discovery of new 1H-pyrazolo[3,4-d]pyrimidine derivatives as anticancer agents targeting EGFRWT and EGFRT790M. J. Enzyme. Inhib. Med. Chem. 2022, 37, 2283–2303. [Google Scholar] [CrossRef]
- Malekan, M.; Ebrahimzadeh, M.A. Vascular endothelial growth factor receptors [VEGFR] as target in breast cancer treatment: Current status in preclinical and clinical studies and future directions. Curr. Top. Med. Chem. 2022, 22, 891–920. [Google Scholar] [CrossRef]
- Reddy, V.G.; Reddy, T.S.; Jadala, C.; Reddy, M.S.; Sultana, F.; Akunuri, R.; Bhargava, S.K.; Wlodkowic, D.; Srihari, P.; Kamal, A. Pyrazolo-benzothiazole hybrids: Synthesis, anticancer properties and evaluation of antiangiogenic activity using in vitro VEGFR-2 kinase and in vivo transgenic zebrafish model. Eur. J. Med. Chem. 2019, 182, 111609. [Google Scholar] [CrossRef]
- Badithapuram, V.; Nukala, K.S.; Dasari, G.; Thirukovela, S.N.; Bandari, S. Synthesis of some new phthalazine-piperazine-pyrazole conjugates; in vitro anti-cancer, ADMET and molecular docking studies. ChemistrySelect 2023, 8, e202204329. [Google Scholar] [CrossRef]
- Dawood, D.H.; Nossier, E.S.; Ali, M.M.; Mahmoud, A.E. Synthesis and molecular docking study of new pyrazole derivatives as potent anti-breast cancer agents targeting VEGFR-2 kinase. Bioorg. Chem. 2020, 101, 103916. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, L.; Hei, R.; Li, X.; Cai, H.; Wu, X.; Zheng, Q.; Cai, C. CDK inhibitors in cancer therapy, an overview of recent development. Am. J. Cancer Res. 2021, 11, 1913–1935. [Google Scholar]
- Harras, M.F.; Sabour, R. Design, synthesis and biological evaluation of novel 1,3,4-trisubstituted pyrazole derivatives as potential chemotherapeutic agents for hepatocellular carcinoma. Bioorg. Chem. 2018, 78, 149–157. [Google Scholar] [CrossRef]
- Ali, G.M.E.; Ibrahim, D.A.; Elmetwali, A.M.; Ismail, N.S.M. Design, synthesis and biological evaluation of certain CDK2 inhibitors based on pyrazole and pyrazolo[1,5-a] pyrimidine scaffold with apoptotic activity. Bioorg. Chem. 2019, 86, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kuthyala, S.; Sheikh, S.; Prabhu, A.; Rekha, P.D.; Karikannar, N.G.; Shankar, M.K. Synthesis, characterization, and anticancer studies of some pyrazole-based hybrid heteroatomics. ChemistrySelect 2020, 5, 10827–10834. [Google Scholar] [CrossRef]
- Hassan, A.S.; Moustafa, G.O.; Awad, H.M.; Nossier, E.S.; Mady, M.F. Design, synthesis, anticancer evaluation, enzymatic assays, and a molecular modeling study of novel pyrazole-indole hybrids. ACS Omega 2021, 6, 12361–12374. [Google Scholar] [CrossRef]
- Metwally, N.H.; Mohamed, M.S.; Deeb, E.A. Synthesis, anticancer evaluation, CDK2 inhibition, and apoptotic activity assessment with molecular docking modeling of new class of pyrazolo[1,5-a]pyrimidines. Res. Chem. Intermed. 2021, 47, 5027–5060. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, W.; Wu, C.; Wan, L.; Zhao, Y.; Zhang, C.; Gu, W.; Wang, S. Pyrazole ring-containing isolongifolanone derivatives as potential CDK2 inhibitors: Evaluation of anticancer activity and investigation of action mechanism. Biomed. Pharmacother. 2021, 139, 111663. [Google Scholar] [CrossRef]
- Khedr, M.A.; Zaghary, W.A.; Elsherif, G.E.; Azzam, R.A.; Elgemeie, G.H. Purine analogs: Synthesis, evaluation and molecular dynamics of pyrazolopyrimidines based benzothiazole as anticancer and antimicrobial CDK inhibitors. Nucleos. Nucleot. Nucl. 2023, 42, 77–104. [Google Scholar] [CrossRef]
- Noorolyai, S.; Shajari, N.; Baghbani, E.; Sadreddini, S.; Baradaran, B. The relation between PI3K/AKT signalling pathway and cancer. Gene 2019, 698, 120–128. [Google Scholar] [CrossRef]
- Guo, Y.J.; Pan, W.W.; Liu, S.B.; Shen, Z.F.; Xu, Y.; Hu, L.L. ERK/MAPK signalling pathway and tumorigenesis. Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demiroglu-Zergeroglu, A.; Ayvali, N.; Turhal, G.; Ceylan, H.; Baytas, S.N. Investigation of potent anticarcinogenic activity of 1, 3-diarylpyrazole acrylamide derivatives in vitro. J. Pharm. Pharmacol. 2018, 70, 1619–1629. [Google Scholar] [CrossRef] [PubMed]
- Metwally, N.H.; Deeb, E.A. Synthesis, anticancer assessment on human breast, liver and colon carcinoma cell lines and molecular modeling study using novel pyrazolo[4,3-c]pyridine derivatives. Bioorg. Chem. 2018, 77, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Thangarasu, P.; Manikandan, A.; Thamaraiselvi, S. Discovery, synthesis and molecular corroborations of medicinally important novel pyrazoles; drug efficacy determinations through in silico, in vitro and cytotoxicity validations. Bioorg. Chem. 2019, 86, 410–419. [Google Scholar] [CrossRef]
- Gunderwala, A.; Cope, N.; Wang, Z. Mechanism and inhibition of BRAF kinase. Curr. Opin. Chem. Biol. 2022, 71, 102205. [Google Scholar] [CrossRef]
- El-Gamal, M.I.; Park, B.J.; Oh, C.H. Synthesis, in vitro antiproliferative activity, and kinase inhibitory effects of pyrazole-containing diarylureas and diarylamides. Eur. J. Med. Chem. 2018, 156, 230–239. [Google Scholar] [CrossRef]
- Tan, B.Q.; Huang, Y.Y.; Zhang, B.; Lin, N.M. The effect of ibrutinib on radiosensitivity in pancreatic cancer cells by targeting EGFR/AKT/mTOR signaling pathway. Biomed. Pharmacother. 2020, 128, 110133. [Google Scholar] [CrossRef]
- Ran, F.; Liu, Y.; Liu, M.; Zhang, D.; Wang, P.; Dong, J.; Tang, W.; Zhao, G. Discovery of pyrazolopyrimidine derivatives as potent BTK inhibitors with effective anticancer activity in MCL. Bioorg. Chem. 2019, 89, 102943. [Google Scholar] [CrossRef]
- Chen, J.; Tang, G. PIM-1 kinase: A potential biomarker of triple-negative breast cancer. Onco. Targets Ther. 2019, 12, 6267–6273. [Google Scholar] [CrossRef] [Green Version]
- Philoppes, J.N.; Khedr, M.A.; Hassan, M.H.A.; Kamel, G.; Lamie, P.F. New pyrazolopyrimidine derivatives with anticancer activity: Design, synthesis, PIM-1 inhibition, molecular docking study and molecular dynamics. Bioorg. Chem. 2020, 100, 103944. [Google Scholar] [CrossRef] [PubMed]
- Amoussou, N.G.; Bigot, A.; Roussakis, C.; Robert, J.H. Haspin: A promising target for the design of inhibitors as potent anticancer drugs. Drug Discov. Today 2018, 23, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Opoku-Temeng, C.; Dayal, N.; Sooreshjani, M.A.; Sintim, H.O. 3H-pyrazolo[4,3-f]quinoline haspin kinase inhibitors and anticancer properties. Bioorg. Chem. 2018, 78, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.M.; Adnane, L.; Newell, P.; Villanueva, A.; Llovet, J.M.; Lynch, M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol. Cancer Ther. 2008, 7, 3129–3140. [Google Scholar] [CrossRef] [Green Version]
- Ruzi, Z.; Bozorov, K.; Nie, L.; Zhao, J.; Aisa, H.A. Novel pyrazolo[3,4-d]pyrimidines as potential anticancer agents: Synthesis, VEGFR-2 inhibition, and mechanisms of action. Biomed. Pharmacother. 2022, 156, 113948. [Google Scholar] [CrossRef]
- Saleh, N.M.; El-Gazzar, M.G.; Aly, H.M.; Othman, R.A. Novel anticancer fused pyrazole derivatives as EGFR and VEGFR-2 dual TK Inhibitors. Front. Chem. 2020, 7, 917. [Google Scholar] [CrossRef]
- Ghorbanpour, M.; Soltani, B.; Mota, A.; Sardroodi, J.J.; Aghdam, E.M.; Shayanfar, A.; Molavi, O.; Mohammad-Rezaei, R.; Ebadi-Nahari, M.; Ziegler, C.J. Copper (II) complexes with N, S donor pyrazole-based ligands as anticancer agents. Biometals 2022, 35, 1095–1111. [Google Scholar] [CrossRef]
- Bhukya, B.; Korra, R.; Guguloth, H. Synthesis of novel amide/amino acid functionalized pyrazolo[3,4-b]pyridine derivatives; their anticancer activity and docking studies. J. Heterocycl. Chem. 2023, 60, 872–0878. [Google Scholar] [CrossRef]
- Zaki, R.M.; Wani, M.Y.; Mohammed, A.; El-Said, W.A. Design, synthesis and evaluation of novel Se-alkylated pyrazoles and their cyclized analogs as potential anticancer agents. J. Mol. Struct. 2023, 1276, 134670. [Google Scholar] [CrossRef]
- Nossier, E.S.; Abd El-Karim, S.S.; Khalifa, N.M.; El-Sayed, A.S.; Hassan, E.S.I.; El-Hallouty, S.M. Kinase inhibitory activities and molecular docking of a novel series of anticancer pyrazole derivatives. Molecules 2018, 23, 3074. [Google Scholar] [CrossRef] [Green Version]
- Portugal, J.; Barceló, F. Noncovalent binding to DNA: Still a target in developing anticancer agents. Curr. Med. Chem. 2016, 23, 4108–4134. [Google Scholar] [CrossRef] [Green Version]
- Paitandi, R.P.; Sharma, V.; Singh, V.D.; Dwivedi, B.K.; Mobin, S.M.; Pandey, D.S. Pyrazole appended quinoline-BODIPY based arene ruthenium complexes: Their anticancer activity and potential applications in cellular imaging. Dalton Trans. 2018, 47, 17500–17514. [Google Scholar] [CrossRef] [PubMed]
- El-Gohary, N.S.; Gabr, M.T.; Shaaban, M.I. Synthesis, molecular modeling and biological evaluation of new pyrazolo[3,4-b]pyridine analogs as potential antimicrobial, antiquorum-sensing and anticancer agents. Bioorg. Chem. 2019, 89, 102976. [Google Scholar] [CrossRef]
- Omran, D.M.; Ghaly, M.A.; El-Messery, S.M.; Badria, F.A.; Abdel-Latif, E.; Shehata, I.A. Targeting hepatocellular carcinoma: Synthesis of new pyrazole-based derivatives, biological evaluation, DNA binding, and molecular modeling studies. Bioorg. Chem. 2019, 88, 102917. [Google Scholar] [CrossRef]
- Delgado, J.L.; Hsieh, C.M.; Chan, N.L.; Hiasa, H. Topoisomerases as anticancer targets. Biochem. J. 2018, 475, 373–398. [Google Scholar] [CrossRef] [PubMed]
- Nagaraju, B.; Kovvuri, J.; Kumar, C.G.; Routhu, S.R.; Shareef, M.A.; Kadagathur, M.; Adiyala, P.R.; Alavala, S.; Nagesh, N.; Kamal, A. Synthesis and biological evaluation of pyrazole linked benzothiazole-β-naphthol derivatives as topoisomerase I inhibitors with DNA binding ability. Bioorg. Med. Chem. 2019, 27, 708–720. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.Q.; Shen, W.Y.; Yang, Q.Y.; Chen, Z.F.; Liang, H. Ru(III) complexes with pyrazolopyrimidines as anticancer agents: Bioactivities and the underlying mechanisms. Dalton Trans. 2022, 51, 1333–1343. [Google Scholar] [CrossRef]
- Nagavath, R.; Nukala, S.K.; Sirassu, N.; Sagam, R.R.; Manchal, R.; Paidakula, S.; Thirukovela, N.S. One-pot synthesis of some new regioselective 4β-pyrazolepodophyllotoxins as DNA topoisomerase-II targeting anticancer agents. J. Mol. Struct. 2022, 1250, 131724. [Google Scholar] [CrossRef]
- Zebbiche, Z.; Şekerci, G.; Boulebd, H.; Küçükbay, F.; Tekin, S.; Tekin, Z.; Küçükbay, H.; Sandal, S.; Boumoud, B. Preparation, DFT calculations, docking studies, antioxidant, and anticancer properties of new pyrazole and pyridine derivatives. J. Biochem. Mol. Toxicol. 2022, 36, e23135. [Google Scholar] [CrossRef]
- Garcia-Saez, I.; Skoufias, D.A. Eg5 targeting agents: From new anti-mitotic based inhibitor discovery to cancer therapy and resistance. Biochem. Pharmacol. 2021, 184, 114364. [Google Scholar] [CrossRef]
- Muthuraja, P.; Veeramani, V.; Prakash, S.; Himesh, M.; Venkatasubramanian, U.; Manisankar, P. Structure-activity relationship of pyrazolo pyrimidine derivatives as inhibitors of mitotic kinesin Eg5 and anticancer agents. Bioorg. Chem. 2019, 84, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Kannan, S.; Partridge, A.W.; Lane, D.P.; Verma, C.S. The dual interactions of p53 with MDM2 and p300: Implications for the design of MDM2 inhibitors. Int. J. Mol. Sci. 2019, 20, 5996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, M.; Poojary, B.; Kalal, B.S.; Gurubasavaraja Swamy, P.M.; Kabilan, S.; Kumar, V.; Shruthi, N.; Alias Anand, S.A.; Pai, V.R. Synthesis and evaluation of thiazolidinone-pyrazole conjugates as anticancer and antimicrobial agents. Future Med. Chem. 2018, 10, 1017–1036. [Google Scholar] [CrossRef] [PubMed]
- Goradel, N.H.; Najafi, M.; Salehi, E.; Farhood, B.; Mortezaee, K. Cyclooxygenase-2 in cancer: A review. J. Cell Physiol. 2019, 234, 5683–5699. [Google Scholar] [CrossRef]
- Ren, S.Z.; Wang, Z.C.; Zhu, D.; Zhu, X.H.; Shen, F.Q.; Wu, S.Y.; Chen, J.J.; Xu, C.; Zhu, H.L. Design, synthesis and biological evaluation of novel ferrocene-pyrazole derivatives containing nitric oxide donors as COX-2 inhibitors for cancer therapy. Eur. J. Med. Chem. 2018, 157, 909–924. [Google Scholar] [CrossRef]
- Yamali, C.; Gul, H.I.; Ece, A.; Bua, S.; Angeli, A.; Sakagami, H.; Sahin, E.; Supuran, C.T. Synthesis, biological evaluation and in silico modelling studies of 1,3,5-trisubstituted pyrazoles carrying benzenesulfonamide as potential anticancer agents and selective cancer-associated hCA IX isoenzyme inhibitors. Bioorg. Chem. 2019, 92, 103222. [Google Scholar] [CrossRef]
- Allam, M.; Bhavani, A.K.D.; Mudiraj, A.; Ranjan, N.; Thippana, M.; Babu, P.P. Synthesis of pyrazolo[3,4-d]pyrimidin-4(5H)-ones tethered to 1,2,3-triazoles and their evaluation as potential anticancer agents. Eur. J. Med. Chem. 2018, 156, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Ge, S.; Guo, J.; Chen, S.; Huang, M.; Yang, J.; Sun, S.; Ling, Y.; Shi, Y. Development of novel bis-pyrazole derivatives as antitumor agents with potent apoptosis induction effects and DNA damage. Eur. J. Med. Chem. 2018, 143, 1066–1076. [Google Scholar] [CrossRef]
- Shaaban, O.G.; Abd El Razik, H.A.; Shams El-Dine, S.E.A.; Ashour, F.A.; El-Tombary, A.A.; Afifi, O.S.; Abu-Serie, M.M. Purines and triazolo[4,3-e]purines containing pyrazole moiety as potential anticancer and antioxidant agents. Future Med. Chem. 2018, 10, 1449–1464. [Google Scholar] [CrossRef]
- Dai, H.; Huang, M.; Qian, J.; Liu, J.; Meng, C.; Li, Y.; Ming, G.; Zhang, T.; Wang, S.; Shi, Y.; et al. Excellent antitumor and antimetastatic activities based on novel coumarin/pyrazole oxime hybrids. Eur. J. Med. Chem. 2019, 166, 470–479. [Google Scholar] [CrossRef]
- Farooq, M.; Sharma, A.; Almarhoon, Z.; Al-Dhfyan, A.; El-Faham, A.; Taha, N.A.; Wadaan, M.A.M.; Torre, B.G.; Albericio, F. Design and synthesis of mono-and di-pyrazolyl-s-triazine derivatives, their anticancer profile in human cancer cell lines, and in vivo toxicity in zebrafish embryos. Bioorg. Chem. 2019, 87, 457–464. [Google Scholar] [CrossRef]
- Mótyán, G.; Gopisetty, M.K.; Kiss-Faludy, R.E.; Kulmány, Á.; Zupkó, I.; Frank, É.; Kiricsi, M. Anti-cancer activity of novel dihydrotestosterone-derived ring A-condensed pyrazoles on androgen non-responsive prostate cancer cell lines. Int. J. Mol. Sci. 2019, 20, 2170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nitulescu, G.M.; Matei, L.; Aldea, I.M.; Draghici, C.; Olaru, O.T.; Bleotu, C. Ultrasound-assisted synthesis and anticancer evaluation of new pyrazole derivatives as cell cycle inhibitors. Arab. J. Chem. 2019, 12, 816–824. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Xu, L.; Cao, M.; Wang, Z.; Xiao, D.; Xu, S.; Deng, J.; Hu, X.; He, C.; Tao, T.; et al. Anticancer properties of novel pyrazole-containing biguanide derivatives with activating the adenosine monophosphate-activated protein kinase signaling pathway. Arch. Pharm. 2019, 352, e1900075. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.D.; Siva, B.; Vadlamudi, S.; Bathula, S.R.; Dutta, H.; Babu, S.K. Design, synthesis, and biological evaluation of pyrazole-linked aloe emodin derivatives as potential anticancer agents. RSC Med. Chem. 2021, 12, 791–796. [Google Scholar] [CrossRef] [PubMed]
- Othman, E.M.; Bekhit, A.A.; Anany, M.A.; Dandekar, T.; Ragab, H.M.; Wahid, A. Design, synthesis, and anticancer screening for repurposed pyrazolo[3,4-d]pyrimidine derivatives on four mammalian cancer cell lines. Molecules 2021, 26, 2961. [Google Scholar] [CrossRef] [PubMed]
- Gobbo, A.; Pereira, S.A.P.; Biancalana, L.; Zacchini, S.; Saraiva, M.L.M.F.S.; Dyson, P.J.; Marchetti, F. Anticancer ruthenium(II) tris(pyrazolyl)methane complexes with bioactive co-ligands. Dalton Trans. 2022, 51, 17050–17063. [Google Scholar] [CrossRef]
- Hess, J.D.; Macias, L.H.; Gutierrez, D.A.; Moran-Santibanez, K.; Contreras, L.; Medina, S.; Villanueva, P.J.; Kirken, R.A.; Varela-Ramirez, A.; Penichet, M.L.; et al. Identification of a unique cytotoxic thieno[2,3-c]pyrazole derivative with potent and selective anticancer effects in vitro. Biology 2022, 11, 930. [Google Scholar] [CrossRef]
- Kamel, M.G.; Sroor, F.M.; Othman, A.M.; Mahrous, K.F.; Saleh, F.M.; Hassaneen, H.M.; Abdallah, T.A.; Abdelhamid, I.A.; Teleb, M.A.M. Structure-based design of novel pyrazolyl-chalcones as anti-cancer and antimicrobial agents: Synthesis and in vitro studies. Monatsh. Chem. 2022, 153, 211–221. [Google Scholar] [CrossRef]
- Adeniyi, A.A.; Ajibade, P.A. The anticancer activities of some nitrogen donor ligands containing bis-pyrazole, bipyridine, and phenanthroline moiety using docking methods. Bioinorg. Chem. Appl. 2018, 2018, 5796287. [Google Scholar] [CrossRef] [Green Version]
- El-Kashef, H.; El-Emary, T.; Verhaeghe, P.; Vanelle, P.; Samy, M. Anticancer and anti-inflammatory activities of some new pyrazolo[3,4-b]pyrazines. Molecules 2018, 23, 2657. [Google Scholar] [CrossRef] [Green Version]
- Nassar, I.F.; El Farargy, A.F.; Abdelrazek, F.M. Synthesis and anticancer activity of some new fused pyrazoles and their glycoside derivatives. J. Heterocycl. Chem. 2018, 55, 1709–1719. [Google Scholar] [CrossRef]
- Verma, G.; Chashoo, G.; Ali, A.; Khan, M.F.; Akhtar, W.; Ali, I.; Akhtar, M.; Alam, M.M.; Shaquiquzzaman, M. Synthesis of pyrazole acrylic acid based oxadiazole and amide derivatives as antimalarial and anticancer agents. Bioorg. Chem. 2018, 77, 106–124. [Google Scholar] [CrossRef] [PubMed]
- Abdelgawad, N.; Ismail, M.F.; Hekal, M.H.; Marzouk, M.I. Design, synthesis and evaluation of some novel heterocycles bearing pyrazole moiety as potential anticancer agents. J. Heterocycl. Chem. 2019, 56, 1771–1779. [Google Scholar] [CrossRef]
- Ahmed, M.H.; El-Hashash, M.A.; Marzouk, M.I.; El-Naggar, A.M. Design, synthesis, and biological evaluation of novel pyrazole, oxazole, and pyridine derivatives as potential anticancer agents using mixed chalcone. J. Heterocycl. Chem. 2019, 56, 114–123. [Google Scholar] [CrossRef] [Green Version]
- Gezegen, H.; Tutar, U.; Hepokur, C.; Ceylan, M. Synthesis and biological evaluation of novel indenopyrazole derivatives. J. Biochem. Mol. Toxic. 2019, 33, e22285. [Google Scholar] [CrossRef]
- Hamza, E.K.; Hamdy, N.A.; Zarie, E.S.; Fakhr, I.M.I.; Elwahy, A.H.M.; Awad, H.M. Synthesis and in vitro evaluation of novel tetralin-pyrazolo[3,4-b]pyridine hybrids as potential anticancer agents. J. Heterocycl. Chem. 2020, 57, 182–196. [Google Scholar] [CrossRef]
- Naik, N.S.; Shastri, L.A.; Shastri, S.L.; Chougala, B.M.; Shaikh, F.; Madar, J.M.; Kulkarni, R.C.; Dodamani, S.; Jalalpure, S.; Joshi, S.D.; et al. Synthesis of polyfunctionalized fused pyrazolo-pyridines: Characterization, anticancer activity, protein binding and molecular docking studies. ChemistrySelect 2019, 4, 285–297. [Google Scholar] [CrossRef]
- Yadav, Y.; Sharma, D.; Kaushik, K.; Kumar, V.; Jha, A.; Prasad, A.K.; Len, C.; Malhotra, S.V.; Wengel, J.; Parmar, V.S. Synthetic, structural, and anticancer activity evaluation studies on novel pyrazolylnucleosides. Molecules 2019, 24, 3922. [Google Scholar] [CrossRef] [Green Version]
- Akhmetova, V.R.; Akhmadiev, N.S.; Abdullin, M.F.; Dzhemileva, L.U.; D’yakonov, V.A. Synthesis of new N,N’-Pd(Pt) complexes based on sulfanyl pyrazoles, and investigation of their in vitro anticancer activity. RSC Adv. 2020, 10, 15116–15123. [Google Scholar] [CrossRef]
- Al-Ghorbani, M.; Gouda, M.A. Synthesis and in vitro anticancer activity of some novel cyclohepta[b]thiophene-3-carboxamides bearing pyrazole moiety. J. Heterocycl. Chem. 2020, 57, 3213–3221. [Google Scholar] [CrossRef]
- Bakhotmah, D.A.; Ali, T.E.; Assiri, M.A.; Yahia, I.S. Synthesis of some novel 2-{pyrano[2,3-c]pyrazoles-4-ylidene}malononitrile fused with pyrazole, pyridine, pyrimidine, diazepine, chromone, pyrano[2,3-c]pyrazole and pyrano[2,3-d]pyrimidine systems as anticancer agents. Polycycl. Aromat. Comp. 2022, 42, 2136–2150. [Google Scholar] [CrossRef]
- Bansal, K.K.; Bhardwaj, J.K.; Saraf, P.; Thakur, V.K.; Sharma, P.C. Synthesis of thiazole clubbed pyrazole derivatives as apoptosis inducers and anti-infective agents. Mater. Today Chem. 2020, 17, 100335. [Google Scholar] [CrossRef]
- Bondock, S.; Alqahtani, S.; Fouda, A.M. Synthesis and anticancer evaluation of some new pyrazolo[3,4-d][1,2,3]triazin-4-ones, pyrazolo[1,5-a]pyrimidines, and imidazo[1,2-b]pyrazoles clubbed with carbazole. J. Heterocycl. Chem. 2021, 58, 56–73. [Google Scholar] [CrossRef]
- Fathy, U.; Azzam, M.A.; Mahdy, F.; El-Maghraby, S.; Allam, R.M. Synthesis and in vitro anticancer activity of some novel tetrahydroquinoline derivatives bearing pyrazole and hydrazide moiety. J. Heterocycl. Chem. 2020, 57, 2108–2120. [Google Scholar] [CrossRef]
- Hassan, A.Y.; Mohamed, M.A.; Abdel-Aziem, A.; Hussain, A.O. Synthesis and anticancer activity of some fused heterocyclic compounds containing pyrazole ring. Polycycl. Aromat. Comp. 2020, 40, 1280–1290. [Google Scholar] [CrossRef]
- Hassan, A.Y.; Saleh, N.M.; Kadh, M.S.; Abou-Amra, E.S. New fused pyrazolopyrimidine derivatives; heterocyclic styling, synthesis, molecular docking and anticancer evaluation. J. Heterocycl. Chem. 2020, 57, 2704–2721. [Google Scholar] [CrossRef]
- Ismail, M.M.F.; Abdulwahab, H.G.; Elnagdi, M.H. Design, synthesis, and in vitro anticancer screening of novel pyrazolinyl-pyrazole/1,2,3-triazole hybrids. J. Heterocycl. Chem. 2020, 57, 3584–3596. [Google Scholar] [CrossRef]
- Kankala, S.; Rama, K.R.; Kesari, C.; Bjorkling, F.; Nerella, S.; Gundepaka, P.; Guguloth, H.; Thota, N. Synthesis of novel fluorophenylpyrazole-picolinamide derivatives and determination of their anticancer activity. Synth. Commun. 2020, 50, 2997–3006. [Google Scholar] [CrossRef]
- Salem, M.S.; El-Helw, E.A.E.; Derbala, H.A.Y. Development of chromone-pyrazole-based anticancer agents. Russ. J. Bioorg. Chem. 2020, 46, 77–84. [Google Scholar] [CrossRef]
- Molnár, B.; Gopisetty, M.K.; Adamecz, D.I.; Kiricsi, M.; Frank, É. Multistep synthesis and in vitro anticancer evaluation of 2-pyrazolyl-estradiol derivatives, pyrazolocoumarin-estradiol hybrids and analogous compounds. Molecules 2020, 25, 4039. [Google Scholar] [CrossRef]
- Motyan, G.; Baji, A.; Marc, M.A.; Gopisetty, M.K.; Adamecz, D.I.; Kiricsi, M.; Enyedy, E.A.; Frank, E. Microwave-assisted synthesis, proton dissociation processes, and anticancer evaluation of novel D-ring-fused steroidal 5-amino-1-arylpyrazoles. Appl. Sci. 2020, 10, 229. [Google Scholar] [CrossRef] [Green Version]
- Pham, V.T.B.; Nguyen, T.V.; Nguyen, H.V.; Nguyen, T.T.; Hoang, H.M. Curcuminoids versus pyrazole-modified analogues: Synthesis and cytotoxicity against HepG2 cancer cell line. ChemistrySelect 2020, 5, 11681–11684. [Google Scholar] [CrossRef]
- Kalavadiya, P.L.; Kapupara, V.H.; Gojiya, D.G.; Bhatt, T.D.; Hadiyal, S.D.; Joshi, H.S. Ultrasonic-assisted synthesis of pyrazolo[3,4-d]pyrimidin-4-ol tethered with 1,2,3-triazoles and their anticancer activity. Russ. J. Bioorg. Chem. 2020, 46, 803–813. [Google Scholar] [CrossRef]
- Ravula, S.; Bobbala, R.R.; Kolli, B. Synthesis of novel isoxazole functionalized pyrazolo[3,4-b]pyridine derivatives; their anticancer activity. J. Heterocycl. Chem. 2020, 57, 2535–2538. [Google Scholar] [CrossRef]
- Alsayari, A.; Muhsinah, A.B.; Asiri, Y.I.; Al-Aizari, F.A.; Kheder, N.A.; Almarhoon, Z.M.; Ghabbour, H.A.; Mabkhot, Y.N. Synthesis, characterization, and biological evaluation of some novel pyrazolo[5,1-b]thiazole derivatives as potential antimicrobial and anticancer agents. Molecules 2021, 26, 5383. [Google Scholar] [CrossRef] [PubMed]
- Chinthaparthi, R.R.; Chittiboena, V.L.; Jorepalli, S.; Gangireddy, C.S.R. Green synthesis and anticancer activity of tetrahydrodipyrazolo[3,4-b:4’,3’-e]pyridines catalyzed by phospho sulfonic acid. J. Heterocycl. Chem. 2021, 58, 1104–1116. [Google Scholar] [CrossRef]
- Huang, Y.; Hu, H.; Yan, R.; Lin, L.; Song, M.; Yao, X. Synthesis and evaluation of antimicrobial and anticancer activities of 3-phenyl-1-phenylsulfonyl pyrazoles containing an aminoguanidine moiety. Arch. Pharm. 2021, 354, e2000165. [Google Scholar] [CrossRef] [PubMed]
- Othman, I.M.M.; Gad-Elkareem, M.A.M.; Radwan, H.A.; Badraoui, R.; Aouadi, K.; Snoussi, M.; Kadri, A. Synthesis, structure-activity relationship and in silico studies of novel pyrazolothiazole and thiazolopyridine derivatives as prospective antimicrobial and anticancer agents. ChemistrySelect 2021, 6, 7860–7872. [Google Scholar] [CrossRef]
- Suryanarayana, K.; Robert, A.R.; Kerru, N.; Pooventhiran, T.; Thomas, R.; Maddila, S.; Jonnalagadda, S.B. Design, synthesis, anticancer activity and molecular docking analysis of novel dinitrophenylpyrazole bearing 1,2,3-triazoles. J. Mol. Struct. 2021, 1243, 130865. [Google Scholar] [CrossRef]
- Aliwaini, S.; Abu Thaher, B.; Al-Masri, I.; Shurrab, N.; El-Kurdi, S.; Schollmeyer, D.; Qeshta, B.; Ghunaim, M.; Csuk, R.; Laufer, S.; et al. Design, synthesis and biological evaluation of novel pyrazolo[1,2,4]triazolopyrimidine derivatives as potential anticancer agents. Molecules 2021, 26, 4065. [Google Scholar] [CrossRef] [PubMed]
- Alshammari, M.M.; Soury, R.; Alenezi, K.M.; Mushtque, M.; Rizvi, M.M.A.; Haque, A. Synthesis, characterization, anticancer and in silico studies of a pyrazole-tethered thiazolidine-2,4-dione derivative. J. Biomol. Struct. Dyn. 2022, 40, 13075–13082. [Google Scholar] [CrossRef] [PubMed]
- Bhogireddy, D.N.; Surapureddi, S.R.; Syed, T.; Prashanth, T.; Tadiboina, B.R. Synthesis and biological evaluation of aryl derivatives of isoxazole pyrazolo[1,5-a]pyrimidines as anticancer agents. Synth. Commun. 2022, 52, 861–874. [Google Scholar] [CrossRef]
- Karrouchi, K.; Sert, Y.; Ansar, M.; Radi, S.; El Bali, B.; Imad, R.; Alam, A.; Irshad, R.; Wajid, S.; Altaf, M. Synthesis, α-glucosidase inhibition, anticancer, DFT and molecular docking Investigations of pyrazole Hydrazone Derivatives. Polycycl. Aromat. Comp. 2023, 43, 5021–5040. [Google Scholar] [CrossRef]
- Kumar, V.H.; Tamminana, R. Copper-catalyzed multicomponent green reaction approach: Synthesis of dihydropyrano [2,3-c] pyrazoles and evaluation of their anti-cancer activity. J. Heterocycl. Chem. 2023, 60, 18–26. [Google Scholar] [CrossRef]
- Mamidala, S.; Aravilli, R.K.; Vaarla, K.; Peddi, S.R.; Gondru, R.; Manga, V.; Vedula, R.R. A facile one-pot, three-component synthesis of a new series of thiazolyl pyrazoles: Anticancer evaluation, ADME and molecular docking studies. Polycycl. Aromat. Comp. 2023, 43, 1332–1348. [Google Scholar] [CrossRef]
- Parikh, P.H.; Timaniya, J.B.; Patel, M.J.; Patel, K.P. Microwave-assisted synthesis of pyrano[2,3-c]pyrazole derivatives and their anti-microbial, anti-malarial, anti-tubercular, and anti-cancer activities. J. Mol. Struct. 2022, 1249, 131605. [Google Scholar] [CrossRef]
- Sadeghian, Z.; Bayat, M.; Safari, F. Synthesis and in vitro anticancer activity evaluation of spiro[indolo[2,1-b]quinazoline-pyrano[2,3-c]pyrazole] via sequential four-component reaction. J. Mol. Struct. 2022, 1250, 131759. [Google Scholar] [CrossRef]
- Saleh, R.O.; Achmad, H.; Daminov, B.T.; Kzar, H.H.; Mahdi, A.B.; Hammid, A.T.; Abid, M.K.; Opulencia, M.J.C.; Mustafa, Y.F.; Sharma, H. Synthesis of bioactive yttrium-metal-organic framework as efficient nanocatalyst in synthesis of novel pyrazolopyranopyrimidine derivatives and evaluation of anticancer activity. Front. Chem. 2022, 10, 928047. [Google Scholar] [CrossRef]
- Xie, D.; Yang, J.; Niu, X.; Wang, Z.; Wu, Z. Synthesis and bioactivity evaluation of 5-trifluoromethyl-1H -pyrazole-4-carboxamide derivatives as potential anticancer and antifungal agents. J. Heterocycl. Chem. 2022, 59, 1759–1767. [Google Scholar] [CrossRef]
- Xu, Q.; Jia, J.; Wu, Y.; Hu, B.; Xin, J.; Liu, Y.; Gao, W.; Li, D. Ag2O-induced regioselective huisgen cycloaddition for the synthesis of fully substituted pyrazoles as potential anticancer agents. J. Org. Chem. 2022, 87, 14496–14506. [Google Scholar] [CrossRef] [PubMed]
- Al Otaibi, A.A.; Alshammari, S.L.; Dhahi Alsukaibi, A.K.; Jamal, A.; Rajendrasozhan, S.; Alenezi, K.M.; Hussain, A.; Khan, I.; Mushtaque, M.; Haque, A. Synthesis, anticancer activity, molecular docking and molecular dynamics studies of some pyrazole-chalcone hybrid. J. Biomol. Struct. Dyn. 2023, 59, 1–11. [Google Scholar]
- Asif, M.; Aqil, F.; Alasmary, F.A.; Almalki, A.S.; Khan, A.R.; Nasibullah, M. Lewis base-catalyzed synthesis of highly functionalized spirooxindole-pyranopyrazoles and their in vitro anticancer studies. Med. Chem. Res. 2023, 32, 1001–1015. [Google Scholar] [CrossRef]
- Azher, O.A.; Hossan, A.; Pashameah, R.A.; Alsoliemy, A.; Alharbi, A.; Habeebullah, T.M.; El-Metwaly, N.M. Synthesis, anticancer evaluation, and molecular modeling study of new 2-(phenylamino)pyrazolo[1,5-a]pyrimidine analogues. Arab. J. Chem. 2023, 16, 104437. [Google Scholar] [CrossRef]
- Elmorsy, M.R.; Abdel-Latif, E.; Gaffer, H.E.; Mahmoud, S.E.; Fadda, A.A. Anticancer evaluation and molecular docking of new pyridopyrazolo-triazine and pyridopyrazolo-triazole derivatives. Sci. Rep. 2023, 13, 2782. [Google Scholar] [CrossRef]
- Hossan, A.; Alrefaei, A.F.; Katouah, H.A.; Bayazeed, A.; Asghar, B.H.; Shaaban, F.; El-Metwaly, N.M. Synthesis, anticancer activity, and molecular docking of new pyrazolo[1,5-a]pyrimidine derivatives. J. Saudi Chem. Soc. 2023, 27, 101599. [Google Scholar] [CrossRef]





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Structure | Principal Indications | Mechanism of Action |
|---|---|---|---|
| Lonazolac [10] |  | Rheumatoid arthritis; osteoarthritis | Lonazolac elicits its therapeutic effects primarily through preferential inhibition of COX-2, leading to reduced prostaglandin synthesis and blocking of inflammatory processes that mediate pain and fever. |
| Celecoxib [11] |  | Rheumatoid arthritis; osteoarthritis | Celecoxib is a highly selective inhibitor of COX-2, which leads to reduced production of proinflammatory prostaglandins such as PGE2. |
| Crizotinib [12] |  | Advanced non-small cell lung cancer | Crizotinib is a highly selective inhibitor of ALK and ROS1 tyrosine kinases. It blocks downstream signaling pathways involved in cellular proliferation and anti-apoptosis. |
| Difenamizole [13] |  | Acute and chronic arthritis; soft tissue inflammation | Difenamizole is a selective inhibitor of tyrosine phosphorylation of COX-2. It suppresses the phosphorylation of tyrosine residues on COX-2, thereby inhibiting its activity. This reduces levels of the inflammatory mediators endoperoxides and prostaglandins produced from the cyclooxygenase pathway. |
| Rimonabant [14] |  | Overweight and obesity | Rimonabant is a selective antagonist of the cannabinoid CB1 receptor. It selectively blocks CB1 receptors in both the central nervous system and peripheral tissues. |
| Apixaban [15] |  | prevention of venous thromboembolism | Apixaban is a selective and reversible inhibitor of coagulation factor Xa. It inhibits factor Xa activity, thereby blocking thrombin generation and blood coagulation. |
| Fezolamine [16] |  | Hypertension; pain | Fezolamine is a non-selective α adrenergic receptor antagonist. It inhibits vasoconstriction mediated by the catecholamines adrenaline and noradrenaline. |
| Sidenafil [17] |  | Erectile dysfunction; pulmonary arterial hypertension | Sildenafil selectively inhibits phosphodiesterase type 5 (PDE5), the enzyme responsible for breaking down cyclic guanosine monophosphate (cGMP). |
| Betazole [18] |  | Diagnosis of impaired gastric acid secretion | Betazole is a highly selective H2 receptor agonist. It binds to and activates H2 receptors with high affinity, stimulating parietal cells to release gastric acid. |
| Fomepizole [19] |  | Severe ethanol intoxication | Fomepizole is a potent competitive inhibitor of alcohol dehydrogenase. By inhibiting this enzyme, fomepizole blocks the metabolism of ethanol, thereby reducing the formation of the toxic metabolite acetaldehyde. |
| Indiplon [20] |  | Sleep disorders | Indiplon acts as a positive allosteric modulator of GABAA receptors. It increases the receptor’s affinity for GABA, facilitating inhibitory synaptic transmission mediated by GABA. |
| Zaleplon [21] |  | Circadian rhythm sleep disorders | Zaleplon selectively binds to the α1 subunit of the GABAA receptor. By increasing the receptor’s affinity for GABA, it facilitates inhibitory synaptic transmission mediated by GABA binding to the GABAA receptor. |
| Tepoxalin [22] |  | Rheumatoid arthritis | Tepoxalin is a dual inhibitor of cyclooxygenase (COX) and 5-lipoxygenase (5-LOX). It inhibits the production of pro-inflammatory mediators including prostaglandins and leukotrienes. |
| Deracoxib [23] |  | Pain and inflammation associated with osteoarthritis in dogs | Deracoxib is a highly selective inhibitor of cyclooxygenase-2 (COX-2). It inhibits the COX-2 mediated synthesis of prostaglandins. |
| Pyrazofurin [24] |  | Acute and chronic myeloid leukemia | Pyrazofurin is an inhibitor of purine nucleotide synthesis. It inhibits the enzyme orotate phosphoribosyltransferase (OMPdecase) in tumor cells, blocking the synthesis of pyrimidine nucleotides and inhibiting DNA and RNA synthesis. |
| Surinabant [25] |  | Was previously developed for treating tobacco dependence and obesity (currently has no approved therapeutic indications) | Surinabant is a selective inverse agonist of the cannabinoid CB1 receptor. It binds to CB1 receptors and inhibits their constitutive activity. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Wu, C.; Zhang, N.; Fan, R.; Ye, Y.; Xu, J. Recent Advances in the Development of Pyrazole Derivatives as Anticancer Agents. Int. J. Mol. Sci. 2023, 24, 12724. https://doi.org/10.3390/ijms241612724
Zhang Y, Wu C, Zhang N, Fan R, Ye Y, Xu J. Recent Advances in the Development of Pyrazole Derivatives as Anticancer Agents. International Journal of Molecular Sciences. 2023; 24(16):12724. https://doi.org/10.3390/ijms241612724
Chicago/Turabian StyleZhang, Yingqian, Chenyuan Wu, Nana Zhang, Rui Fan, Yang Ye, and Jun Xu. 2023. "Recent Advances in the Development of Pyrazole Derivatives as Anticancer Agents" International Journal of Molecular Sciences 24, no. 16: 12724. https://doi.org/10.3390/ijms241612724
APA StyleZhang, Y., Wu, C., Zhang, N., Fan, R., Ye, Y., & Xu, J. (2023). Recent Advances in the Development of Pyrazole Derivatives as Anticancer Agents. International Journal of Molecular Sciences, 24(16), 12724. https://doi.org/10.3390/ijms241612724