
Detection and Monitoring of Tumor-Derived Mutations in Circulating Tumor DNA Using the UltraSEEK Lung Panel on the MassARRAY System in Metastatic Non-Small Cell Lung Cancer Patients
 , , , , , , , , , and
, , , , , , , , , and 
Abstract
:1. Introduction
2. Results
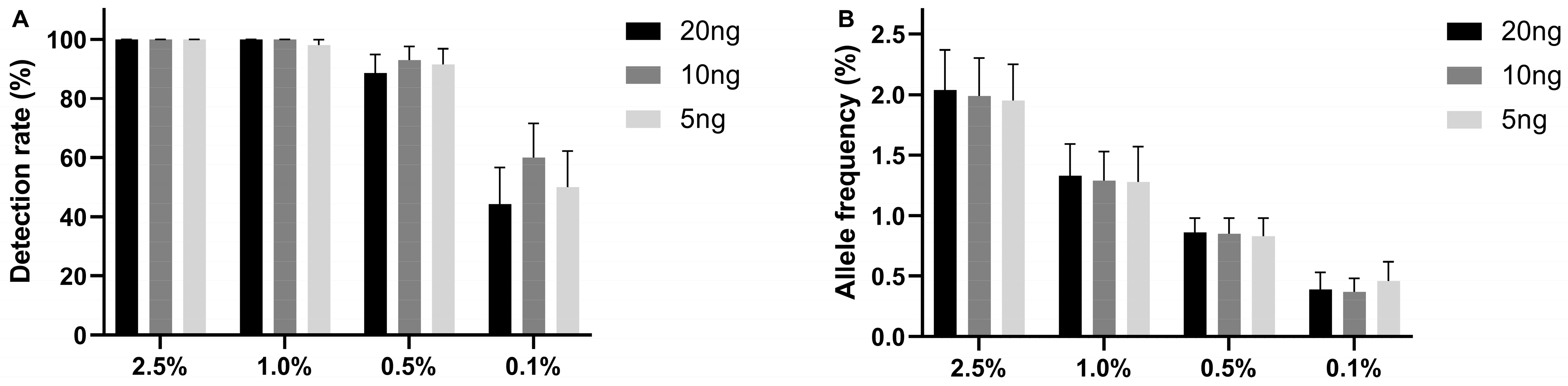
2.1. The Limit of Detection of the UltraSEEK® Lung Panel Using Reference Material
2.2. Comparison of Detection of Mutant ctDNA in Patient-Derived Plasma between UltraSEEK® and ddPCR
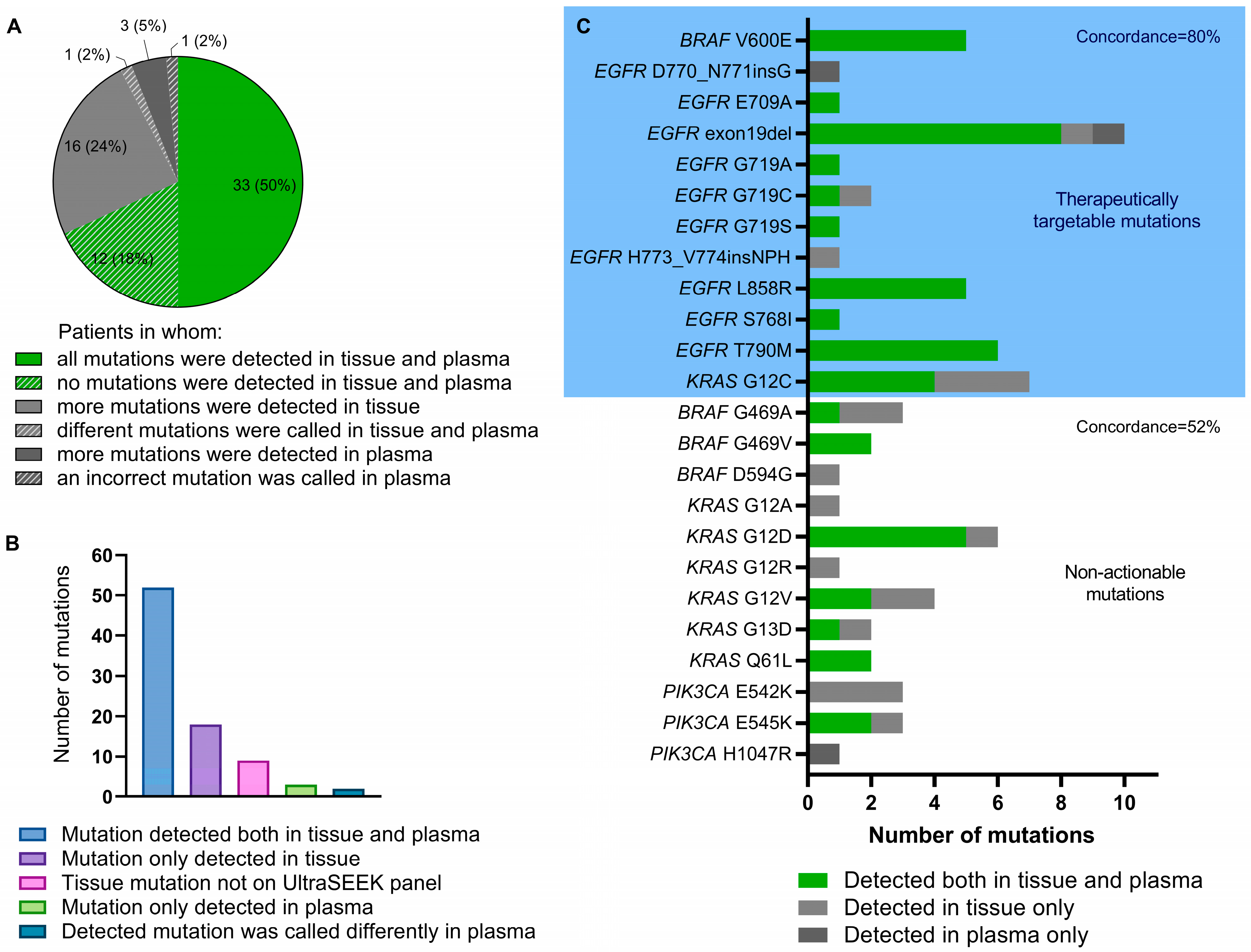
2.3. Detection of Tumor Tissue-Derived Variants in the Baseline Plasma Sample Using the UltraSEEK® Lung Panel
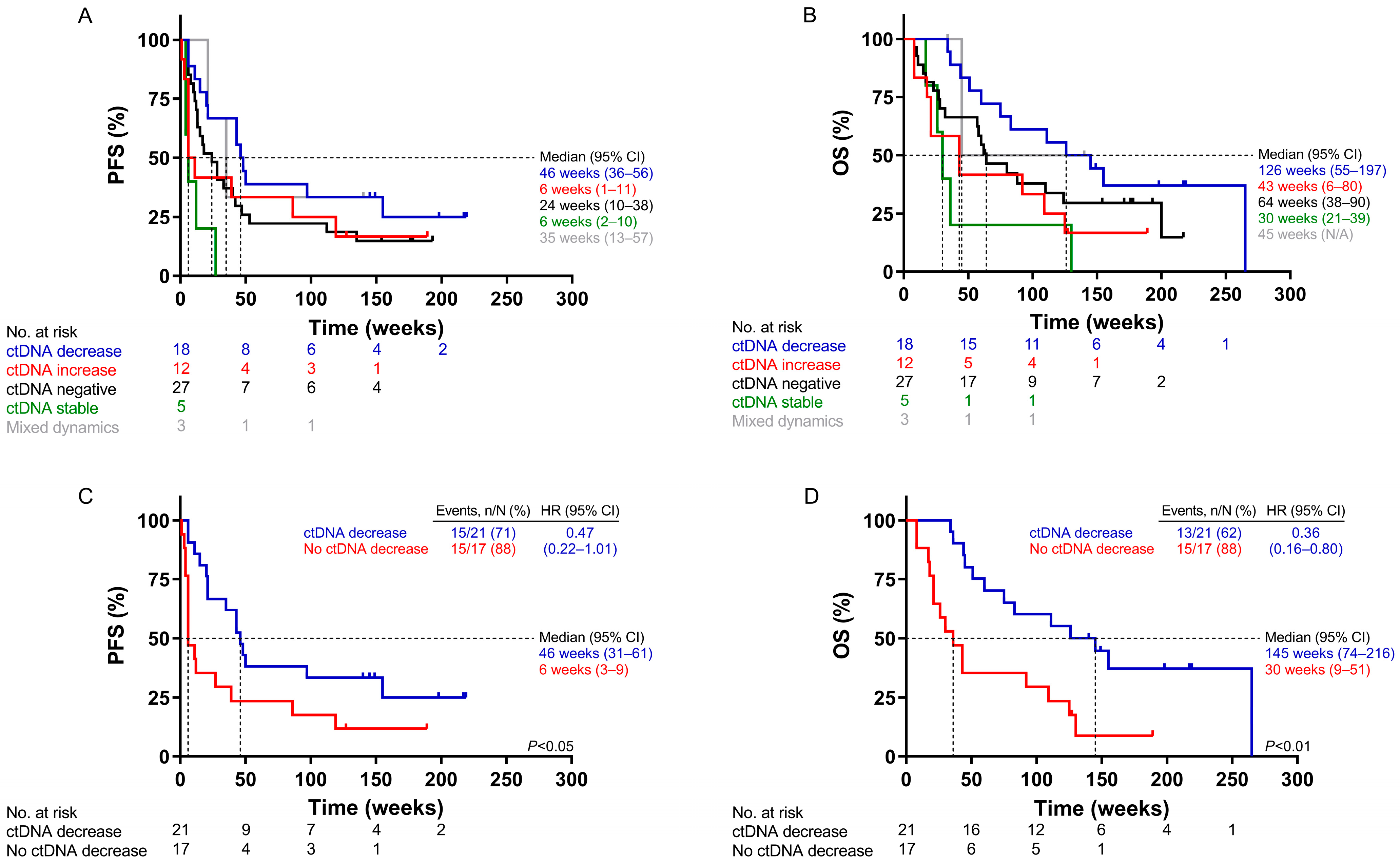
2.4. Survival Analysis According to ctDNA Dynamics Using UltraSEEK®
2.5. Plasma-Based Determination of Disease Progression and Acquired Treatment Resistance
3. Discussion
4. Materials and Methods
4.1. Sample Collection
4.2. Molecular Analysis
4.3. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statements
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Heitzer, E.; Haque, I.S.; Roberts, C.E.S.; Speicher, M.R. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat. Rev. Genet. 2019, 20, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Boonstra, P.A.; Wind, T.T.; van Kruchten, M.; Schuuring, E.; Hospers, G.A.P.; van der Wekken, A.J.; de Groot, D.-J.; Schröder, C.P.; Fehrmann, R.S.N.; Reyners, A.K.L. Clinical utility of circulating tumor DNA as a response and follow-up marker in cancer therapy. Cancer Metastasis Rev. 2020, 39, 999–1013. [Google Scholar] [CrossRef] [PubMed]
- NCCN Clinical Practice Guidelines in Oncology: Non-Small Cell Lung Cancer. Available online: https://www.nccn.org/login?ReturnURL=https://www.nccn.org/professionals/physician_gls/PDF/nscl.pdf (accessed on 14 June 2023).
- Planchard, D.; Popat, S.; Kerr, K.; Novello, S.; Smit, E.F.; Faivre-Finn, C.; Mok, T.S.; Reck, M.; Van Schil, P.E.; Hellmann, M.D.; et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29 (Suppl. S4), iv192–iv237. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.C.; Carpenter, E.L.; Silva, B.A.; Rosenstein, J.; Chien, A.L.; Quinn, K.; Espenschied, C.R.; Mak, A.; Kiedrowski, L.A.; Lefterova, M.; et al. Serial Monitoring of Circulating Tumor DNA by Next-Generation Gene Sequencing as a Biomarker of Response and Survival in Patients with Advanced NSCLC Receiving Pembrolizumab-Based Therapy. JCO Precis. Oncol. 2021, 5, 510–524. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, S.B.; Narayan, A.; Kole, A.J.; Decker, R.H.; Teysir, J.; Carriero, N.J.; Lee, A.; Nemati, R.; Nath, S.K.; Mane, S.M.; et al. Early assessment of lung cancer immunotherapy response via circulating tumor DNA. Clin. Cancer Res. 2018, 24, 1872–1880. [Google Scholar] [CrossRef] [PubMed]
- Leest, P.v.d.; Hiddinga, B.; Miedema, A.; Aguirre Azpurua, M.L.; Rifaela, N.; Elst, A.t.; Timens, W.; Groen, H.J.M.; Kempen, L.C.v.; Hiltermann, T.J.N.; et al. Circulating tumor DNA as a biomarker for monitoring early treatment responses of patients with advanced lung adenocarcinoma receiving immune checkpoint inhibitors. Mol. Oncol. 2021, 15, 2910–2922. [Google Scholar] [CrossRef]
- Weber, S.; van der Leest, P.; Donker, H.C.; Schlange, T.; Timens, W.; Tamminga, M.; Hasenleithner, S.O.; Graf, R.; Moser, T.; Spiegl, B.; et al. Dynamic Changes of Circulating Tumor DNA Predict Clinical Outcome in Patients with Advanced Non–Small-Cell Lung Cancer Treated with Immune Checkpoint Inhibitors. JCO Precis. Oncol. 2021, 5, 1540–1553. [Google Scholar] [CrossRef] [PubMed]
- Oxnard, G.R.; Paweletz, C.P.; Kuang, Y.; Mach, S.L.; O’Connell, A.; Messineo, M.M.; Luke, J.J.; Butaney, M.; Kirschmeier, P.; Jackman, D.M.; et al. Noninvasive detection of response and resistance in EGFR-mutant lung cancer using quantitative next-generation genotyping of cell-free plasma DNA. Clin. Cancer Res. 2014, 20, 1698–1705. [Google Scholar] [CrossRef]
- Heitzer, E.; Ulz, P.; Geigl, J.B. Circulating tumor DNA as a liquid biopsy for cancer. Clin. Chem. 2015, 61, 112–123. [Google Scholar] [CrossRef]
- Rangachari, D.; To, C.; Shpilsky, J.E.; VanderLaan, P.A.; Kobayashi, S.S.; Mushajiang, M.; Lau, C.J.; Paweletz, C.P.; Oxnard, G.R.; Jänne, P.A.; et al. EGFR-Mutated Lung Cancers Resistant to Osimertinib through EGFR C797S Respond to First-Generation Reversible EGFR Inhibitors but Eventually Acquire EGFR T790M/C797S in Preclinical Models and Clinical Samples. J. Thorac. Oncol. 2019, 14, 1995–2002. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.-L.; Ahn, M.-J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.M.E.; et al. Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef]
- Merker, J.D.; Oxnard, G.R.; Compton, C.; Diehn, M.; Hurley, P.; Lazar, A.J.; Lindeman, N.; Lockwood, C.M.; Rai, A.J.; Schilsky, R.L.; et al. Circulating Tumor DNA Analysis in Patients with Cancer: American Society of Clinical Oncology and College of American Pathologists Joint Review. J. Clin. Oncol. 2018, 36, 1631–1641. [Google Scholar] [CrossRef] [PubMed]
- Lampignano, R.; Neumann, M.H.D.; Weber, S.; Kloten, V.; Herdean, A.; Voss, T.; Groelz, D.; Babayan, A.; Tibbesma, M.; Schlumpberger, M.; et al. Multicenter Evaluation of Circulating Cell-Free DNA Extraction and Downstream Analyses for the Development of Standardized (Pre)analytical Work Flows. Clin. Chem. 2020, 66, 149–160. [Google Scholar] [CrossRef]
- Wan, J.C.M.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Leest, P.v.d.; Ketelaar, E.M.; Noesel, C.J.M.v.; Broek, D.v.d.; Boerdonk, R.A.A.v.; Deiman, B.; Rifaela, N.; Geize, R.v.d.; Huijsmans, C.J.J.; Speel, E.J.M.; et al. Dutch National Round Robin Trial on Plasma-Derived Circulating Cell-Free DNA Extraction Methods Routinely Used in Clinical Pathology for Molecular Tumor Profiling. Clin. Chem. 2022, 68, 963–972. [Google Scholar] [CrossRef] [PubMed]
- van der Leest, P.; Boonstra, P.A.; ter Elst, A.; van Kempen, L.C.; Tibbesma, M.; Koopmans, J.; Miedema, A.; Tamminga, M.; Groen, H.J.M.; Reyners, A.K.L.; et al. Comparison of Circulating Cell-Free DNA Extraction Methods for Downstream Analysis in Cancer Patients. Cancers 2020, 12, 1222. [Google Scholar] [CrossRef]
- Lamy, P.-J.; Castan, F.; Lozano, N.; Montélion, C.; Audran, P.; Bibeau, F.; Roques, S.; Montels, F.; Laberenne, A.-C. Next-Generation Genotyping by Digital PCR to Detect and Quantify the BRAF V600E Mutation in Melanoma Biopsies. J. Mol. Diagn. 2015, 17, 366–373. [Google Scholar] [CrossRef]
- Lee, J.H.; Menzies, A.M.; Carlino, M.S.; McEvoy, A.C.; Sandhu, S.; Weppler, A.M.; Diefenbach, R.J.; Dawson, S.-J.; Kefford, R.F.; Millward, M.J.; et al. Longitudinal Monitoring of ctDNA in Patients with Melanoma and Brain Metastases Treated with Immune Checkpoint Inhibitors. Clin. Cancer Res. 2020, 26, 4064–4071. [Google Scholar] [CrossRef]
- Steeghs, E.M.P.; Kroeze, L.I.; Tops, B.B.J.; Van Kempen, L.C.; Ter Elst, A.; Raaij, A.W.M.K.-V.; Hendriks-Cornelissen, S.J.B.; Hermsen, M.J.W.; Jansen, E.A.M.; Nederlof, P.M.; et al. Comprehensive routine diagnostic screening to identify predictive mutations, gene amplifications, and microsatellite instability in FFPE tumor material. BMC Cancer 2020, 20, 291. [Google Scholar] [CrossRef]
- Aggarwal, C.; Thompson, J.C.; Black, T.A.; Katz, S.I.; Fan, R.; Yee, S.S.; Chien, A.L.; Evans, T.L.; Bauml, J.M.; Alley, E.W.; et al. Clinical Implications of Plasma-Based Genotyping with the Delivery of Personalized Therapy in Metastatic Non-Small Cell Lung Cancer. JAMA Oncol. 2019, 5, 173–180. [Google Scholar] [CrossRef]
- Ignatiadis, M.; Sledge, G.W.; Jeffrey, S.S. Liquid biopsy enters the clinic—implementation issues and future challenges. Nat. Rev. Clin. Oncol. 2021, 18, 297–312. [Google Scholar] [CrossRef]
- Heitzer, E.; Broek, D.v.D.; Denis, M.; Hofman, P.; Hubank, M.; Mouliere, F.; Paz-Ares, L.; Schuuring, E.; Sültmann, H.; Vainer, G.; et al. Recommendations for a practical implementation of circulating tumor DNA mutation testing in metastatic non-small-cell lung cancer. ESMO Open 2022, 7, 100399. [Google Scholar] [CrossRef]
- Weber, S.; Spiegl, B.; Perakis, S.O.; Ulz, C.M.; Abuja, P.M.; Kashofer, K.; van der Leest, P.; Azpurua, M.A.; Tamminga, M.; Brudzewsky, D.; et al. Technical Evaluation of Commercial Mutation Analysis Platforms and Reference Materials for Liquid Biopsy Profiling. Cancers 2020, 12, 1588. [Google Scholar] [CrossRef]
- Kramer, A.; Schuuring, E.; Vessies, D.C.; van der Leest, P.; Geerlings, M.J.; Rozendal, P.; Lanfermeijer, M.; Linders, T.C.; van Kempen, L.C.; Fijneman, R.J.A.; et al. A Micro-Costing Framework for Circulating Tumor DNA Testing in Dutch Clinical Practice. J. Mol. Diagn. 2023, 25, 36–45. [Google Scholar] [CrossRef]
- List of Cleared or Approved Companion Diagnostic Devices (In Vitro and Imaging Tools). Available online: https://www.fda.gov/medical-devices/in-vitro-diagnostics/list-cleared-or-approved-companion-diagnostic-devices-in-vitro-and-imaging-tools (accessed on 14 June 2023).
- Lamy, P.-J.; van der Leest, P.; Lozano, N.; Becht, C.; Duboeuf, F.; Groen, H.J.M.; Hilgers, W.; Pourel, N.; Rifaela, N.; Schuuring, E.; et al. Mass Spectrometry as a Highly Sensitive Method for Specific Circulating Tumor DNA Analysis in NSCLC: A Comparison Study. Cancers 2020, 12, 3002. [Google Scholar] [CrossRef]
- Kulasinghe, A.; O’Leary, C.; Monkman, J.; Bharti, V.; Irwin, D.; Dutta, S.; Richard, D.J.; Hughes, B.; Ladwa, R.; O’Byrne, K. The identification of circulating tumour DNA using MassARRAY technology in non-small-cell lung cancer (NSCLC). Lung Cancer 2021, 160, 73–77. [Google Scholar] [CrossRef]
- Belloum, Y.; Janning, M.; Mohme, M.; Simon, R.; Kropidlowski, J.; Sartori, A.; Irwin, D.; Westphal, M.; Lamszus, K.; Loges, S.; et al. Discovery of Targetable Genetic Alterations in NSCLC Patients with Different Metastatic Patterns Using a MassARRAY-Based Circulating Tumor DNA Assay. Cells 2020, 9, 2337. [Google Scholar] [CrossRef] [PubMed]
- Gray, E.S.; Witkowski, T.; Pereira, M.; Calapre, L.; Herron, K.; Irwin, D.; Chapman, B.; Khattak, M.A.; Raleigh, J.; Hatzimihalis, A.; et al. Genomic Analysis of Circulating Tumor DNA Using a Melanoma-Specific UltraSEEK Oncogene Panel. J. Mol. Diagn. 2019, 21, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Mehrotra, M.; Singh, R.R.; Loghavi, S.; Duose, D.Y.; Barkoh, B.A.; Behrens, C.; Patel, K.P.; Routbort, M.J.; Kopetz, S.; Broaddus, R.R.; et al. Detection of somatic mutations in cell-free DNA in plasma and correlation with overall survival in patients with solid tumors. Oncotarget 2018, 9, 10259–10271. [Google Scholar] [CrossRef]
- Kapeleris, J.; Müller Bark, J.; Ranjit, S.; Irwin, D.; Hartel, G.; Warkiani, M.E.; Leo, P.; O’Leary, C.; Ladwa, R.; O’Byrne, K.; et al. Prognostic value of integrating circulating tumour cells and cell-free DNA in non-small cell lung cancer. Heliyon 2022, 8, e09971. [Google Scholar] [CrossRef] [PubMed]
- Remon, J.; Lacroix, L.; Jovelet, C.; Caramella, C.; Howarth, K.; Plagnol, V.; Rosenfeld, N.; Morris, C.; Mezquita, L.; Pannet, C.; et al. Real-World Utility of an Amplicon-Based Next-Generation Sequencing Liquid Biopsy for Broad Molecular Profiling in Patients with Advanced Non–Small-Cell Lung Cancer. JCO Precis. Oncol. 2019, 3, 1–14. [Google Scholar] [CrossRef]
- Nagano, T.; Tachihara, M.; Nishimura, Y. Mechanism of Resistance to Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitors and a Potential Treatment Strategy. Cells 2018, 7, 212. [Google Scholar] [CrossRef]
- Schmid, S.; Li, J.J.N.; Leighl, N.B. Mechanisms of osimertinib resistance and emerging treatment options. Lung Cancer 2020, 147, 123–129. [Google Scholar] [CrossRef]
- Paik, P.K.; Arcila, M.E.; Fara, M.; Sima, C.S.; Miller, V.A.; Kris, M.G.; Ladanyi, M.; Riely, G.J. Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations. J. Clin. Oncol. 2011, 29, 2046–2051. [Google Scholar] [CrossRef]
- Anguera, G.; Majem, M. BRAF inhibitors in metastatic non-small cell lung cancer. J. Thorac. Dis. 2018, 10, 589–592. [Google Scholar] [CrossRef] [PubMed]
- Weber, B.; Meldgaard, P.; Hager, H.; Wu, L.; Wei, W.; Tsai, J.; Khalil, A.; Nexo, E.; Sorensen, B.S. Detection of EGFR mutations in plasma and biopsies from non-small cell lung cancer patients by allele-specific PCR assays. BMC Cancer 2014, 14, 294. [Google Scholar] [CrossRef] [PubMed]
- Syed, Y.Y. therascreen ® EGFR RGQ PCR Kit: A Companion Diagnostic for Afatinib and Gefitinib in Non-Small Cell Lung Cancer. Mol. Diagn. Ther. 2016, 20, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Shen, L.; Zheng, D. Diagnostic value of circulating free DNA for the detection of EGFR mutation status in NSCLC: A systematic review and meta-analysis. Sci. Rep. 2014, 4, 6269. [Google Scholar] [CrossRef]
- Passaro, A.; Brahmer, J.; Antonia, S.; Mok, T.; Peters, S. Managing Resistance to Immune Checkpoint Inhibitors in Lung Cancer: Treatment and Novel Strategies. J. Clin. Oncol. 2022, 40, 598–610. [Google Scholar] [CrossRef]
- Du, X.; Shao, Y.; Qin, H.-F.; Tai, Y.-H.; Gao, H.-J. ALK-rearrangement in non-small-cell lung cancer (NSCLC). Thorac. Cancer 2018, 9, 423–430. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Vicente, D.; Tafreshi, A.; Robinson, A.; Soto Parra, H.; Mazières, J.; Hermes, B.; Cicin, I.; Medgyasszay, B.; Rodríguez-Cid, J.; et al. A Randomized, Placebo-Controlled Trial of Pembrolizumab Plus Chemotherapy in Patients with Metastatic Squamous NSCLC: Protocol-Specified Final Analysis of KEYNOTE-407. J. Thorac. Oncol. 2020, 15, 1657–1669. [Google Scholar] [CrossRef] [PubMed]
- Guibert, N.; Jones, G.; Beeler, J.F.; Plagnol, V.; Morris, C.; Mourlanette, J.; Delaunay, M.; Keller, L.; Rouquette, I.; Favre, G.; et al. Targeted sequencing of plasma cell-free DNA to predict response to PD1 inhibitors in advanced non-small cell lung cancer. Lung Cancer 2019, 137, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Zulato, E.; Attili, I.; Pavan, A.; Nardo, G.; Del Bianco, P.; Boscolo Bragadin, A.; Verza, M.; Pasqualini, L.; Pasello, G.; Fassan, M.; et al. Early assessment of KRAS mutation in cfDNA correlates with risk of progression and death in advanced non-small-cell lung cancer. Br. J. Cancer 2020, 123, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Boonstra, P.A.; Elst, A.t.; Tibbesma, M.; Bosman, L.J.; Mathijssen, R.; Atrafi, F.; Coevorden, F.v.; Steeghs, N.; Farag, S.; Gelderblom, H.; et al. A single digital droplet PCR assay to detect multiple KIT exon 11 mutations in tumor and plasma from patients with gastrointestinal stromal tumors. Oncotarget 2018, 9, 13870–13883. [Google Scholar] [CrossRef]
- Gray, J.E.; Okamoto, I.; Sriuranpong, V.; Vansteenkiste, J.; Imamura, F.; Lee, J.S.; Pang, Y.-K.; Cobo, M.; Kasahara, K.; Cheng, Y.; et al. Tissue and Plasma EGFR Mutation Analysis in the FLAURA Trial: Osimertinib versus Comparator EGFR Tyrosine Kinase Inhibitor as First-Line Treatment in Patients with EGFR-Mutated Advanced Non-Small Cell Lung Cancer. Clin. Cancer Res. 2019, 25, 6644–6652. [Google Scholar] [CrossRef]
- Mosko, M.J.; Nakorchevsky, A.A.; Flores, E.; Metzler, H.; Ehrich, M.; van den Boom, D.J.; Sherwood, J.L.; Nygren, A.O.H. Ultrasensitive Detection of Multiplexed Somatic Mutations Using MALDI-TOF Mass Spectrometry. J. Mol. Diagn. 2016, 18, 23–31. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Total Cohort | UMCG Cohort | UKE Cohort | |
|---|---|---|---|
| Patients | 72 | 60 | 12 |
| Median age | 65 (38–85) | 63 (38–85) | 68 (57–79) |
| Sex | |||
| Male | 37 (51%) | 29 (48%) | 8 (67%) |
| Female | 35 (49%) | 31 (52%) | 4 (33%) |
| ECOG PS | |||
| 0 | 23 (32%) | 21 (35%) | 2 (17%) |
| 1 | 44 (61%) | 37 (62%) | 7 (58%) |
| 2 | 5 (7%) | 2 (3%) | 3 (25%) |
| Histology | |||
| Adenocarcinoma | 64 (89%) | 56 (93%) | 8 (67%) |
| Squamous cell carcinoma | 8 (11%) | 4 (7%) | 4 (33%) |
| Smoking status | |||
| Active smoker | 33 (46%) | 29 (48%) | 4 (33%) |
| Former smoker | 27 (38%) | 23 (38%) | 4 (33%) |
| Never smoker | 11 (15%) | 8 (13%) | 3 (25%) |
| Unknown | 1 (1%) | 1 (8%) | |
| Current treatment | |||
| Chemotherapy | 3 (4%) | 1 (2%) | 2 (17%) |
| Carboplatine/pemetrexed | 1 (1%) | 1 (8%) | |
| Cisplatine/pemetrexed | 1 (1%) | 1 (2%) | |
| Docetaxel/ramucirumab | 1 (1%) | 1 (8%) | |
| Chemo-immunotherapy | 2 (3%) | 1 (2%) | 1 (8%) |
| Carboplatine/paclitaxel/bevacizumab | 1 (1%) | 1 (2%) | |
| Carboplatine/pemetrexed/pembrolizumab | 1 (1%) | 1 (8%) | |
| Immunotherapy | 54 (75%) | 45 (75%) | 9 (75%) |
| Atezolizumab | 1 (1%) | 1 (2%) | |
| Nivolumab | 40 (56%) | 37 (62%) | 3 (25%) |
| Pembrolizumab | 13 (18%) | 7 (12%) | 6 (50%) |
| Targeted therapy | 13 (18%) | 13 (22%) | |
| Afatinib | 7 (10%) | 7 (12%) | |
| Gefitinib | 1 (1%) | 1 (2%) | |
| Osimertinib | 5 (7%) | 5 (8%) | |
| Previous lines of therapies | |||
| 0 | 19 (26%) | 11 (18%) | 8 (67%) |
| 1 | 32 (44%) | 30 (50%) | 2 (17%) |
| 2 | 15 (21%) | 15 (25%) | |
| ≥3 | 6 (8%) | 4 (7%) | 2 (17%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leest, P.v.d.; Janning, M.; Rifaela, N.; Azpurua, M.L.A.; Kropidlowski, J.; Loges, S.; Lozano, N.; Sartori, A.; Irwin, D.; Lamy, P.-J.; et al. Detection and Monitoring of Tumor-Derived Mutations in Circulating Tumor DNA Using the UltraSEEK Lung Panel on the MassARRAY System in Metastatic Non-Small Cell Lung Cancer Patients. Int. J. Mol. Sci. 2023, 24, 13390. https://doi.org/10.3390/ijms241713390
Leest Pvd, Janning M, Rifaela N, Azpurua MLA, Kropidlowski J, Loges S, Lozano N, Sartori A, Irwin D, Lamy P-J, et al. Detection and Monitoring of Tumor-Derived Mutations in Circulating Tumor DNA Using the UltraSEEK Lung Panel on the MassARRAY System in Metastatic Non-Small Cell Lung Cancer Patients. International Journal of Molecular Sciences. 2023; 24(17):13390. https://doi.org/10.3390/ijms241713390
Chicago/Turabian StyleLeest, Paul van der, Melanie Janning, Naomi Rifaela, Maria L. Aguirre Azpurua, Jolanthe Kropidlowski, Sonja Loges, Nicolas Lozano, Alexander Sartori, Darryl Irwin, Pierre-Jean Lamy, and et al. 2023. "Detection and Monitoring of Tumor-Derived Mutations in Circulating Tumor DNA Using the UltraSEEK Lung Panel on the MassARRAY System in Metastatic Non-Small Cell Lung Cancer Patients" International Journal of Molecular Sciences 24, no. 17: 13390. https://doi.org/10.3390/ijms241713390
APA StyleLeest, P. v. d., Janning, M., Rifaela, N., Azpurua, M. L. A., Kropidlowski, J., Loges, S., Lozano, N., Sartori, A., Irwin, D., Lamy, P. -J., Hiltermann, T. J. N., Groen, H. J. M., Pantel, K., Kempen, L. C. v., Wikman, H., & Schuuring, E. (2023). Detection and Monitoring of Tumor-Derived Mutations in Circulating Tumor DNA Using the UltraSEEK Lung Panel on the MassARRAY System in Metastatic Non-Small Cell Lung Cancer Patients. International Journal of Molecular Sciences, 24(17), 13390. https://doi.org/10.3390/ijms241713390