
Improved Theory of the Effective Dipole Moments and Absolute Line Strengths of the XY2 Asymmetric Top Molecules in the X2B1 Doublet Electronic States

 ,
,  , , , and
, , , and
Abstract
:1. Introduction
2. Absolute Intensity of an Isolated Line of the XY2 () Molecule in a Singlet Electronic State: Rotational Transitions
3. Absolute Intensity of an Isolated Line of the XY2 () Molecule in a Singlet Electronic State: Ro-Vibrational Transitions
4. Absolute Intensity of an Isolated Line of the XY2 () Molecule in Doublet Electronic State: Spin–Rotational Transitions in the Model That Neglects Spin–Rotational Interactions in the Effective Dipole Moment Operator
5. Absolute Intensity of an Isolated Line of the XY2 () Molecule in Doublet Electronic State: Spin–Rotational Transitions: -Operator Depends on Molecular Vibrations
5.1. Parallel Ro-Vibrational Bands
5.2. Perpendicular Ro-Vibrational Bands
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Flaud, J.; Camy-Peyret, C. Vibration-rotation intensities in H2O—Type molecules application to the 2ν2, ν1, ν3 band of H216O. J. Mol. Spectrosc. 1975, 55, 278–310. [Google Scholar] [CrossRef]
- Loéte, M. Devéloppement complet du moment dipolaire des molécules tétraé. Application aux bandes triplement dégénéréet a la diade ν2 et ν4. Can. J. Phys. 1983, 61, 1242–1259. [Google Scholar] [CrossRef]
- Boudon, V.; Grigoryan, T.; Philipot, F.; Richard, C.; Tchana, F.K.; Manceron, L.; Rizopoulos, A.; Auwera, J.V.; Encrenaz, T. Line positions and intensities for the ν3 band of 5 isotopologues of germane for planetary applications. J. Quant. Spectrosc. Radiat. Transf. 2018, 205, 174–183. [Google Scholar] [CrossRef]
- Tarrago, J.; Ulenikov, O.; Poussigue, G. Dipole moment matrix for vibration–rotation transitions in C3v molecules. J. Phys. Paris 1984, 45, 1429–1447. [Google Scholar] [CrossRef]
- Saveliev, V.N.; Ulenikov, O.N. Calculation of vibration–rotation line intensities of polyatomic molecules based on the formalism of irreducible tensorial sets. J. Phys. B At. Mol. Phys. 1987, 20, 67–83. [Google Scholar] [CrossRef]
- Herzberg, G. Molecular Spectra and Molecular Structure, Volume 2: Infrared and Raman and Spectra of Polyatomic Molecules, 1st ed.; D. Van Nostrand Company: New York, NY, USA, 1945. [Google Scholar]
- Nielsen, H.H. The Vibration-Rotation Energies of Molecules. Rev. Mod. Phys. 1951, 23, 90–136. [Google Scholar] [CrossRef]
- Papouek, D.; Aliev, M.R. Molecular Vibrational—Rotational Spectra; Elsevier: Amsterdam, The Netherlands, 1982. [Google Scholar]
- Ulenikov, O.; Bekhtereva, E.; Gromova, O.; Raspopova, N.; Sydow, C.; Bauerecker, S. Extended analysis of the ν3 band of HD32S: Line positions, energies, and line strengths. J. Quant. Spectrosc. Radiat. Transf. 2019, 230, 131–141. [Google Scholar] [CrossRef]
- Ulenikov, O.; Bekhtereva, E.; Gromova, O.; Raspopova, N.; Belova, A.; Maul, C.; Sydow, C.; Bauerecker, S. Experimental line strengths of the 5ν2 band of H232S in comparison with the results of "variational” calculation and HITRAN database. J. Quant. Spectrosc. Radiat. Transf. 2020, 243, 106812. [Google Scholar] [CrossRef]
- Ro–vibrational analysis of the first hexad of hydrogen sulfide: Line position and strength analysis of the 4ν2 band of H232S and H234S for HITRAN applications. J. Quant. Spectrosc. Radiat. Transf. 2020, 255, 107236. [CrossRef]
- Ulenikov, O.; Bekhtereva, E.; Gromova, O.; Konova, Y.; Aslapovskaya, Y.; Sydow, C.; Berezkin, K.; Bauerecker, S. Quantitative analysis of ro–vibrational spectra of ethylene: Line strengths of the ν12 and ν3 bands of 12C2H2D2–cis. J. Quant. Spectrosc. Radiat. Transf. 2021, 261, 107434. [Google Scholar] [CrossRef]
- Ulenikov, O.; Bekhtereva, E.; Gromova, O.; Belova, A.; Morzhikova, Y.; Sydow, C.; Maul, C.; Bauerecker, S. Line strength analysis of the second overtone 3ν2 band of D232S. J. Quant. Spectrosc. Radiat. Transf. 2021, 270, 107686. [Google Scholar] [CrossRef]
- Malathy Devi, V.; Das, P.P.; Bano, A.; Narahari Rao, K.; Flaud, J.M.; Camy-Peyret, C.; Chevillard, J.P. Diode laser measurements of intensities, N2-broadening, and self–broadening coefficients of lines of the ν2 band of 14N16O2. J. Mol. Spectrosc. 1981, 88, 251–258. [Google Scholar] [CrossRef]
- Perrin, A.; Flaud, J.M.; Camy-Peyret, C.; Carli, B.; Carlotti, M. The far infrared spectrum of 14N16O2 electron spin-rotation and hyperfine fermi contact resonances in the ground state. Mol. Phys. 1988, 63, 791–810. [Google Scholar] [CrossRef]
- Perrin, A.; Flaud, J.M.; Camy-Peyret, C.; Vasserot, A.M.; Guelachvili, G.; Goldman, A.; Murcray, F.; Blatherwick, R. The ν1, 2ν2, and ν3 interacting bands of 14N16O2: Line positions and intensities. J. Mol. Spectrosc. 1992, 154, 391–406. [Google Scholar] [CrossRef]
- Ortigoso, J.; Escribano, R.; Burkholder, J.B.; Lafferty, W.J. Intensities and dipole moment derivatives of the fundamental bands of 35ClO2 and an intensity analysis of the ν1 band. J. Mol. Spectrosc. 1992, 156, 89–97. [Google Scholar] [CrossRef]
- Curl, R.; Kinsey, J.L.; Baker, J.G.; Baird, J.C.; Bird, G.R.; Heidelberg, R.F.; Sugden, T.; Jenkins, D.; Kenney, C. Microwave spectrum of chlorine dioxide. I. Rotational assignment. Phys. Rev. 1961, 121, 1119–1123. [Google Scholar] [CrossRef]
- Curl, R., Jr.; Heidelberg, R.F.; Kinsey, J.L. Microwave spectrum of chlorine dioxide. II. Analysis of hyperfine structure and the spectrum of 35Cl16O18O. Phys. Rev. 1962, 125, 1993–1999. [Google Scholar] [CrossRef]
- Curl, R., Jr. Microwave spectrum of chlorine dioxide. III. Interpretation of the hyperfine coupling constants obtained in terms of the electronic structure. J. Chem. Phys. 1962, 37, 779–784. [Google Scholar] [CrossRef]
- Pillai, M.G.K.; Curl, R., Jr. Microwave spectrum of chlorine dioxide. IV. Determination of centrifugal distortion effects and potential constants. J. Chem. Phys. 1962, 37, 2921–2926. [Google Scholar] [CrossRef]
- Tolles, W.; Kinsey, J.L.; Curl, R.; Heidelberg, R.F. Microwave spectrum of chlorine dioxide. V. The Stark and Zeeman effects. J. Chem. Phys. 1962, 37, 927–930. [Google Scholar] [CrossRef]
- Mariella, R.P., Jr.; Curl, R., Jr. Microwave spectrum of chlorine dioxide. VI. v2 = 1 State. J. Chem. Phys. 1970, 52, 757–763. [Google Scholar] [CrossRef]
- Jones, H.; Brown, J.M. Infrared–microwave double–resonance spectroscopy of the ClO2 radical: A textbook example. J. Mol. Spectrosc. 1981, 90, 222–248. [Google Scholar] [CrossRef]
- Tanoura, M.; Chiba, K.; Tanaka, K.; Tanaka, T. Microwave spectroscopy of chlorine dioxide: Centrifugal distortion, spin-rotation interaction, and hyperfine interaction constants of 35ClO2 and 37ClO2. J. Mol. Spectrosc. 1982, 95, 157–181. [Google Scholar] [CrossRef]
- Miyazaki, K.; Tanoura, M.; Tanaka, K.; Tanaka, T. Microwave spectrum of chlorine dioxide in excited vibrational states. J. Mol. Spectrosc. 1986, 116, 435–449. [Google Scholar] [CrossRef]
- Müller, H.S.; Sørensen, G.; Birk, M.; Friedl, R.R. The Rotational Spectrum and Anharmonic Force Field of Chlorine Dioxide, OClO. J. Mol. Spectrosc. 1997, 186, 177–188. [Google Scholar] [CrossRef]
- Hamada, Y.; Tsuboi, M. High Resolution Infrared Spectrum of Chlorine Dioxide: The ν2 Fundamental Band. Bull. Chem. Soc. Jpn. 1979, 52, 383–385. [Google Scholar] [CrossRef] [Green Version]
- Hamada, Y.; Tsuboi, M. High–resolution infrared spectrum of chlorine dioxide: The ν1 fundamental band. J. Mol. Spectrosc. 1980, 83, 373–390. [Google Scholar] [CrossRef]
- Tanaka, K.; Tanaka, T. CO2 and N2O laser Stark spectroscopy of the ν1 band of the ClO2 radical. J. Mol. Spectrosc. 1983, 98, 425–452. [Google Scholar] [CrossRef]
- Ortigoso, J.; Escribano, R.; Burkholder, J.B.; Howard, C.J.; Lafferty, W.J. High–resolution infrared spectrum of the ν1 band of OClO. J. Mol. Spectrosc. 1991, 148, 346–370. [Google Scholar] [CrossRef]
- Ortigoso, J.; Escribano, R.; Burkholder, J.B.; Lafferty, W.J. The ν2 and ν3 bands and ground state constants of OClO. J. Mol. Spectrosc. 1992, 155, 25–43. [Google Scholar] [CrossRef]
- Ortigoso, J.; Escribano, R.; Burkholder, J.; Lafferty, W. Infrared Spectrum of (OClO) in the 2000 cm−1 Region: The 2ν1 and ν1+ν3 Bands. J. Mol. Spectrosc. 1993, 158, 347–356. [Google Scholar] [CrossRef] [Green Version]
- Ulenikov, O.N.; Bekhtereva, E.S.; Gromova, O.V.; Quack, M.; Berezkin, K.B.; Sydow, C.; Bauerecker, S. High resolution ro–vibrational analysis of molecules in doublet electronic states: The ν1 fundamental of chlorine dioxide (16O35Cl16O) in the X2B1 electronic ground state. Phys.Chem.Chem.Phys. 2021, 23, 4580–4596. [Google Scholar] [CrossRef]
- Ulenikov, O.; Bekhtereva, E.; Gromova, O.; Kakaulin, A.; Sydow, C.; Berezkin, K.; Bauerecker, S. High resolution spectroscopy of the v1 + v3 band of the 35Cl16O2 free radical: Spin–rotation–vibration interactions. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2022, 278, 121379. [Google Scholar] [CrossRef]
- Ulenikov, O.N.; Bekhtereva, E.S.; Gromova, O.V.; Kakaulin, A.N.; Merkulova, M.A.; Sydow, C.; Berezkin, K.B.; Bauerecker, S. High resolution spectroscopy of asymmetric top molecules in nonsinglet electronic states: The v3 fundamental of chlorine dioxide (16O35Cl16O) free radical in the X2B1 electronic ground state. Phys. Chem. Chem. Phys. 2023, 25, 6270–6287. [Google Scholar] [CrossRef] [PubMed]
- Griffits, D.J. Introduction to Quantum Mechanics; Prentice Hall, Inc.: Hoboken, NJ, USA, 1995. [Google Scholar]
- Schmitt, M.; Meerts, L. Structures and dipole moments of molecules in their electronically excited states. In Structures and Dipole Moments of Molecules in Their Electronically Excited States, in Frontiers and Advances in Molecular Spectroscopy; Laane, J., Ed.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 143–193. [Google Scholar] [CrossRef]
- Varshalovitch, D.A.; Moskalev, A.N.; Khersonsky, V.K. Quantum Theory of Angular Momentum; Nauka: Leningrad, Russia, 1975. [Google Scholar]
- Van Vleck, J. The coupling of angular momentum vectors in molecules. Rev. Mod. Phys. 1951, 23, 213–227. [Google Scholar] [CrossRef]
- Fano, U.; Racah, G.D. Irreducible Tensorial Sets; Academic Press: New York, NY, USA, 1959. [Google Scholar]
- Wigner, E.P. Quantum Theory of Angular Momentum; Academic Press: New York, NY, USA, 1965. [Google Scholar]
- Ulenikov, O.; Bekhtereva, E.; Gromova, O.; Fomchenko, A.; Morzhikova, Y.; Sidko, S.; Sydow, C.; Bauerecker, S. Effective dipole moment model for axially symmetric C3v molecules: Application to the precise study of absolute line strengths of the ν6 fundamental of CH3Cl. Int. J. Mol. Sci. 2023, 24, 12122. [Google Scholar] [CrossRef]
- Cheglokov, A.; Ulenikov, O.; Zhilyakov, A.; Cherepanov, V.; Makushkin, Y.; Malikova, A. On the determination of spectroscopic constants as functions of intramolecular parameters. J. Phys. B At. Mol. Opt. Phys. 1989, 22, 997–1015. [Google Scholar] [CrossRef]


{kind=link}
| j | n | ; | ||
|---|---|---|---|---|
| 1 | 1 | |||
| 2 | 1 | |||
| 3 | 1 | |||
| 4 | 1 | |||
| 5 | 1 | |||
| 6 | 1 | |||
| 7 | 1 | |||
| 8 | 3 | |||
| j | n | ; | ||
|---|---|---|---|---|
| 1 | 0 | |||
| 2 | 0 | |||
| 3 | 0 | |||
| 4 | 0 | 0 | ||
| 0 | ||||
| 5 | 0 | |||
| 6 | 2 | |||
| 7 | 2 | |||
| 8 | 2 | |||
| J | Value | |||
|---|---|---|---|---|
| = = | J = | |||
| = = | J = | |||
| = = | J = | |||
| = = | J = | 0 | ||
| = | J = | |||
| = | J = | |||
| = | J = | |||
| = | J = | |||
| = = | J = | |||
| = = | J = | |||
| = = | J = | |||
| = = | J = | 0 |
| J | L | M | ||
|---|---|---|---|---|
| 0 | 0 | N | N | |
| 1 | ||||
| −1 | ||||
| 1 | 0 | N | ||
| 1 | N | |||
| −1 | 0 | N | ||
| −1 | N | |||
| 2 | 1 | |||
| −2 | -1 |
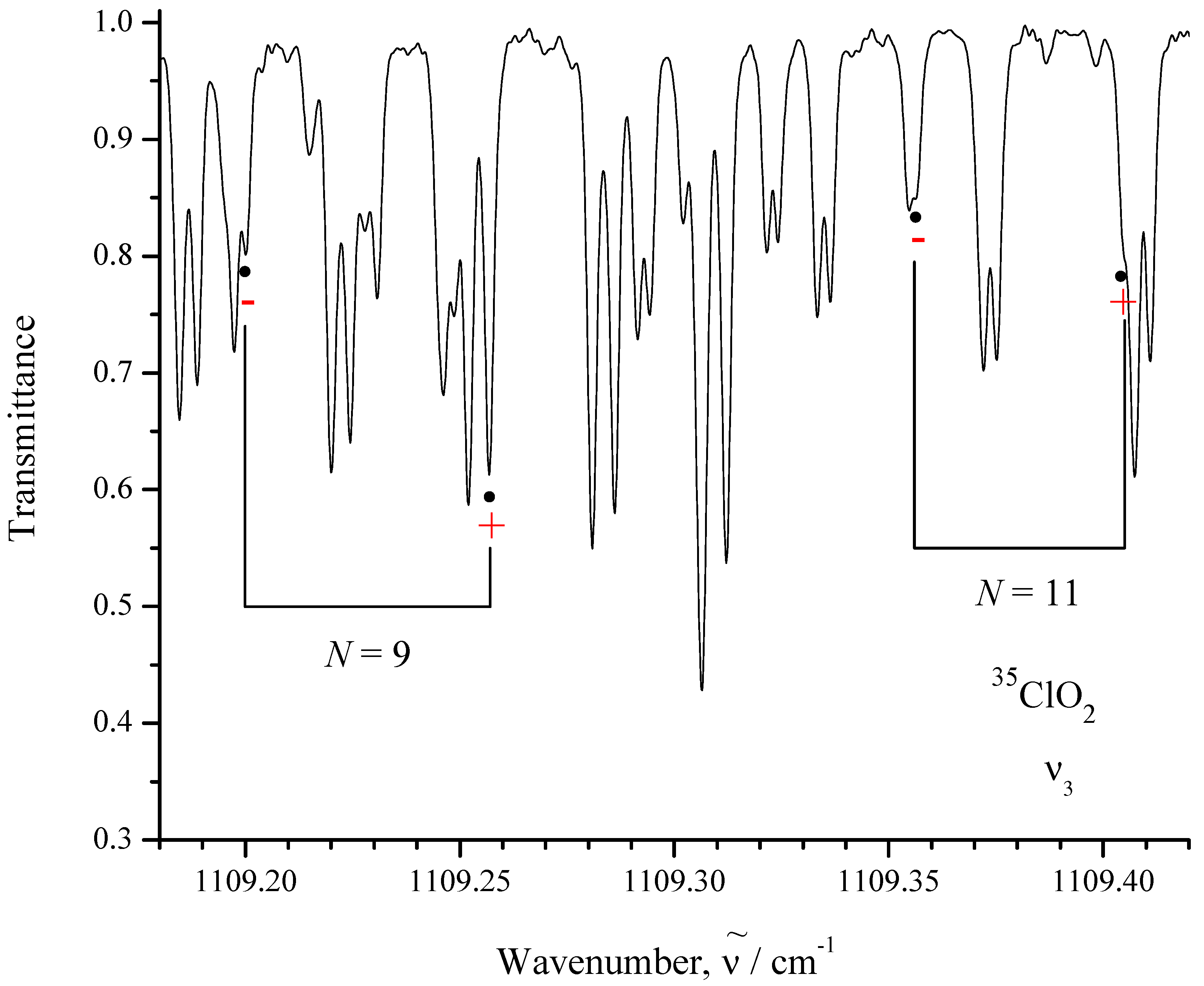
| Transition | Transmitt. | ||||
|---|---|---|---|---|---|
| − | in cm−1 | in cm−1 | in % | ||
| 1 | 2 | 3 | 4 | ||
| − | 1106.7267 | 1106.7262 | 92 | ||
| − | 1106.7824 | 1106.7828 | 92 | ||
| − | 1107.7978 | 1107.7976 | 95 | ||
| − | 1107.8319 | 1107.8315 | 89 | ||
| − | 1108.6547 | 1108.6546 | 86 | ||
| − | 1108.6882 | covered | 62 | ||
| − | 1109.1992 | 1109.2000 | 80 | ||
| − | 1109.2575 | covered | 61 | ||
| − | 1109.3568 | 1109.3563 | 85 | ||
| − | 1109.4050 | 1109.4050 | 80 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ulenikov, O.; Bekhtereva, E.; Gromova, O.; Kakaulin, A.; Sydow, C.; Bauerecker, S. Improved Theory of the Effective Dipole Moments and Absolute Line Strengths of the XY2 Asymmetric Top Molecules in the X2B1 Doublet Electronic States. Int. J. Mol. Sci. 2023, 24, 12734. https://doi.org/10.3390/ijms241612734
Ulenikov O, Bekhtereva E, Gromova O, Kakaulin A, Sydow C, Bauerecker S. Improved Theory of the Effective Dipole Moments and Absolute Line Strengths of the XY2 Asymmetric Top Molecules in the X2B1 Doublet Electronic States. International Journal of Molecular Sciences. 2023; 24(16):12734. https://doi.org/10.3390/ijms241612734
Chicago/Turabian StyleUlenikov, Oleg, Elena Bekhtereva, Olga Gromova, Aleksei Kakaulin, Christian Sydow, and Sigurd Bauerecker. 2023. "Improved Theory of the Effective Dipole Moments and Absolute Line Strengths of the XY2 Asymmetric Top Molecules in the X2B1 Doublet Electronic States" International Journal of Molecular Sciences 24, no. 16: 12734. https://doi.org/10.3390/ijms241612734
APA StyleUlenikov, O., Bekhtereva, E., Gromova, O., Kakaulin, A., Sydow, C., & Bauerecker, S. (2023). Improved Theory of the Effective Dipole Moments and Absolute Line Strengths of the XY2 Asymmetric Top Molecules in the X2B1 Doublet Electronic States. International Journal of Molecular Sciences, 24(16), 12734. https://doi.org/10.3390/ijms241612734