

n-3 Polyunsaturated Fatty Acids Modulate LPS-Induced ARDS and the Lung–Brain Axis of Communication in Wild-Type versus Fat-1 Mice Genetically Modified for Leukotriene B4 Receptor 1 or Chemerin Receptor 23 Knockout
 ,
,  ,
, 
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
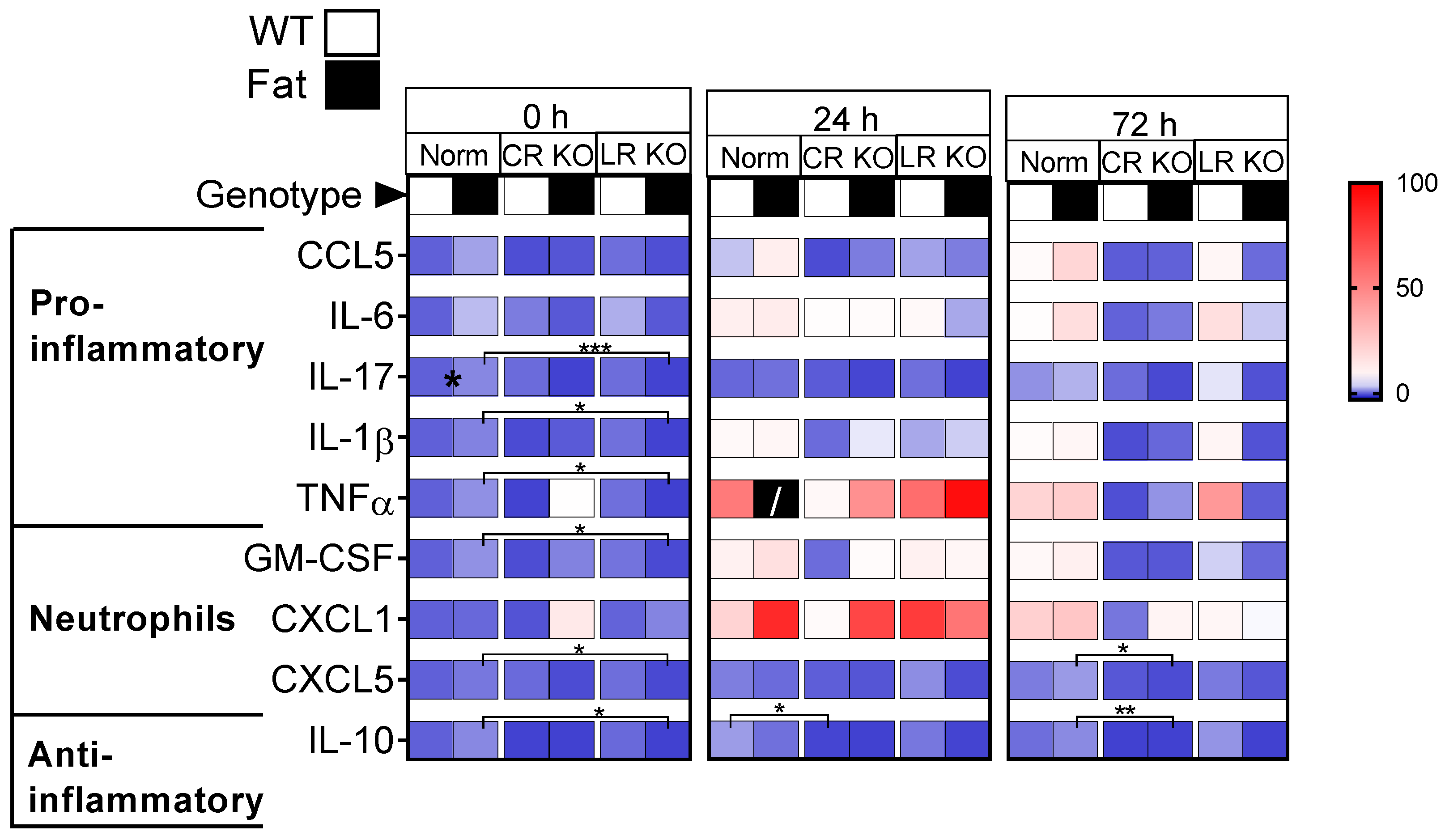
2.1. Protein Analyses of Inflammatory Mediators in the Lung: n-3 PUFAs and RvE1 Receptors Altered LPS-Induced Pro-Inflammatory Cytokines, Anti-Inflammatory IL-10, and Neutrophil Markers at 0 h, 24 h, and 72 h p.i. in CR and LR KO Mice
2.2. Analyses of Inflammatory Marker Proteins in the Liver: n-3 PUFAs and RvE1 Receptors Altered LPS-Induced Pro-Inflammatory Cytokines, Anti-Inflammatory IL-10, and Neutrophil Markers at 24 h p.i. in Norm, CR, and LR KO Mice
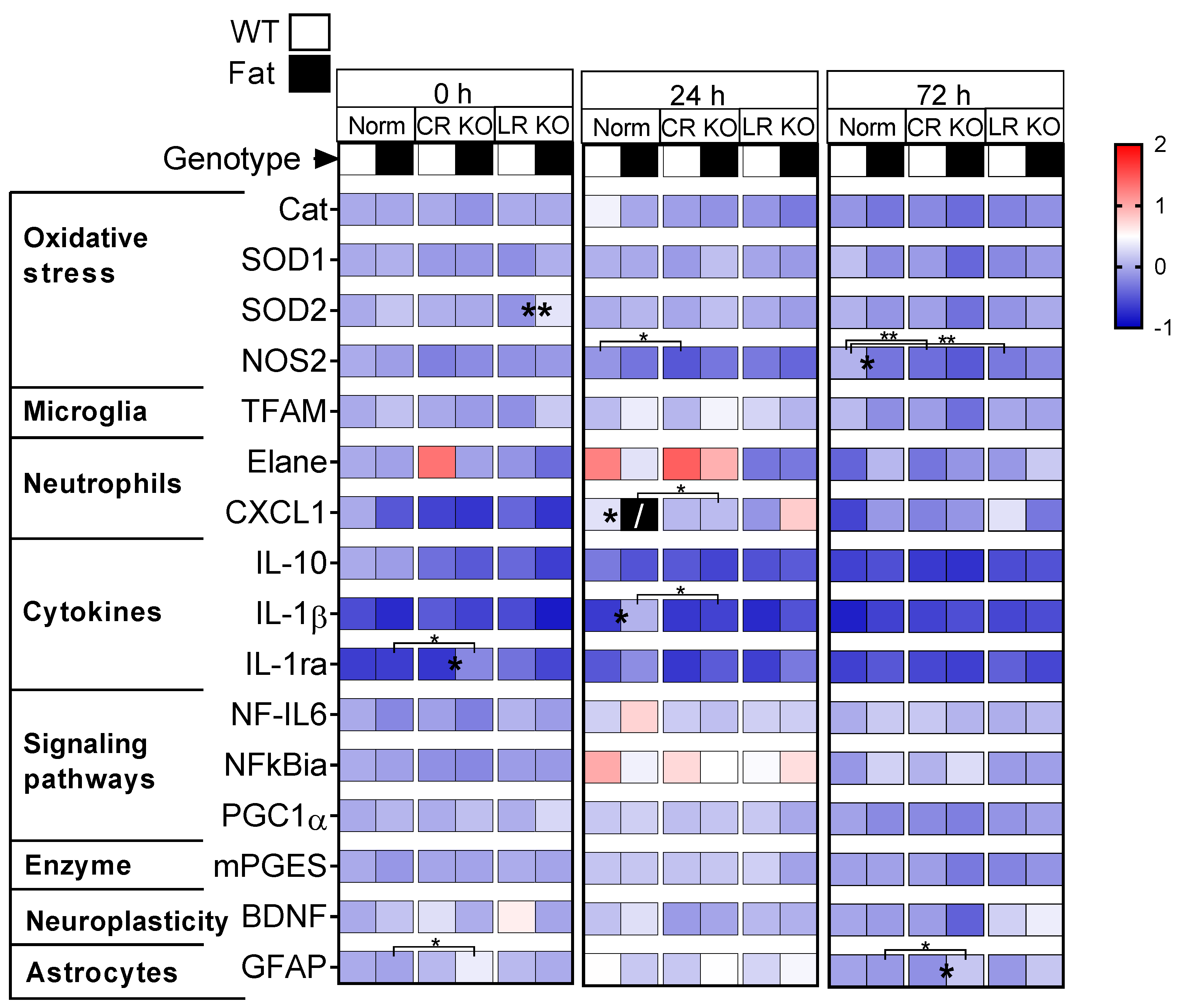
2.3. mRNA Analyses of Hypothalamic Inflammatory Marker Proteins: n-3 PUFAs and RvE1 Receptors Altered LPS-Induced Glial Activation, Neutrophil Markers, Pro-Inflammatory IL-1ß, Anti-Inflammatory IL-1ra and IL-10, Signaling Pathways, and the Enzyme Precursor for Prostaglandin Synthesis mPGES in Norm, CR, and LR KO Mice
2.4. Protein Analyses of Hypothalamic Oxidative Stress Markers and Immunohistochemical Detection of Total Astrocyte Area in the Paraventricular Nucleus: RvE1 Receptor Deficiency Increased Astrocytic Activation at 24 h p.i but Did Not Alter Mediators for Oxidative Stress in Norm or CR KO Mice
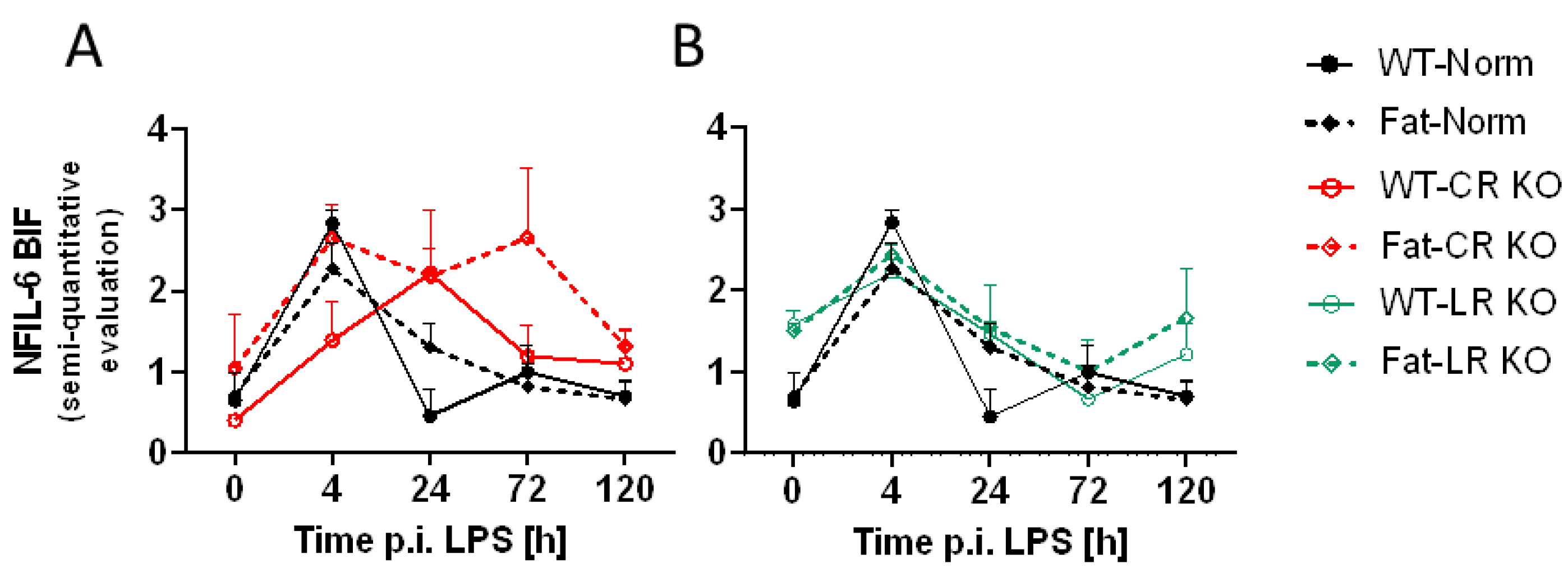
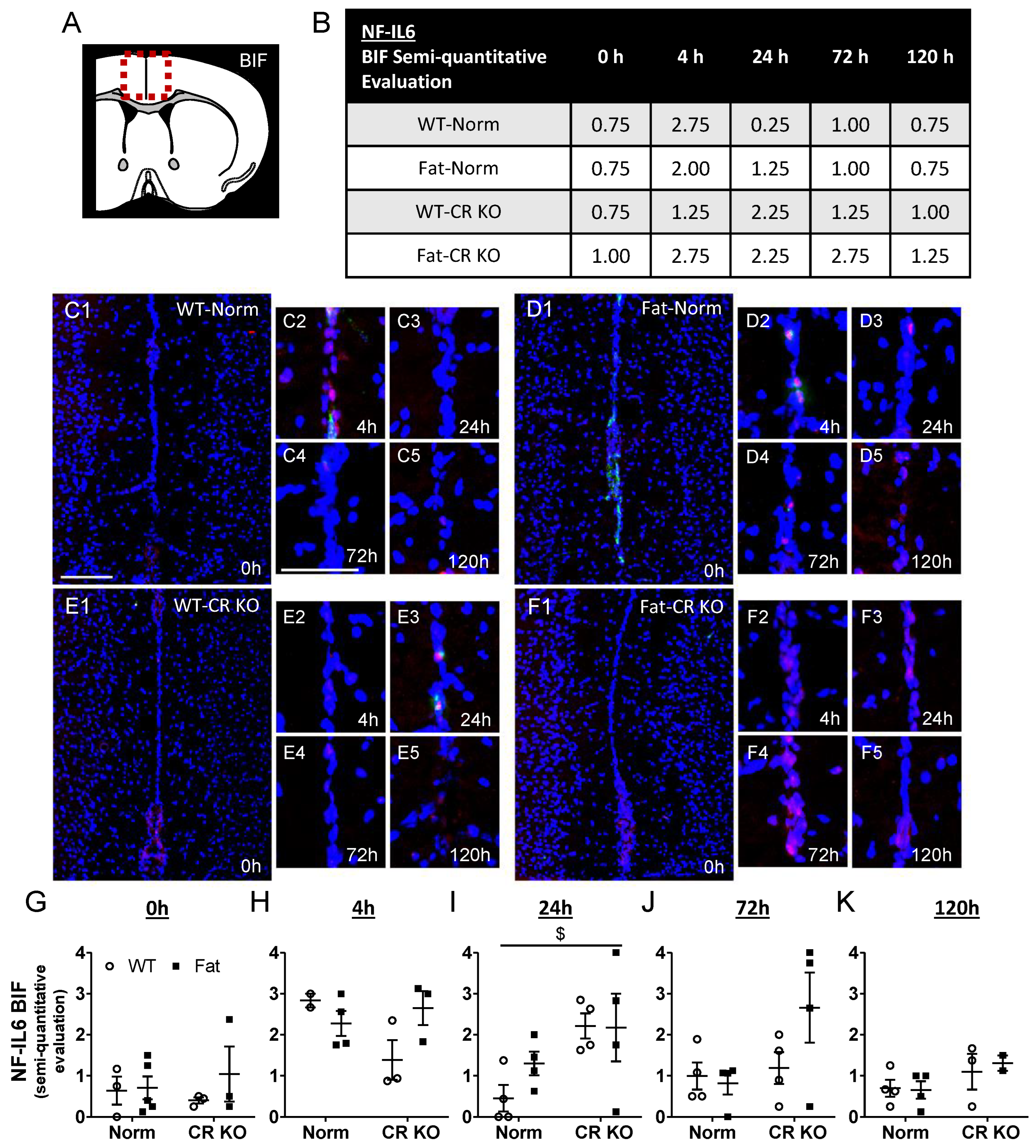
2.5. Immunohistochemical Detection of Inflammatory Transcription Factor Expression at the OVLT, a Brain Structure with a Leaky BBB, and Bifurcation, a Brain Structure with a Complete BBB: Preliminary Results Indicate That n-3 PUFAs and RvE1 Receptors May Alter LPS-Induced NF-IL6 Immunoreactivity at the BIF in Norm and CR KO Mice
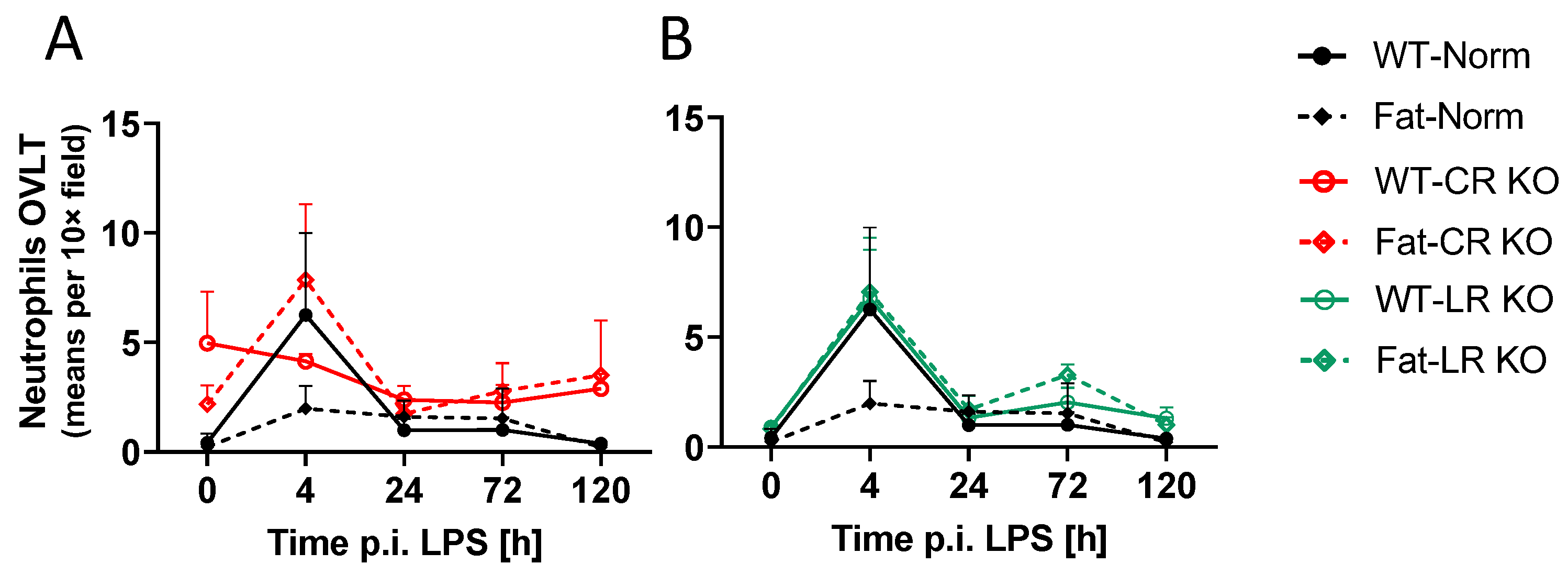
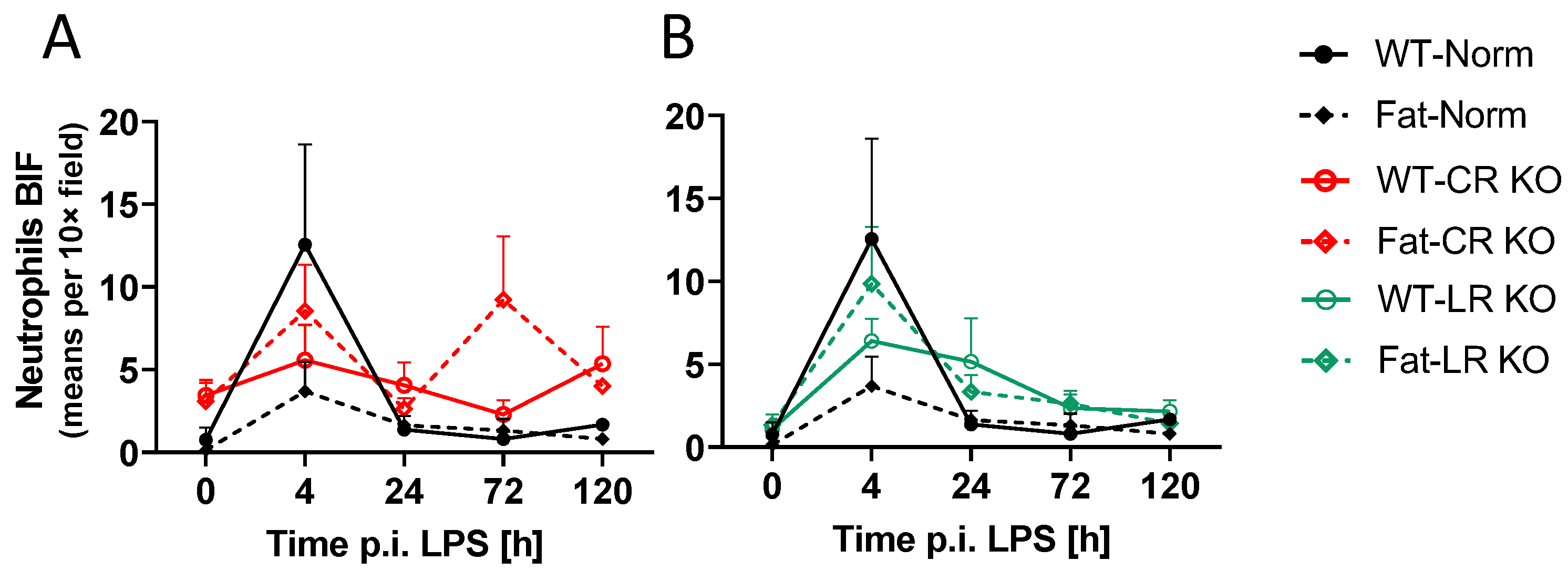
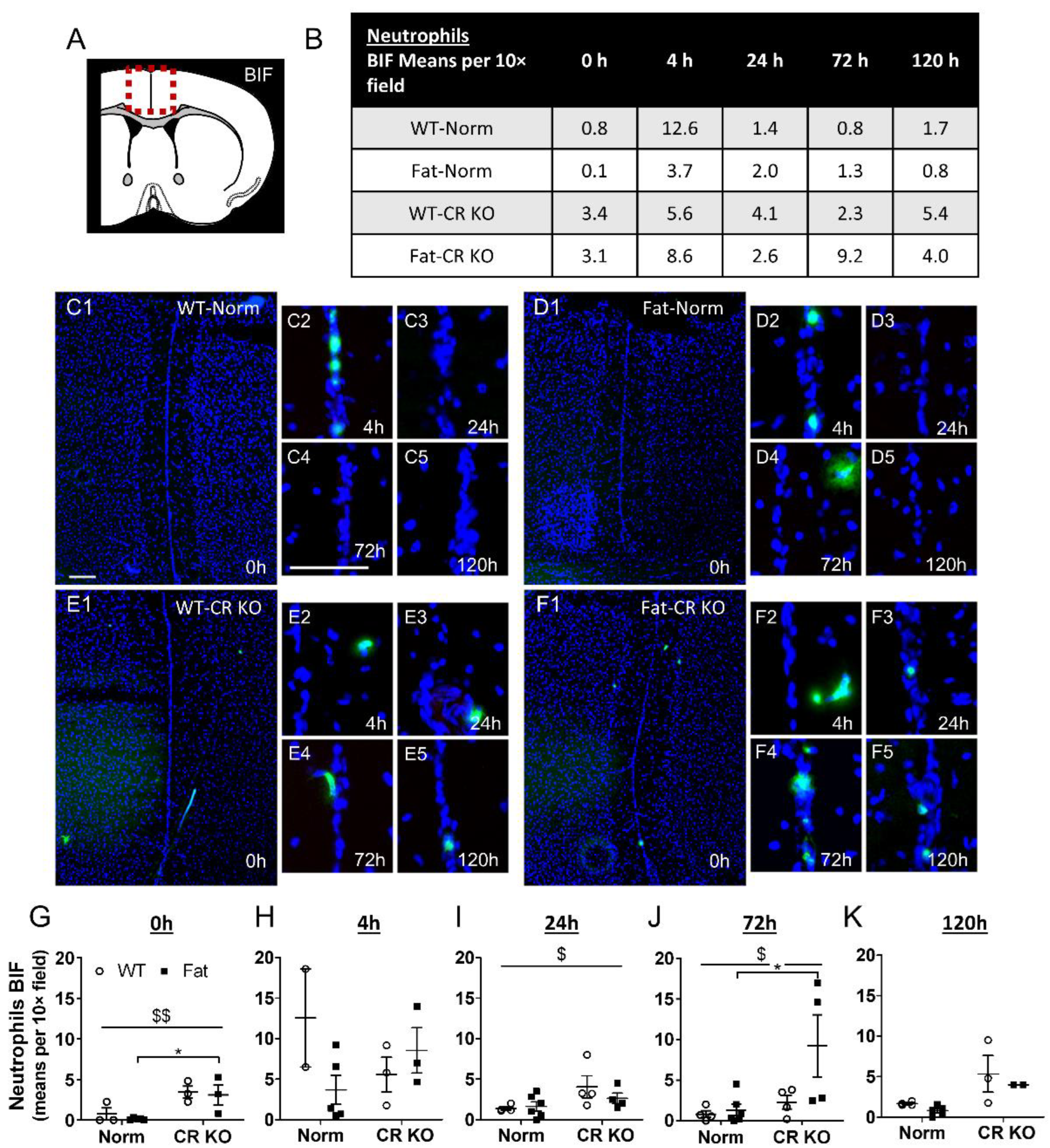
2.6. Analyses of Immune Cell Recruitment to the OVLT and Bifurcation: Preliminary Results Indicate That LPS Stimulated Neutrophil Recruitment in CR KO Mice May Be Altered in the OVLT by n-3 PUFAs at 4 h p.i. and by CR at 0 h and 120 h p.i. in the Bifurcation. LPS Stimulated Neutrophil Recruitment in CR KO Mice Were Altered by n-3 PUFAs at 0 h and 72 h p.i. and by CR at 0 h, 24 h and 72 h p.i.
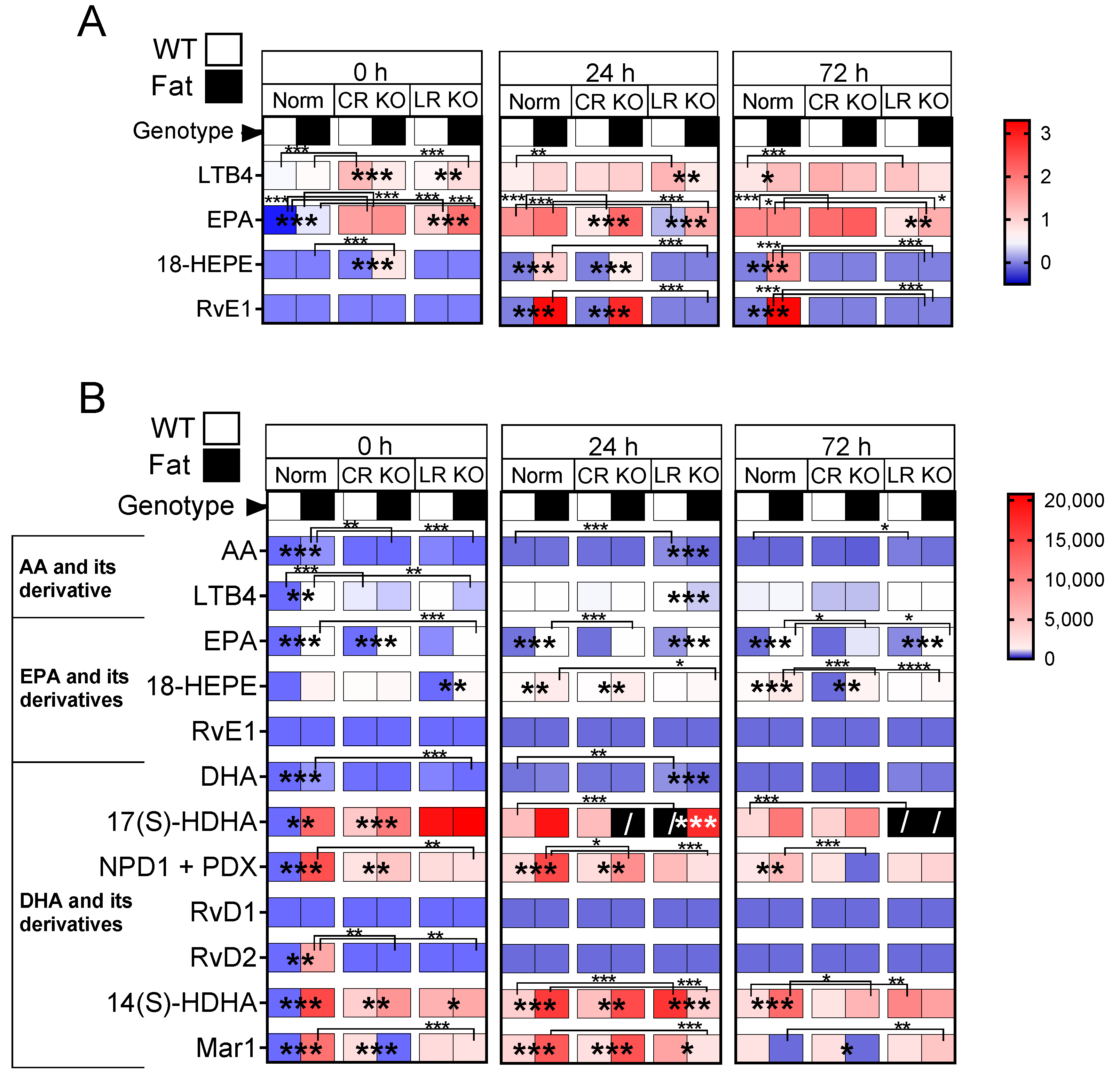
2.7. Analyses of Lipid Mediators in the Lung and Brain: n-3 PUFAs and RvE1 Receptors Altered Lipid Mediators at 0 h, 24 h, and 72 h p.i. in CR and LR KO Mice
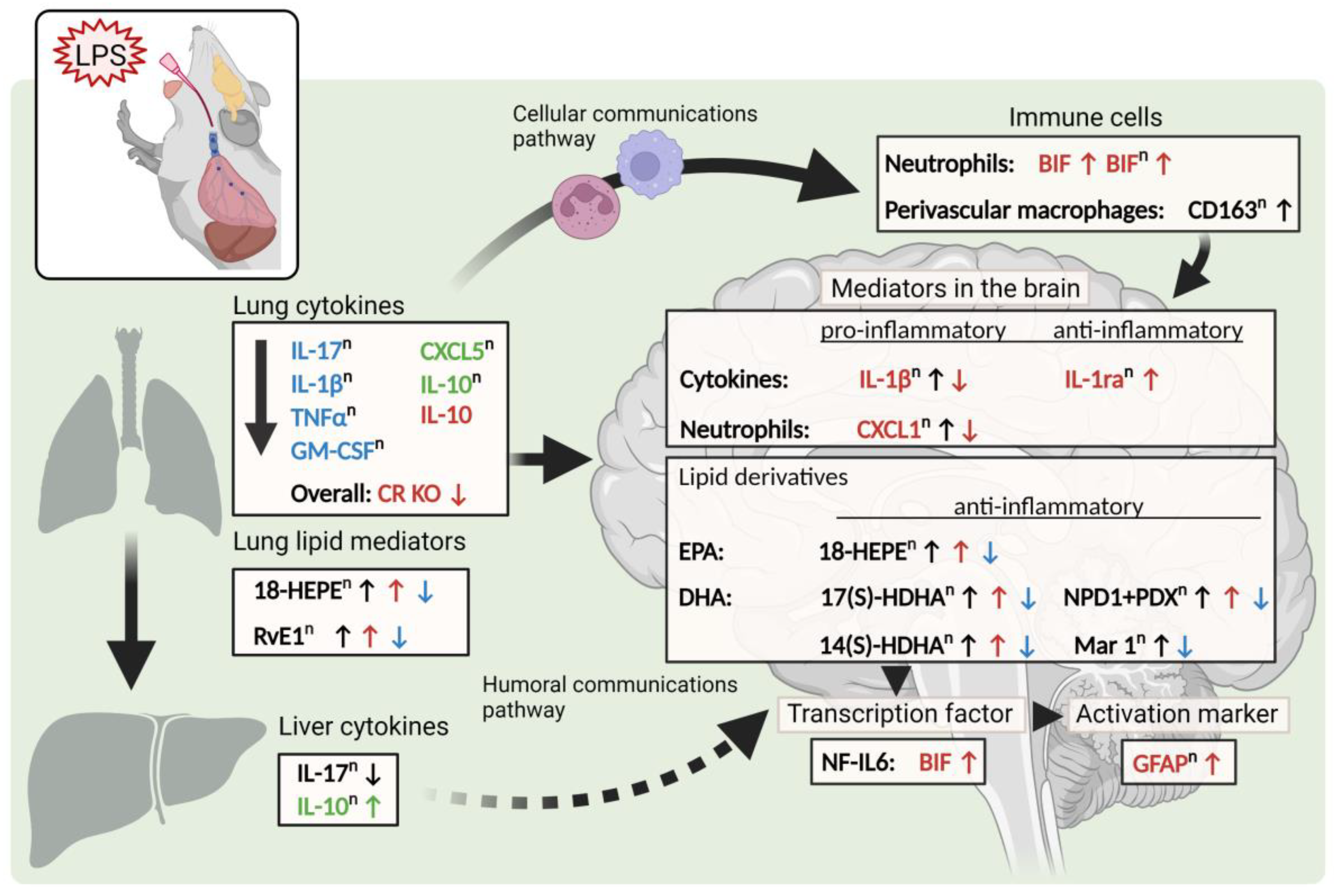
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Experimental Protocol
4.3. Immunohistochemistry
4.3.1. General Protocol
4.3.2. NF-IL6
4.3.3. MPO
4.3.4. GFAP
4.3.5. Microscopic Analysis
4.4. Protein Carbonyl Content
4.5. Protein Analysis
4.6. Cytokine Measurements
4.7. Eicosanoid Extraction and LC-MS/MS-Based Mass Spectrometric Analysis
4.8. Real Time (RT)-PCR
4.9. Data Analysis and Statistics
4.9.1. Evaluation of Cytokine Measurements
4.9.2. Evaluation of RT-PCR
4.9.3. Evaluation of Western Blot Protein Analysis
4.9.4. Evaluation of Immunohistochemistry
NF-IL6 and MPO
GFAP
4.9.5. Evaluation of LC-MS/MS
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bazinet, R.P.; Layé, S. Polyunsaturated fatty acids and their metabolites in brain function and disease. Nat. Rev. Neurosci. 2014, 15, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Layé, S.; Nadjar, A.; Joffre, C.; Bazinet, R.P. Anti-Inflammatory Effects of Omega-3 Fatty Acids in the Brain: Physiological Mechanisms and Relevance to Pharmacology. Pharmacol. Rev. 2018, 70, 12–38. [Google Scholar] [CrossRef] [PubMed]
- Madore, C.; Leyrolle, Q.; Morel, L.; Rossitto, M.; Greenhalgh, A.D.; Delpech, J.C.; Martinat, M.; Bosch-Bouju, C.; Bourel, J.; Rani, B.; et al. Essential omega-3 fatty acids tune microglial phagocytosis of synaptic elements in the mouse developing brain. Nat. Commun. 2020, 11, 6133. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Petasis, N.A. Resolvins and protectins in inflammation resolution. Chem. Rev. 2011, 111, 5922–5943. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Levy, B.D. Resolvins in inflammation: Emergence of the pro-resolving superfamily of mediators. J. Clin. Investig. 2018, 128, 2657–2669. [Google Scholar] [CrossRef] [PubMed]
- Qu, Q.; Xuan, W.; Fan, G.-H. Roles of resolvins in the resolution of acute inflammation. Cell Biol. Int. 2015, 39, 3–22. [Google Scholar] [CrossRef]
- Mayer, K.; Sommer, N.; Hache, K.; Hecker, A.; Reiche, S.; Schneck, E.; Weissmann, N.; Seeger, W.; Hecker, M. Resolvin E1 Improves Mitochondrial Function in Human Alveolar Epithelial Cells during Severe Inflammation. Lipids 2019, 54, 53–65. [Google Scholar] [CrossRef]
- Schwab, J.M.; Chiang, N.; Arita, M.; Serhan, C.N. Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature 2007, 447, 869–874. [Google Scholar] [CrossRef]
- Serhan, C.N.; Yang, R.; Martinod, K.; Kasuga, K.; Pillai, P.S.; Porter, T.F.; Oh, S.F.; Spite, M. Maresins: Novel macrophage mediators with potent antiinflammatory and proresolving actions. J. Exp. Med. 2009, 206, 15–23. [Google Scholar] [CrossRef]
- Serhan, C.N.; Clish, C.B.; Brannon, J.; Colgan, S.P.; Chiang, N.; Gronert, K. Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from omega-3 fatty acids via cyclooxygenase 2-nonsteroidal antiinflammatory drugs and transcellular processing. J. Exp. Med. 2000, 192, 1197–1204. [Google Scholar] [CrossRef]
- Haworth, O.; Cernadas, M.; Yang, R.; Serhan, C.N.; Levy, B.D. Resolvin E1 regulates interleukin 23, interferon-γ and lipoxin A4 to promote the resolution of allergic airway inflammation. Nat. Immunol. 2008, 9, 873–879. [Google Scholar] [CrossRef] [PubMed]
- Arita, M.; Bianchini, F.; Aliberti, J.; Sher, A.; Chiang, N.; Hong, S.; Yang, R.; Petasis, N.A.; Serhan, C.N. Stereochemical assignment, antiinflammatory properties, and receptor for the omega-3 lipid mediator resolvin E1. J. Exp. Med. 2005, 201, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Arita, M.; Yoshida, M.; Hong, S.; Tjonahen, E.; Glickman, J.N.; Petasis, N.A.; Blumberg, R.S.; Serhan, C.N. Resolvin E1, an endogenous lipid mediator derived from omega-3 eicosapentaenoic acid, protects against 2,4,6-trinitrobenzene sulfonic acid-induced colitis. Proc. Natl. Acad. Sci. USA 2005, 102, 7671–7676. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Gronert, K.; Devchand, P.R.; Moussignac, R.; Serhan, C.N. Novel docosatrienes and 17S-resolvins generated from docosahexaenoic acid in murine brain, human blood, and glial cells. Autacoids in anti-inflammation. J. Biol. Chem. 2003, 278, 14677–14687. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, N.; Burkett, P.R.; Dalli, J.; Abdulnour, R.-E.E.; Colas, R.; Ramon, S.; Phipps, R.P.; Petasis, N.A.; Kuchroo, V.K.; Serhan, C.N.; et al. Cutting edge: Maresin-1 engages regulatory T cells to limit type 2 innate lymphoid cell activation and promote resolution of lung inflammation. J. Immunol. 2015, 194, 863–867. [Google Scholar] [CrossRef]
- Serhan, C.N.; Dalli, J.; Karamnov, S.; Choi, A.; Park, C.; Xu, Z.; Ji, R.; Zhu, M.; Petasis, N.A. Macrophage proresolving mediator maresin 1 stimulates tissue regeneration and controls pain. FASEB J. 2012, 26, 1755–1765. [Google Scholar] [CrossRef]
- Xie, W.; Wang, H.; Wang, L.; Yao, C.; Yuan, R.; Wu, Q. Resolvin D1 reduces deterioration of tight junction proteins by upregulating HO-1 in LPS-induced mice. Lab. Investig. 2013, 93, 991–1000. [Google Scholar] [CrossRef]
- Aïd, S.; Vancassel, S.; Poumès-Ballihaut, C.; Chalon, S.; Guesnet, P.; Lavialle, M. Effect of a diet-induced n-3 PUFA depletion on cholinergic parameters in the rat hippocampus. J. Lipid Res. 2003, 44, 1545–1551. [Google Scholar] [CrossRef]
- Serhan, C.N.; Fredman, G.; Yang, R.; Karamnov, S.; Belayev, L.S.; Bazan, N.G.; Zhu, M.; Winkler, J.W.; Petasis, N.A. Novel proresolving aspirin-triggered DHA pathway. Chem. Biol. 2011, 18, 976–987. [Google Scholar] [CrossRef]
- Chalon, S. Omega-3 fatty acids and monoamine neurotransmission. Prostaglandins Leukot. Essent. Fat. Acids 2006, 75, 259–269. [Google Scholar] [CrossRef]
- Arita, M.; Ohira, T.; Sun, Y.-P.; Elangovan, S.; Chiang, N.; Serhan, C.N. Resolvin E1 selectively interacts with leukotriene B4 receptor BLT1 and ChemR23 to regulate inflammation. J. Immunol. 2007, 178, 3912–3917. [Google Scholar] [CrossRef] [PubMed]
- Campbell, E.L.; Louis, N.A.; Tomassetti, S.E.; Canny, G.O.; Arita, M.; Serhan, C.N.; Colgan, S.P. Resolvin E1 promotes mucosal surface clearance of neutrophils: A new paradigm for inflammatory resolution. FASEB J. 2007, 21, 3162–3170. [Google Scholar] [CrossRef] [PubMed]
- Haworth, O.; Cernadas, M.; Levy, B.D. NK cells are effectors for resolvin E1 in the timely resolution of allergic airway inflammation. J. Immunol. 2011, 186, 6129–6135. [Google Scholar] [CrossRef]
- Liu, G.; Gong, Y.; Zhang, R.; Piao, L.; Li, X.; Liu, Q.; Yan, S.; Shen, Y.; Guo, S.; Zhu, M.; et al. Resolvin E1 attenuates injury-induced vascular neointimal formation by inhibition of inflammatory responses and vascular smooth muscle cell migration. FASEB J. 2018, 32, 5413–5425. [Google Scholar] [CrossRef] [PubMed]
- Deyama, S.; Shimoda, K.; Suzuki, H.; Ishikawa, Y.; Ishimura, K.; Fukuda, H.; Hitora-Imamura, N.; Ide, S.; Satoh, M.; Kaneda, K.; et al. Resolvin E1/E2 ameliorate lipopolysaccharide-induced depression-like behaviors via ChemR23. Psychopharmacology 2018, 235, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Zhang, W.-Y.; Li, H.; Zhang, P.-H.; Tian, C.; Wu, C.-H.; Zhao, A.-N.; Chen, M.-L.; Guo, Y.-F.; Cho, Y.-C.; et al. Pro-Resolving Mediator Resolvin E1 Restores Alveolar Fluid Clearance in Acute Respiratory Distress Syndrome. Shock 2022, 57, 565–575. [Google Scholar] [CrossRef]
- Gantz, I.; Konda, Y.; Yang, Y.K.; Miller, D.E.; Dierick, H.A.; Yamada, T. Molecular cloning of a novel receptor (CMKLR1) with homology to the chemotactic factor receptors. Cytogenet. Cell Genet. 1996, 74, 286–290. [Google Scholar] [CrossRef]
- Samson, M.; Edinger, A.L.; Stordeur, P.; Rucker, J.; Verhasselt, V.; Sharron, M.; Govaerts, C.; Mollereau, C.; Vassart, G.; Doms, R.W.; et al. ChemR23, a putative chemoattractant receptor, is expressed in monocyte-derived dendritic cells and macrophages and is a coreceptor for SIV and some primary HIV-1 strains. Eur. J. Immunol. 1998, 28, 1689–1700. [Google Scholar] [CrossRef]
- Goralski, K.B.; McCarthy, T.C.; Hanniman, E.A.; Zabel, B.A.; Butcher, E.C.; Parlee, S.D.; Muruganandan, S.; Sinal, C.J. Chemerin, a novel adipokine that regulates adipogenesis and adipocyte metabolism. J. Biol. Chem. 2007, 282, 28175–28188. [Google Scholar] [CrossRef]
- Parolini, S.; Santoro, A.; Marcenaro, E.; Luini, W.; Massardi, L.; Facchetti, F.; Communi, D.; Parmentier, M.; Majorana, A.; Sironi, M.; et al. The role of chemerin in the colocalization of NK and dendritic cell subsets into inflamed tissues. Blood 2007, 109, 3625–3632. [Google Scholar] [CrossRef]
- Kaur, J.; Adya, R.; Tan, B.K.; Chen, J.; Randeva, H.S. Identification of chemerin receptor (ChemR23) in human endothelial cells: Chemerin-induced endothelial angiogenesis. Biochem. Biophys. Res. Commun. 2010, 391, 1762–1768. [Google Scholar] [CrossRef] [PubMed]
- Lämmermann, T.; Afonso, P.V.; Angermann, B.R.; Wang, J.M.; Kastenmüller, W.; Parent, C.A.; Germain, R.N. Neutrophil swarms require LTB4 and integrins at sites of cell death in vivo. Nature 2013, 498, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Hiram, R.; Rizcallah, E.; Marouan, S.; Sirois, C.; Sirois, M.; Morin, C.; Fortin, S.; Rousseau, E. Resolvin E1 normalizes contractility, Ca2+ sensitivity and smooth muscle cell migration rate in TNF-α- and IL-6-pretreated human pulmonary arteries. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L776–L788. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.F.; Dona, M.; Fredman, G.; Krishnamoorthy, S.; Irimia, D.; Serhan, C.N. Resolvin E2 formation and impact in inflammation resolution. J. Immunol. 2012, 188, 4527–4534. [Google Scholar] [CrossRef]
- Lukiw, W.J.; Cui, J.-G.; Marcheselli, V.L.; Bodker, M.; Botkjaer, A.; Gotlinger, K.; Serhan, C.N.; Bazan, N.G. A role for docosahexaenoic acid-derived neuroprotectin D1 in neural cell survival and Alzheimer disease. J. Clin. Investig. 2005, 115, 2774–2783. [Google Scholar] [CrossRef]
- Rey, C.; Nadjar, A.; Buaud, B.; Vaysse, C.; Aubert, A.; Pallet, V.; Layé, S.; Joffre, C. Resolvin D1 and E1 promote resolution of inflammation in microglial cells in vitro. Brain Behav. Immun. 2016, 55, 249–259. [Google Scholar] [CrossRef]
- Schaefer, M.B.; Ott, J.; Mohr, A.; Bi, M.H.; Grosz, A.; Weissmann, N.; Ishii, S.; Grimminger, F.; Seeger, W.; Mayer, K. Immunomodulation by n-3- versus n-6-rich lipid emulsions in murine acute lung injury--role of platelet-activating factor receptor. Crit. Care Med. 2007, 35, 544–554. [Google Scholar] [CrossRef]
- Mayer, K.; Kiessling, A.; Ott, J.; Schaefer, M.B.; Hecker, M.; Henneke, I.; Schulz, R.; Günther, A.; Wang, J.; Wu, L.; et al. Acute lung injury is reduced in fat-1 mice endogenously synthesizing n-3 fatty acids. Am. J. Respir. Crit. Care Med. 2009, 179, 474–483. [Google Scholar] [CrossRef]
- Seki, H.; Fukunaga, K.; Arita, M.; Arai, H.; Nakanishi, H.; Taguchi, R.; Miyasho, T.; Takamiya, R.; Asano, K.; Ishizaka, A.; et al. The anti-inflammatory and proresolving mediator resolvin E1 protects mice from bacterial pneumonia and acute lung injury. J. Immunol. 2010, 184, 836–843. [Google Scholar] [CrossRef]
- Ferguson, N.D.; Fan, E.; Camporota, L.; Antonelli, M.; Anzueto, A.; Beale, R.; Brochard, L.; Brower, R.; Esteban, A.; Gattinoni, L.; et al. The Berlin definition of ARDS: An expanded rationale, justification, and supplementary material. Intensive Care Med. 2012, 38, 1573–1582. [Google Scholar] [CrossRef]
- Heuer, J.F.; Sauter, P.; Pelosi, P.; Herrmann, P.; Brück, W.; Perske, C.; Schöndube, F.; Crozier, T.A.; Bleckmann, A.; Beißbarth, T.; et al. Effects of pulmonary acid aspiration on the lungs and extra-pulmonary organs: A randomized study in pigs. Crit. Care 2012, 16, R35. [Google Scholar] [CrossRef]
- Steimback, P.W.; Oliveira, G.P.; Rzezinski, A.F.; Silva, P.L.; Garcia, C.S.N.B.; Rangel, G.; Morales, M.M.; Lapa E Silva, J.R.; Capelozzi, V.L.; Pelosi, P.; et al. Effects of frequency and inspiratory plateau pressure during recruitment manoeuvres on lung and distal organs in acute lung injury. Intensive Care Med. 2009, 35, 1120–1128. [Google Scholar] [CrossRef]
- Manouchehrian, O.; Ramos, M.; Bachiller, S.; Lundgaard, I.; Deierborg, T. Acute systemic LPS-exposure impairs perivascular CSF distribution in mice. J. Neuroinflamm. 2021, 18, 34. [Google Scholar] [CrossRef] [PubMed]
- Boitsova, E.B.; Morgun, A.V.; Osipova, E.D.; Pozhilenkova, E.A.; Martinova, G.P.; Frolova, O.V.; Olovannikova, R.Y.; Tohidpour, A.; Gorina, Y.V.; Panina, Y.A.; et al. The inhibitory effect of LPS on the expression of GPR81 lactate receptor in blood-brain barrier model in vitro. J. Neuroinflamm. 2018, 15, 196. [Google Scholar] [CrossRef]
- Hopkins, R.O.; Suchyta, M.R.; Snow, G.L.; Jephson, A.; Weaver, L.K.; Orme, J.F. Blood glucose dysregulation and cognitive outcome in ARDS survivors. Brain Inj. 2010, 24, 1478–1484. [Google Scholar] [CrossRef] [PubMed]
- Fries, M.; Bickenbach, J.; Henzler, D.; Beckers, S.; Dembinski, R.; Sellhaus, B.; Rossaint, R.; Kuhlen, R. S-100 protein and neurohistopathologic changes in a porcine model of acute lung injury. Anesthesiology 2005, 102, 761–767. [Google Scholar] [CrossRef]
- Hopkins, R.O.; Weaver, L.K.; Chan, K.J.; Orme, J.F. Quality of life, emotional, and cognitive function following acute respiratory distress syndrome. J. Int. Neuropsychol. Soc. JINS 2004, 10, 1005–1017. [Google Scholar] [CrossRef]
- Hopkins, R.O.; Gale, S.D.; Weaver, L.K. Brain atrophy and cognitive impairment in survivors of Acute Respiratory Distress Syndrome. Brain Inj. 2006, 20, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Bondue, B.; Vosters, O.; de Nadai, P.; Glineur, S.; de Henau, O.; Luangsay, S.; van Gool, F.; Communi, D.; de Vuyst, P.; Desmecht, D.; et al. ChemR23 dampens lung inflammation and enhances anti-viral immunity in a mouse model of acute viral pneumonia. PLoS Pathog. 2011, 7, e1002358. [Google Scholar] [CrossRef]
- Kang, J.X.; Wang, J.; Wu, L.; Kang, Z.B. Transgenic mice: Fat-1 mice convert n-6 to n-3 fatty acids. Nature 2004, 427, 504. [Google Scholar] [CrossRef] [PubMed]
- Rummel, C. Inflammatory transcription factors as activation markers and functional readouts in immune-to-brain communication. Brain Behav. Immun. 2016, 54, 1–14. [Google Scholar] [CrossRef]
- Roth, J.; Harré, E.-M.; Rummel, C.; Gerstberger, R.; Hübschle, T. Signaling the brain in systemic inflammation: Role of sensory circumventricular organs. Front. Biosci.(Landmark Ed.) 2004, 9, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Harden, L.M.; Rummel, C.; Luheshi, G.N.; Poole, S.; Gerstberger, R.; Roth, J. Interleukin-10 modulates the synthesis of inflammatory mediators in the sensory circumventricular organs: Implications for the regulation of fever and sickness behaviors. J. Neuroinflamm. 2013, 10, 22. [Google Scholar] [CrossRef] [PubMed]
- Pflieger, F.J.; Hernandez, J.; Schweighöfer, H.; Herden, C.; Rosengarten, B.; Rummel, C. The role of neutrophil granulocytes in immune-to-brain communication. Temperature 2018, 5, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Kiss, L.; Röder, Y.; Bier, J.; Weissmann, N.; Seeger, W.; Grimminger, F. Direct eicosanoid profiling of the hypoxic lung by comprehensive analysis via capillary liquid chromatography with dual online photodiode-array and tandem mass-spectrometric detection. Anal. Bioanal. Chem. 2008, 390, 697–714. [Google Scholar] [CrossRef]
- Domscheit, H.; Hegeman, M.A.; Carvalho, N.; Spieth, P.M. Molecular Dynamics of Lipopolysaccharide-Induced Lung Injury in Rodents. Front. Physiol. 2020, 11, 36. [Google Scholar] [CrossRef]
- Liu, S.; Su, X.; Pan, P.; Zhang, L.; Hu, Y.; Tan, H.; Wu, D.; Liu, B.; Li, H.; Li, H.; et al. Neutrophil extracellular traps are indirectly triggered by lipopolysaccharide and contribute to acute lung injury. Sci. Rep. 2016, 6, 37252. [Google Scholar] [CrossRef]
- Kelly, D.P.; Scarpulla, R.C. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 2004, 18, 357–368. [Google Scholar] [CrossRef]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef]
- Voynow, J.A.; Shinbashi, M. Neutrophil Elastase and Chronic Lung Disease. Biomolecules 2021, 11, 1065. [Google Scholar] [CrossRef] [PubMed]
- Corraliza, I. Recruiting specialized macrophages across the borders to restore brain functions. Front. Cell. Neurosci. 2014, 8, 262. [Google Scholar] [CrossRef] [PubMed]
- Galea, I.; Felton, L.M.; Waters, S.; van Rooijen, N.; Perry, V.H.; Newman, T.A. Immune-to-brain signalling: The role of cerebral CD163-positive macrophages. Neurosci. Lett. 2008, 448, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Graeber, M.B.; Streit, W.J.; Kreutzberg, G.W. Identity of ED2-positive perivascular cells in rat brain. J. Neurosci. Res. 1989, 22, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Damm, J.; Luheshi, G.N.; Gerstberger, R.; Roth, J.; Rummel, C. Spatiotemporal nuclear factor interleukin-6 expression in the rat brain during lipopolysaccharide-induced fever is linked to sustained hypothalamic inflammatory target gene induction. J. Comp. Neurol. 2011, 519, 480–505. [Google Scholar] [CrossRef] [PubMed]
- Schneiders, J.; Fuchs, F.; Damm, J.; Herden, C.; Gerstberger, R.; Soares, D.M.; Roth, J.; Rummel, C. The transcription factor nuclear factor interleukin 6 mediates pro- and anti-inflammatory responses during LPS-induced systemic inflammation in mice. Brain Behav. Immun. 2015, 48, 147–164. [Google Scholar] [CrossRef]
- Nadjar, A.; Bluthé, R.-M.; May, M.J.; Dantzer, R.; Parnet, P. Inactivation of the cerebral NFκB pathway inhibits interleukin-1β-induced sickness behavior and c-Fos expression in various brain nuclei. Neuropsychopharmacology 2005, 30, 1492–1499. [Google Scholar] [CrossRef]
- Koenig, S.; Luheshi, G.N.; Wenz, T.; Gerstberger, R.; Roth, J.; Rummel, C. Leptin is involved in age-dependent changes in response to systemic inflammation in the rat. Brain Behav. Immun. 2014, 36, 128–138. [Google Scholar] [CrossRef]
- Rummel, C.; Matsumura, K.; Luheshi, G.N. Circulating IL-6 contributes to peripheral LPS-induced mPGES-1 expression in the rat brain. Brain Res. Bull. 2011, 86, 319–325. [Google Scholar] [CrossRef]
- Bauer, J.; Sminia, T.; Wouterlood, F.G.; Dijkstra, C.D. Phagocytic activity of macrophages and microglial cells during the course of acute and chronic relapsing experimental autoimmune encephalomyelitis. J. Neurosci. Res. 1994, 38, 365–375. [Google Scholar] [CrossRef]
- Damoiseaux, J.G.; Döpp, E.A.; Calame, W.; Chao, D.; MacPherson, G.G.; Dijkstra, C.D. Rat macrophage lysosomal membrane antigen recognized by monoclonal antibody ED1. Immunology 1994, 83, 140–147. [Google Scholar] [PubMed]
- Bredehöft, J.; Bhandari, D.R.; Pflieger, F.J.; Schulz, S.; Kang, J.X.; Layé, S.; Roth, J.; Gerstberger, R.; Mayer, K.; Spengler, B.; et al. Visualizing and Profiling Lipids in the OVLT of Fat-1 and Wild Type Mouse Brains during LPS-Induced Systemic Inflammation Using AP-SMALDI MSI. ACS Chem. Neurosci. 2019, 10, 4394–4406. [Google Scholar] [CrossRef]
- Ando, K.; Nagata, K.; Yoshida, R.; Kikugawa, K.; Suzuki, M. Effect of n-3 polyunsaturated fatty acid supplementation on lipid peroxidation of rat organs. Lipids 2000, 35, 401–407. [Google Scholar] [CrossRef]
- Kulkarni, A.; Zhao, A.; Yang, B.; Zhang, Y.; Linderborg, K.M. Tissue-Specific Content of Polyunsaturated Fatty Acids in (n-3) Deficiency State of Rats. Foods 2022, 11, 208. [Google Scholar] [CrossRef]
- Joffre, C.; Rey, C.; Layé, S. N-3 Polyunsaturated Fatty Acids and the Resolution of Neuroinflammation. Front. Pharmacol. 2019, 10, 1022. [Google Scholar] [CrossRef] [PubMed]
- Ostermann, A.I.; Waindok, P.; Schmidt, M.J.; Chiu, C.-Y.; Smyl, C.; Rohwer, N.; Weylandt, K.-H.; Schebb, N.H. Modulation of the endogenous omega-3 fatty acid and oxylipin profile in vivo-A comparison of the fat-1 transgenic mouse with C57BL/6 wildtype mice on an omega-3 fatty acid enriched diet. PLoS ONE 2017, 12, e0184470. [Google Scholar] [CrossRef] [PubMed]
- Pawlosky, R.J.; Hibbeln, J.R.; Novotny, J.A.; Salem, N. Physiological compartmental analysis of α-linolenic acid metabolism in adult humans. J. Lipid Res. 2001, 42, 1257–1265. [Google Scholar] [CrossRef]
- Darwish, I.; Mubareka, S.; Liles, W.C. Immunomodulatory therapy for severe influenza. Expert Rev. Anti Infect. Ther. 2011, 9, 807–822. [Google Scholar] [CrossRef]
- Gonzalvo, R.; Martí-Sistac, O.; Blanch, L.; López-Aguilar, J. Bench-to-bedside review: Brain-lung interaction in the critically ill—A pending issue revisited. Crit. Care 2007, 11, 216. [Google Scholar] [CrossRef]
- Sahu, B.; Sandhir, R.; Naura, A.S. Two hit induced acute lung injury impairs cognitive function in mice: A potential model to study cross talk between lung and brain. Brain Behav. Immun. 2018, 73, 633–642. [Google Scholar] [CrossRef]
- Goodman, R.B.; Pugin, J.; Lee, J.S.; Matthay, M.A. Cytokine-mediated inflammation in acute lung injury. Cytokine Growth Factor Rev. 2003, 14, 523–535. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Chen, W.; Lin, F.; Li, W.; Wang, P.; Liao, G.; Zhang, L. Functional Two-Way Crosstalk Between Brain and Lung: The Brain-Lung Axis. Cell. Mol. Neurobiol. 2023, 43, 991–1003. [Google Scholar] [CrossRef]
- Rummel, C.; Del Rey, A.; Bähr, L.; Krüger, K.; Peters, E. 1st European Psychoneuroimmunology Network (EPN) Autumn School: Lung-Brain Axis in Health and Disease. Neuroimmunomodulation 2022, 29 (Suppl. 2), 3–8. [Google Scholar] [CrossRef] [PubMed]
- Pelosi, P.; Rocco, P.R.M. The lung and the brain: A dangerous cross-talk. Crit. Care 2011, 15, 168. [Google Scholar] [CrossRef]
- Nadeem, A.; Siddiqui, N.; Al-Harbi, N.O.; Attia, S.M.; AlSharari, S.D.; Ahmad, S.F. Acute lung injury leads to depression-like symptoms through upregulation of neutrophilic and neuronal NADPH oxidase signaling in a murine model. Int. Immunopharmacol. 2017, 47, 218–226. [Google Scholar] [CrossRef]
- Meduri, G.U.; Annane, D.; Chrousos, G.P.; Marik, P.E.; Sinclair, S.E. Activation and regulation of systemic inflammation in ARDS: Rationale for prolonged glucocorticoid therapy. Chest 2009, 136, 1631–1643. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A. The blood-brain barrier in neuroimmunology: Tales of separation and assimilation. Brain Behav. Immun. 2015, 44, 1–8. [Google Scholar] [CrossRef]
- Carson, M.J.; Doose, J.M.; Melchior, B.; Schmid, C.D.; Ploix, C.C. CNS immune privilege: Hiding in plain sight. Immunol. Rev. 2006, 213, 48–65. [Google Scholar] [CrossRef]
- Wang, Y.; Jin, S.; Sonobe, Y.; Cheng, Y.; Horiuchi, H.; Parajuli, B.; Kawanokuchi, J.; Mizuno, T.; Takeuchi, H.; Suzumura, A. Interleukin-1β induces blood-brain barrier disruption by downregulating Sonic hedgehog in astrocytes. PLoS ONE 2014, 9, e110024. [Google Scholar] [CrossRef]
- Horii, Y.; Nakaya, M.; Ohara, H.; Nishihara, H.; Watari, K.; Nagasaka, A.; Nakaya, T.; Sugiura, Y.; Okuno, T.; Koga, T.; et al. Leukotriene B4 receptor 1 exacerbates inflammation following myocardial infarction. FASEB J. 2020, 34, 8749–8763. [Google Scholar] [CrossRef]
- Harden, L.M.; Rummel, C.; Laburn, H.P.; Damm, J.; Wiegand, F.; Poole, S.; Gerstberger, R.; Roth, J. Critical role for peripherally-derived interleukin-10 in mediating the thermoregulatory manifestations of fever and hypothermia in severe forms of lipopolysaccharide-induced inflammation. Pflug. Arch. Eur. J. Physiol. 2014, 466, 1451–1466. [Google Scholar] [CrossRef] [PubMed]
- Bellenger, J.; Bellenger, S.; Bataille, A.; Massey, K.A.; Nicolaou, A.; Rialland, M.; Tessier, C.; Kang, J.X.; Narce, M. High pancreatic n-3 fatty acids prevent STZ-induced diabetes in fat-1 mice: Inflammatory pathway inhibition. Diabetes 2011, 60, 1090–1099. [Google Scholar] [CrossRef] [PubMed]
- Shokry, A.A.; El-Shiekh, R.A.; Kamel, G.; Bakr, A.F.; Ramadan, A. Bioactive phenolics fraction of Hedera helix L. (Common Ivy Leaf) standardized extract ameliorates LPS-induced acute lung injury in the mouse model through the inhibition of proinflammatory cytokines and oxidative stress. Heliyon 2022, 8, e09477. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.R.; Ma, A.Y.; Liu, Y.; Qu, Y. Effects of inflammatory factors including plasma tumor necrosis factor-α in the clinical treatment of acute respiratory distress syndrome. Oncol. Lett. 2017, 13, 5016–5020. [Google Scholar] [CrossRef]
- El Kebir, D.; Gjorstrup, P.; Filep, J.G. Resolvin E1 promotes phagocytosis-induced neutrophil apoptosis and accelerates resolution of pulmonary inflammation. Proc. Natl. Acad. Sci. USA 2012, 109, 14983–14988. [Google Scholar] [CrossRef]
- Herová, M.; Schmid, M.; Gemperle, C.; Hersberger, M. ChemR23, the receptor for chemerin and resolvin E1, is expressed and functional on M1 but not on M2 macrophages. J. Immunol. 2015, 194, 2330–2337. [Google Scholar] [CrossRef]
- Ware, L.B.; Matthay, M.A. The acute respiratory distress syndrome. N. Engl. J. Med. 2000, 342, 1334–1349. [Google Scholar] [CrossRef]
- Parsey, M.V.; Tuder, R.M.; Abraham, E. Neutrophils are major contributors to intraparenchymal lung IL-1β expression after hemorrhage and endotoxemia. J. Immunol. 1998, 160, 1007–1013. [Google Scholar] [CrossRef]
- Mikacenic, C.; Hansen, E.E.; Radella, F.; Gharib, S.A.; Stapleton, R.D.; Wurfel, M.M. Interleukin-17A Is Associated with Alveolar Inflammation and Poor Outcomes in Acute Respiratory Distress Syndrome. Crit. Care Med. 2016, 44, 496–502. [Google Scholar] [CrossRef]
- Bhatia, M.; Zemans, R.L.; Jeyaseelan, S. Role of chemokines in the pathogenesis of acute lung injury. Am. J. Respir. Cell Mol. Biol. 2012, 46, 566–572. [Google Scholar] [CrossRef]
- Sawant, K.V.; Xu, R.; Cox, R.; Hawkins, H.; Sbrana, E.; Kolli, D.; Garofalo, R.P.; Rajarathnam, K. Chemokine CXCL1-Mediated Neutrophil Trafficking in the Lung: Role of CXCR2 Activation. J. Innate Immun. 2015, 7, 647–658. [Google Scholar] [CrossRef]
- Ballinger, M.N.; Paine, R.; Serezani, C.H.C.; Aronoff, D.M.; Choi, E.S.; Standiford, T.J.; Toews, G.B.; Moore, B.B. Role of granulocyte macrophage colony-stimulating factor during gram-negative lung infection with Pseudomonas aeruginosa. Am. J. Respir. Cell Mol. Biol. 2006, 34, 766–774. [Google Scholar] [CrossRef]
- Jeyaseelan, S.; Chu, H.W.; Young, S.K.; Worthen, G.S. Transcriptional profiling of lipopolysaccharide-induced acute lung injury. Infect. Immun. 2004, 72, 7247–7256. [Google Scholar] [CrossRef]
- Matute-Bello, G.; Liles, W.C.; Radella, F.; Steinberg, K.P.; Ruzinski, J.T.; Hudson, L.D.; Martin, T.R. Modulation of neutrophil apoptosis by granulocyte colony-stimulating factor and granulocyte/macrophage colony-stimulating factor during the course of acute respiratory distress syndrome. Crit. Care Med. 2000, 28, 1–7. [Google Scholar] [CrossRef]
- Li, C.; Yang, P.; Sun, Y.; Li, T.; Wang, C.; Wang, Z.; Zou, Z.; Yan, Y.; Wang, W.; Chen, Z.; et al. IL-17 response mediates acute lung injury induced by the 2009 pandemic influenza A (H1N1) virus. Cell Res. 2012, 22, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Bilal, S.; Haworth, O.; Wu, L.; Weylandt, K.H.; Levy, B.D.; Kang, J.X. Fat-1 transgenic mice with elevated omega-3 fatty acids are protected from allergic airway responses. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2011, 1812, 1164–1169. [Google Scholar] [CrossRef] [PubMed]
- Tiesset, H.; Pierre, M.; Desseyn, J.-L.; Guéry, B.; Beermann, C.; Galabert, C.; Gottrand, F.; Husson, M.-O. Dietary (n-3) polyunsaturated fatty acids affect the kinetics of pro- and antiinflammatory responses in mice with Pseudomonas aeruginosa lung infection. J. Nutr. 2009, 139, 82–89. [Google Scholar] [CrossRef]
- Unoda, K.; Doi, Y.; Nakajima, H.; Yamane, K.; Hosokawa, T.; Ishida, S.; Kimura, F.; Hanafusa, T. Eicosapentaenoic acid (EPA) induces peroxisome proliferator-activated receptors and ameliorates experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2013, 256, 7–12. [Google Scholar] [CrossRef]
- Monk, J.M.; Hou, T.Y.; Turk, H.F.; Weeks, B.; Wu, C.; McMurray, D.N.; Chapkin, R.S. Dietary n-3 polyunsaturated fatty acids (PUFA) decrease obesity-associated Th17 cell-mediated inflammation during colitis. PLoS ONE 2012, 7, e49739. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Wen, J.; Bai, X.-C.; Chen, T.-Y.; Zheng, R.-C.; Zhou, G.-B.; Ma, J.; Feng, J.-Y.; Zhong, B.-L.; Li, Y.-M. Endogenous n-3 polyunsaturated fatty acids protect against imiquimod-induced psoriasis-like inflammation via the IL-17/IL-23 axis. Mol. Med. Rep. 2014, 9, 2097–2104. [Google Scholar] [CrossRef]
- Mastroianni, C.M.; Liuzzi, G.M.; Vullo, V.; Jirillo, E.; Delia, S.; Riccio, P. Detection of cerebrospinal fluid antibodies against myelin basic protein in patients with AIDS dementia complex. Mol. Chem. Neuropathol. 1991, 14, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Roth, J.; Blatteis, C.M. Mechanisms of fever production and lysis: Lessons from experimental LPS fever. Compr. Physiol. 2014, 4, 1563–1604. [Google Scholar] [CrossRef] [PubMed]
- von Bernhardi, R.; Eugenín-von Bernhardi, L.; Eugenín, J. Microglial cell dysregulation in brain aging and neurodegeneration. Front. Aging Neurosci. 2015, 7, 124. [Google Scholar] [CrossRef] [PubMed]
- Hritcu, L.; Ciobica, A.; Stefan, M.; Mihasan, M.; Palamiuc, L.; Nabeshima, T. Spatial memory deficits and oxidative stress damage following exposure to lipopolysaccharide in a rodent model of Parkinson’s disease. Neurosci. Res. 2011, 71, 35–43. [Google Scholar] [CrossRef]
- Garcia, I.J.P.; Kinoshita, P.F.; de Oliveira Braga, I.; Parreira, G.M.; Mignaco, J.A.; Scavone, C.; Barbosa, L.A.; de Lima Santos, H. Ouabain attenuates the oxidative stress induced by lipopolysaccharides in the cerebellum of rats. J. Cell. Biochem. 2018, 119, 2156–2167. [Google Scholar] [CrossRef]
- Hritcu, L.; Ciobica, A. Intranigral lipopolysaccharide administration induced behavioral deficits and oxidative stress damage in laboratory rats: Relevance for Parkinson’s disease. Behav. Brain Res. 2013, 253, 25–31. [Google Scholar] [CrossRef]
- Nandi, A.; Yan, L.-J.; Jana, C.K.; Das, N. Role of Catalase in Oxidative Stress- and Age-Associated Degenerative Diseases. Oxid. Med. Cell. Longev. 2019, 2019, 9613090. [Google Scholar] [CrossRef]
- Younus, H. Therapeutic potentials of superoxide dismutase. Int. J. Health Sci. 2018, 12, 88–93. [Google Scholar]
- Loh, K.P.; Huang, S.H.; de Silva, R.; Tan, B.K.H.; Zhu, Y.Z. Oxidative stress: Apoptosis in neuronal injury. Curr. Alzheimer Res. 2006, 3, 327–337. [Google Scholar] [CrossRef]
- Mariani, E.; Polidori, M.C.; Cherubini, A.; Mecocci, P. Oxidative stress in brain aging, neurodegenerative and vascular diseases: An overview. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2005, 827, 65–75. [Google Scholar] [CrossRef]
- Moreira, P.I.; Siedlak, S.L.; Aliev, G.; Zhu, X.; Cash, A.D.; Smith, M.A.; Perry, G. Oxidative stress mechanisms and potential therapeutics in Alzheimer disease. J. Neural Transm. 2005, 112, 921–932. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhu, M.; Hjorth, E.; Cortés-Toro, V.; Eyjolfsdottir, H.; Graff, C.; Nennesmo, I.; Palmblad, J.; Eriksdotter, M.; Sambamurti, K.; et al. Resolution of inflammation is altered in Alzheimer’s disease. Alzheimer’s Dement. 2015, 11, 40–50.e2. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, N.; Ding, Y.; Zhang, Y.; Li, Q.; Flores, J.; Haghighiabyaneh, M.; Doycheva, D.; Tang, J.; Zhang, J.H. Chemerin suppresses neuroinflammation and improves neurological recovery via CaMKK2/AMPK/Nrf2 pathway after germinal matrix hemorrhage in neonatal rats. Brain Behav. Immun. 2018, 70, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, L.; Zhang, Y.; Yuan, Y.; Miao, Z.; Lu, K.; Zhang, X.; Ni, R.; Zhang, H.; Zhao, Y.; et al. ChemR23 signaling ameliorates cognitive impairments in diabetic mice via dampening oxidative stress and NLRP3 inflammasome activation. Redox Biol. 2022, 58, 102554. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Wang, J. HuR stabilizes TFAM mRNA in an ATM/p38-dependent manner in ionizing irradiated cancer cells. Cancer Sci. 2018, 109, 2446–2457. [Google Scholar] [CrossRef]
- Kang, D.; Kim, S.H.; Hamasaki, N. Mitochondrial transcription factor A (TFAM): Roles in maintenance of mtDNA and cellular functions. Mitochondrion 2007, 7, 39–44. [Google Scholar] [CrossRef]
- Alawdi, S.H.; El-Denshary, E.S.; Safar, M.M.; Eidi, H.; David, M.-O.; Abdel-Wahhab, M.A. Neuroprotective Effect of Nanodiamond in Alzheimer’s Disease Rat Model: A Pivotal Role for Modulating NF-κB and STAT3 Signaling. Mol. Neurobiol. 2017, 54, 1906–1918. [Google Scholar] [CrossRef] [PubMed]
- Marcheselli, V.L.; Hong, S.; Lukiw, W.J.; Tian, X.H.; Gronert, K.; Musto, A.; Hardy, M.; Gimenez, J.M.; Chiang, N.; Serhan, C.N.; et al. Novel docosanoids inhibit brain ischemia-reperfusion-mediated leukocyte infiltration and pro-inflammatory gene expression. J. Biol. Chem. 2003, 278, 43807–43817. [Google Scholar] [CrossRef]
- Caputo, M.P.; Radlowski, E.C.; Lawson, M.A.; Antonson, A.M.; Watson, J.E.; Matt, S.M.; Leyshon, B.J.; Das, A.; Johnson, R.W. Herring roe oil supplementation alters microglial cell gene expression and reduces peripheral inflammation after immune activation in a neonatal piglet model. Brain Behav. Immun. 2019, 81, 455–469. [Google Scholar] [CrossRef]
- Pflieger, F.J.; Wolf, J.; Feldotto, M.; Nockher, A.; Wenderoth, T.; Hernandez, J.; Roth, J.; Ott, D.; Rummel, C. Norepinephrine Inhibits Lipopolysaccharide-Stimulated TNF-α but Not Oxylipin Induction in n-3/n-6 PUFA-Enriched Cultures of Circumventricular Organs. Int. J. Mol. Sci. 2022, 23, 8745. [Google Scholar] [CrossRef]
- Flesher, R.P.; Herbert, C.; Kumar, R.K. Resolvin E1 promotes resolution of inflammation in a mouse model of an acute exacerbation of allergic asthma. Clin. Sci. 2014, 126, 805–814. [Google Scholar] [CrossRef]
- Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef]
- Biesmans, S.; Meert, T.F.; Bouwknecht, J.A.; Acton, P.D.; Davoodi, N.; de Haes, P.; Kuijlaars, J.; Langlois, X.; Matthews, L.J.R.; Ver Donck, L.; et al. Systemic immune activation leads to neuroinflammation and sickness behavior in mice. Mediat. Inflamm. 2013, 2013, 271359. [Google Scholar] [CrossRef]
- Hoogland, I.C.M.; Houbolt, C.; van Westerloo, D.J.; van Gool, W.A.; van de Beek, D. Systemic inflammation and microglial activation: Systematic review of animal experiments. J. Neuroinflamm. 2015, 12, 114. [Google Scholar] [CrossRef]
- Prinz, M.; Priller, J. Microglia and brain macrophages in the molecular age: From origin to neuropsychiatric disease. Nat. Rev. Neurosci. 2014, 15, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Pinteaux, E.; Rothwell, N.J.; Boutin, H. Neuroprotective actions of endogenous interleukin-1 receptor antagonist (IL-1ra) are mediated by glia. Glia 2006, 53, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Ulich, T.R.; Yin, S.M.; Guo, K.Z.; Del Castillo, J.; Eisenberg, S.P.; Thompson, R.C. The intratracheal administration of endotoxin and cytokines. III. The interleukin-1 (IL-1) receptor antagonist inhibits endotoxin- and IL-1-induced acute inflammation. Am. J. Pathol. 1991, 138, 521–524. [Google Scholar] [PubMed]
- Plata-Salamán, C.R.; ffrench-Mullen, J.M.H. Interleukin-1β inhibits Ca2+ channel currents in hippocampal neurons through protein kinase C. Eur. J. Pharmacol. Mol. Pharmacol. 1994, 266, 1–10. [Google Scholar] [CrossRef]
- Seckinger, P.; Williamson, K.; Balavoine, J.F.; Mach, B.; Mazzei, G.; Shaw, A.; Dayer, J.M. A urine inhibitor of interleukin 1 activity affects both interleukin 1 alpha and 1 beta but not tumor necrosis factor alpha. J. Immunol. 1987, 139, 1541–1545. [Google Scholar] [CrossRef]
- Shih, R.-H.; Wang, C.-Y.; Yang, C.-M. NF-κB Signaling Pathways in Neurological Inflammation: A Mini Review. Front. Mol. Neurosci. 2015, 8, 77. [Google Scholar] [CrossRef]
- Villapol, S. Roles of Peroxisome Proliferator-Activated Receptor Gamma on Brain and Peripheral Inflammation. Cell. Mol. Neurobiol. 2018, 38, 121–132. [Google Scholar] [CrossRef]
- Draper, E.; Reynolds, C.M.; Canavan, M.; Mills, K.H.; Loscher, C.E.; Roche, H.M. Omega-3 fatty acids attenuate dendritic cell function via NF-κB independent of PPARγ. J. Nutr. Biochem. 2011, 22, 784–790. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Ward, W.F. PGC-1α: A key regulator of energy metabolism. Adv. Physiol. Educ. 2006, 30, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Tak, P.P.; Firestein, G.S. NF-κB: A key role in inflammatory diseases. J. Clin. Investig. 2001, 107, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Uematsu, S.; Matsumoto, M.; Takeda, K.; Akira, S. Lipopolysaccharide-dependent prostaglandin E2 production is regulated by the glutathione-dependent prostaglandin E2 synthase gene induced by the Toll-like receptor 4/MyD88/NF-IL6 pathway. J. Immunol. 2002, 168, 5811–5816. [Google Scholar] [CrossRef] [PubMed]
- Hoogland, I.C.M.; Westhoff, D.; Engelen-Lee, J.-Y.; Melief, J.; Valls Serón, M.; Houben-Weerts, J.H.M.P.; Huitinga, I.; van Westerloo, D.J.; van der Poll, T.; van Gool, W.A.; et al. Microglial Activation After Systemic Stimulation with Lipopolysaccharide and Escherichia coli. Front. Cell. Neurosci. 2018, 12, 110. [Google Scholar] [CrossRef]
- Biesmans, S.; Acton, P.D.; Cotto, C.; Langlois, X.; Ver Donck, L.; Bouwknecht, J.A.; Aelvoet, S.-A.; Hellings, N.; Meert, T.F.; Nuydens, R. Effect of stress and peripheral immune activation on astrocyte activation in transgenic bioluminescent Gfap-luc mice. Glia 2015, 63, 1126–1137. [Google Scholar] [CrossRef]
- Xie, Z.; Wei, M.; Morgan, T.E.; Fabrizio, P.; Han, D.; Finch, C.E.; Longo, V.D. Peroxynitrite mediates neurotoxicity of amyloid β-peptide1–42- and lipopolysaccharide-activated microglia. J. Neurosci. 2002, 22, 3484–3492. [Google Scholar] [CrossRef]
- Chhor, V.; Le Charpentier, T.; Lebon, S.; Oré, M.-V.; Celador, I.L.; Josserand, J.; Degos, V.; Jacotot, E.; Hagberg, H.; Sävman, K.; et al. Characterization of phenotype markers and neuronotoxic potential of polarised primary microglia in vitro. Brain Behav. Immun. 2013, 32, 70–85. [Google Scholar] [CrossRef]
- Cherry, J.D.; Olschowka, J.A.; O’Banion, M.K. Neuroinflammation and M2 microglia: The good, the bad, and the inflamed. J. Neuroinflamm. 2014, 11, 98. [Google Scholar] [CrossRef]
- Michaud, J.-P.; Hallé, M.; Lampron, A.; Thériault, P.; Préfontaine, P.; Filali, M.; Tribout-Jover, P.; Lanteigne, A.-M.; Jodoin, R.; Cluff, C.; et al. Toll-like receptor 4 stimulation with the detoxified ligand monophosphoryl lipid A improves Alzheimer’s disease-related pathology. Proc. Natl. Acad. Sci. USA 2013, 110, 1941–1946. [Google Scholar] [CrossRef] [PubMed]
- Bousquet, M.; Gue, K.; Emond, V.; Julien, P.; Kang, J.X.; Cicchetti, F.; Calon, F. Transgenic conversion of omega-6 into omega-3 fatty acids in a mouse model of Parkinson’s disease. J. Lipid Res. 2011, 52, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Emre, C.; Hjorth, E.; Bharani, K.; Carroll, S.; Granholm, A.-C.; Schultzberg, M. Receptors for pro-resolving mediators are increased in Alzheimer’s disease brain. Brain Pathol. 2020, 30, 614–640. [Google Scholar] [CrossRef] [PubMed]
- Weissman, C. The metabolic response to stress: An overview and update. Anesthesiology 1990, 73, 308–327. [Google Scholar] [CrossRef]
- Mayer, K.; Fegbeutel, C.; Hattar, K.; Sibelius, U.; Krämer, H.-J.; Heuer, K.-U.; Temmesfeld-Wollbrück, B.; Gokorsch, S.; Grimminger, F.; Seeger, W. Omega-3 vs. omega-6 lipid emulsions exert differential influence on neutrophils in septic shock patients: Impact on plasma fatty acids and lipid mediator generation. Intensive Care Med. 2003, 29, 1472–1481. [Google Scholar] [CrossRef]
- Hong, S.; Porter, T.F.; Lu, Y.; Oh, S.F.; Pillai, P.S.; Serhan, C.N. Resolvin E1 metabolome in local inactivation during inflammation-resolution. J. Immunol. 2008, 180, 3512–3519. [Google Scholar] [CrossRef]
- Schebb, N.H.; Kühn, H.; Kahnt, A.S.; Rund, K.M.; O’Donnell, V.B.; Flamand, N.; Peters-Golden, M.; Jakobsson, P.-J.; Weylandt, K.H.; Rohwer, N.; et al. Formation, Signaling and Occurrence of Specialized Pro-Resolving Lipid Mediators-What is the Evidence so far? Front. Pharmacol. 2022, 13, 838782. [Google Scholar] [CrossRef]
- Orr, S.K.; Palumbo, S.; Bosetti, F.; Mount, H.T.; Kang, J.X.; Greenwood, C.E.; Ma, D.W.L.; Serhan, C.N.; Bazinet, R.P. Unesterified docosahexaenoic acid is protective in neuroinflammation. J. Neurochem. 2013, 127, 378–393. [Google Scholar] [CrossRef]
- Ariel, A.; Li, P.; Wang, W.; Tang, W.; Fredman, G.; Hong, S.; Gotlinger, K.H.; Serhan, C.N. The docosatriene protectin D1 is produced by TH2 skewing and promotes human T cell apoptosis via lipid raft clustering. J. Biol. Chem. 2005, 280, 43079–43086. [Google Scholar] [CrossRef]
- Serhan, C.N.; Gotlinger, K.; Hong, S.; Lu, Y.; Siegelman, J.; Baer, T.; Yang, R.; Colgan, S.P.; Petasis, N.A. Anti-inflammatory actions of neuroprotectin D1/protectin D1 and its natural stereoisomers: Assignments of dihydroxy-containing docosatrienes. J. Immunol. 2006, 176, 1848–1859. [Google Scholar] [CrossRef]
- Chiang, N.; Fredman, G.; Bäckhed, F.; Oh, S.F.; Vickery, T.; Schmidt, B.A.; Serhan, C.N. Infection regulates pro-resolving mediators that lower antibiotic requirements. Nature 2012, 484, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Wang, X.; Hjorth, E.; Colas, R.A.; Schroeder, L.; Granholm, A.-C.; Serhan, C.N.; Schultzberg, M. Pro-Resolving Lipid Mediators Improve Neuronal Survival and Increase Aβ42 Phagocytosis. Mol. Neurobiol. 2016, 53, 2733–2749. [Google Scholar] [CrossRef] [PubMed]
- Russell, W.; Burch, R.L. The Principles of Humane Experimental Technique. Med. J. Aust. 1960, 1, 500. [Google Scholar] [CrossRef]
- Card, J.W.; Carey, M.A.; Bradbury, J.A.; DeGraff, L.M.; Morgan, D.L.; Moorman, M.P.; Flake, G.P.; Zeldin, D.C. Gender differences in murine airway responsiveness and lipopolysaccharide-induced inflammation. J. Immunol. 2006, 177, 621–630. [Google Scholar] [CrossRef]
- Matute-Bello, G.; Frevert, C.W.; Martin, T.R. Animal models of acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 295, L379–L399. [Google Scholar] [CrossRef]
- Gotts, J.E.; Bernard, O.; Chun, L.; Croze, R.H.; Ross, J.T.; Nesseler, N.; Wu, X.; Abbott, J.; Fang, X.; Calfee, C.S.; et al. Clinically relevant model of pneumococcal pneumonia, ARDS, and nonpulmonary organ dysfunction in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 317, L717–L736. [Google Scholar] [CrossRef] [PubMed]
- Bi, M.H.; Ott, J.; Fischer, T.; Hecker, M.; Dietrich, H.; Schaefer, M.B.; Markart, P.; Wang, B.E.; Seeger, W.; Mayer, K. Induction of lymphocyte apoptosis in a murine model of acute lung injury—Modulation by lipid emulsions. Shock 2010, 33, 179–188. [Google Scholar] [CrossRef]
- Hecker, M.; Ott, J.; Sondermann, C.; Schaefer, M.; Obert, M.; Hecker, A.; Morty, R.E.; Vadasz, I.; Herold, S.; Rosengarten, B.; et al. Immunomodulation by fish-oil containing lipid emulsions in murine acute respiratory distress syndrome. Crit. Care 2014, 18, R85. [Google Scholar] [CrossRef]
- Schaefer, M.B.; Pose, A.; Ott, J.; Hecker, M.; Behnk, A.; Schulz, R.; Weissmann, N.; Günther, A.; Seeger, W.; Mayer, K. Peroxisome proliferator-activated receptor-α reduces inflammation and vascular leakage in a murine model of acute lung injury. Eur. Respir. J. 2008, 32, 1344–1353. [Google Scholar] [CrossRef]
- Haqqani, A.S.; Sandhu, J.K.; Birnboim, H.C. A myeloperoxidase-specific assay based upon bromide-dependent chemiluminescence of luminol. Anal. Biochem. 1999, 273, 126–132. [Google Scholar] [CrossRef]
- Donnelly, S.C.; Strieter, R.M.; Reid, P.T.; Kunkel, S.L.; Burdick, M.D.; Armstrong, I.; Mackenzie, A.; Haslett, C. The association between mortality rates and decreased concentrations of interleukin-10 and interleukin-1 receptor antagonist in the lung fluids of patients with the adult respiratory distress syndrome. Ann. Intern. Med. 1996, 125, 191–196. [Google Scholar] [CrossRef]
- Ye, P.; Garvey, P.B.; Zhang, P.; Nelson, S.; Bagby, G.; Summer, W.R.; Schwarzenberger, P.; Shellito, J.E.; Kolls, J.K. Interleukin-17 and lung host defense against Klebsiella pneumoniae infection. Am. J. Respir. Cell Mol. Biol. 2001, 25, 335–340. [Google Scholar] [CrossRef]
- Law, A.H.Y.; Lee, D.C.W.; Cheung, B.K.W.; Yim, H.C.H.; Lau, A.S.Y. Role for nonstructural protein 1 of severe acute respiratory syndrome coronavirus in chemokine dysregulation. J. Virol. 2007, 81, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Crispe, I.N. The liver as a lymphoid organ. Annu. Rev. Immunol. 2009, 27, 147–163. [Google Scholar] [CrossRef] [PubMed]
- Motulsky, H.J.; Brown, R.E. Detecting outliers when fitting data with nonlinear regression—A new method based on robust nonlinear regression and the false discovery rate. BMC Bioinform. 2006, 7, 123. [Google Scholar] [CrossRef]
- Yuan, J.S.; Reed, A.; Chen, F.; Stewart, C.N. Statistical analysis of real-time PCR data. BMC Bioinform. 2006, 7, 85. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernandez, J.; Schäffer, J.; Herden, C.; Pflieger, F.J.; Reiche, S.; Körber, S.; Kitagawa, H.; Welter, J.; Michels, S.; Culmsee, C.; et al. n-3 Polyunsaturated Fatty Acids Modulate LPS-Induced ARDS and the Lung–Brain Axis of Communication in Wild-Type versus Fat-1 Mice Genetically Modified for Leukotriene B4 Receptor 1 or Chemerin Receptor 23 Knockout. Int. J. Mol. Sci. 2023, 24, 13524. https://doi.org/10.3390/ijms241713524
Hernandez J, Schäffer J, Herden C, Pflieger FJ, Reiche S, Körber S, Kitagawa H, Welter J, Michels S, Culmsee C, et al. n-3 Polyunsaturated Fatty Acids Modulate LPS-Induced ARDS and the Lung–Brain Axis of Communication in Wild-Type versus Fat-1 Mice Genetically Modified for Leukotriene B4 Receptor 1 or Chemerin Receptor 23 Knockout. International Journal of Molecular Sciences. 2023; 24(17):13524. https://doi.org/10.3390/ijms241713524
Chicago/Turabian StyleHernandez, Jessica, Julia Schäffer, Christiane Herden, Fabian Johannes Pflieger, Sylvia Reiche, Svenja Körber, Hiromu Kitagawa, Joelle Welter, Susanne Michels, Carsten Culmsee, and et al. 2023. "n-3 Polyunsaturated Fatty Acids Modulate LPS-Induced ARDS and the Lung–Brain Axis of Communication in Wild-Type versus Fat-1 Mice Genetically Modified for Leukotriene B4 Receptor 1 or Chemerin Receptor 23 Knockout" International Journal of Molecular Sciences 24, no. 17: 13524. https://doi.org/10.3390/ijms241713524
APA StyleHernandez, J., Schäffer, J., Herden, C., Pflieger, F. J., Reiche, S., Körber, S., Kitagawa, H., Welter, J., Michels, S., Culmsee, C., Bier, J., Sommer, N., Kang, J. X., Mayer, K., Hecker, M., & Rummel, C. (2023). n-3 Polyunsaturated Fatty Acids Modulate LPS-Induced ARDS and the Lung–Brain Axis of Communication in Wild-Type versus Fat-1 Mice Genetically Modified for Leukotriene B4 Receptor 1 or Chemerin Receptor 23 Knockout. International Journal of Molecular Sciences, 24(17), 13524. https://doi.org/10.3390/ijms241713524