
Guanylation Reactions for the Rational Design of Cancer Therapeutic Agents

 , ,
, ,  , , ,
, , ,  and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
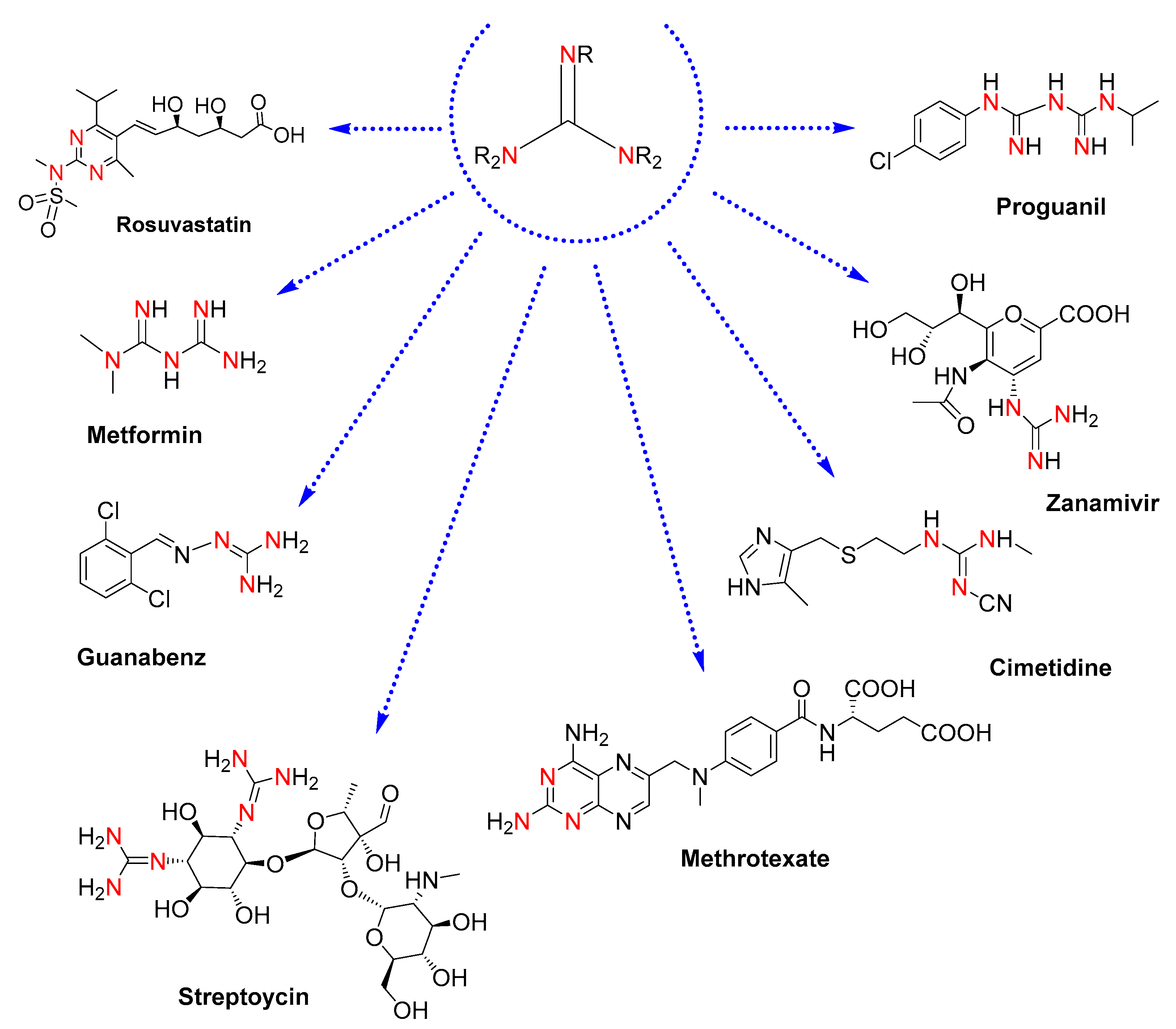
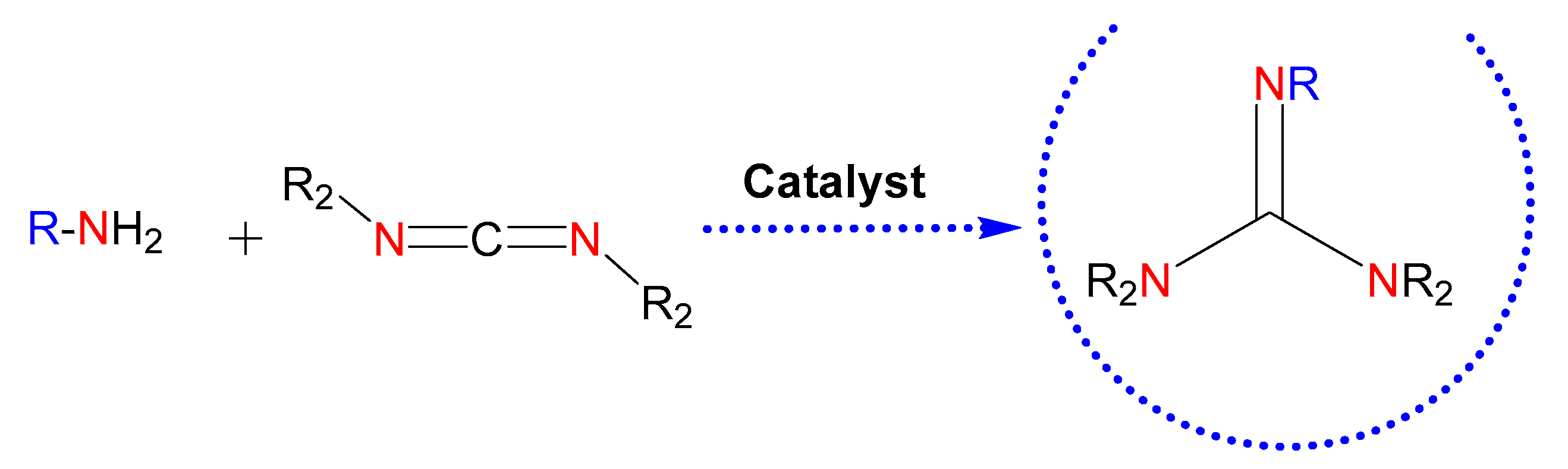
1. Introduction
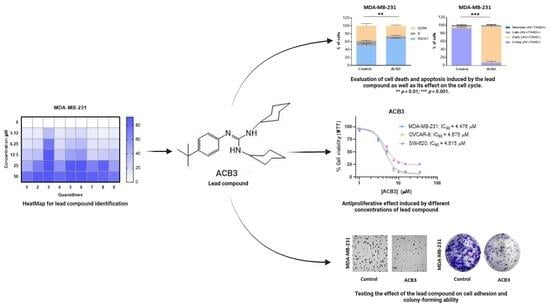
2. Results
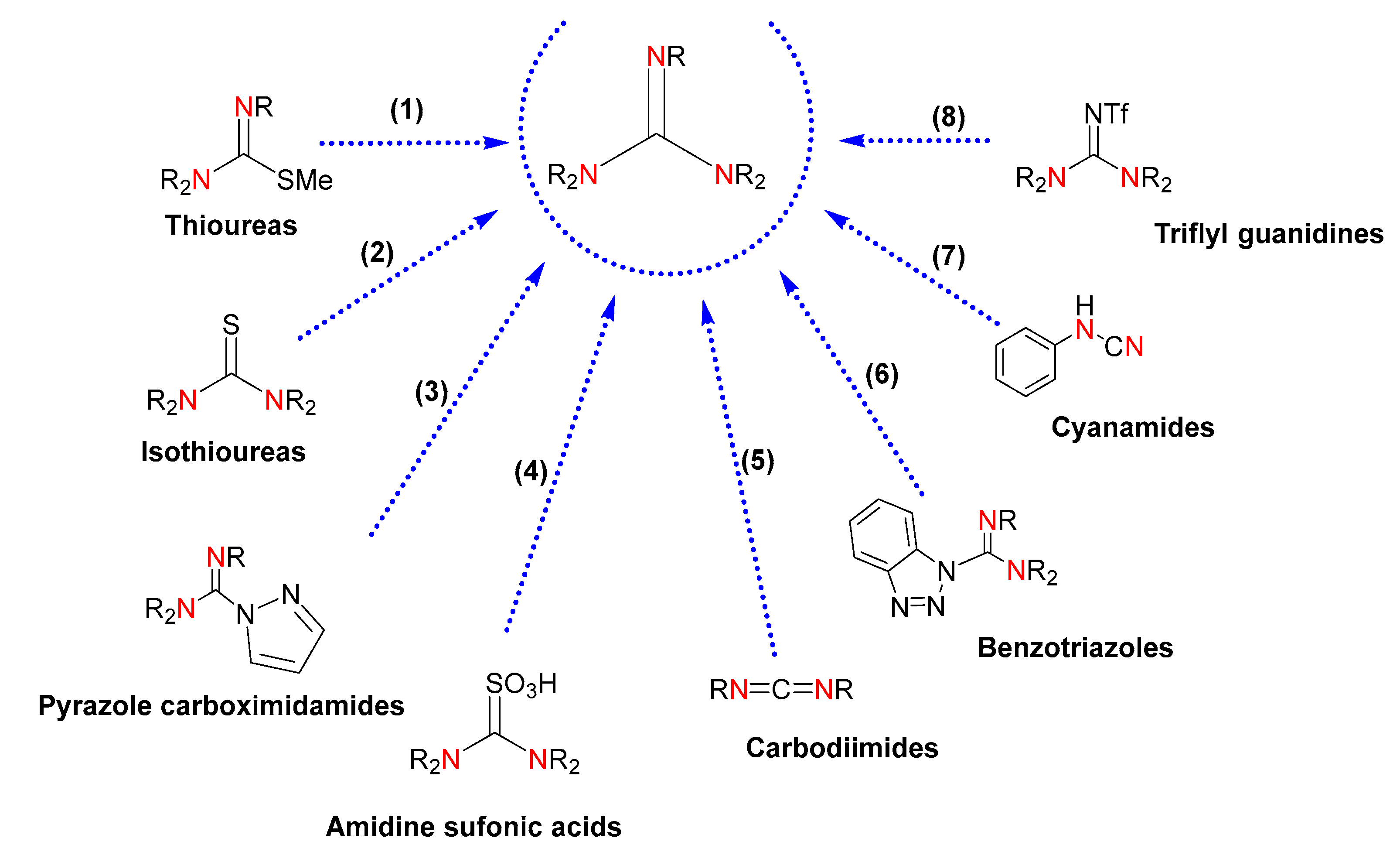
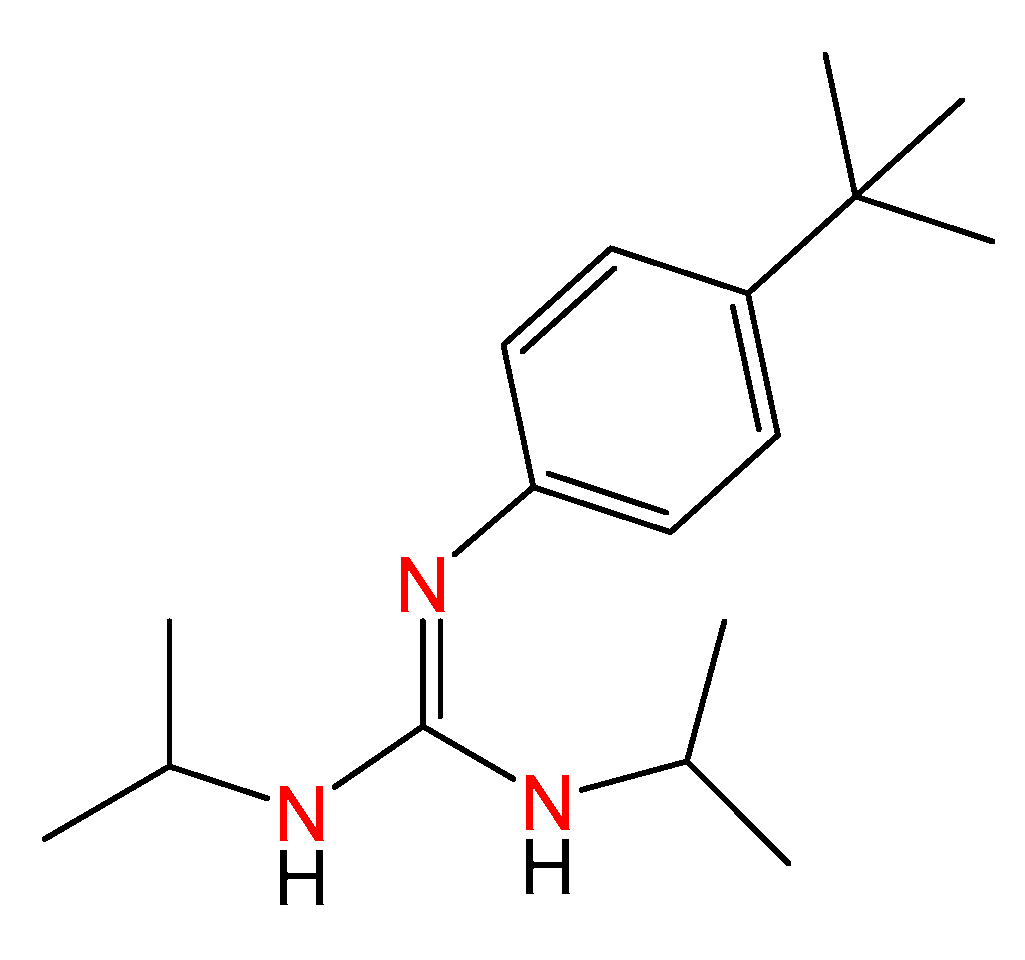
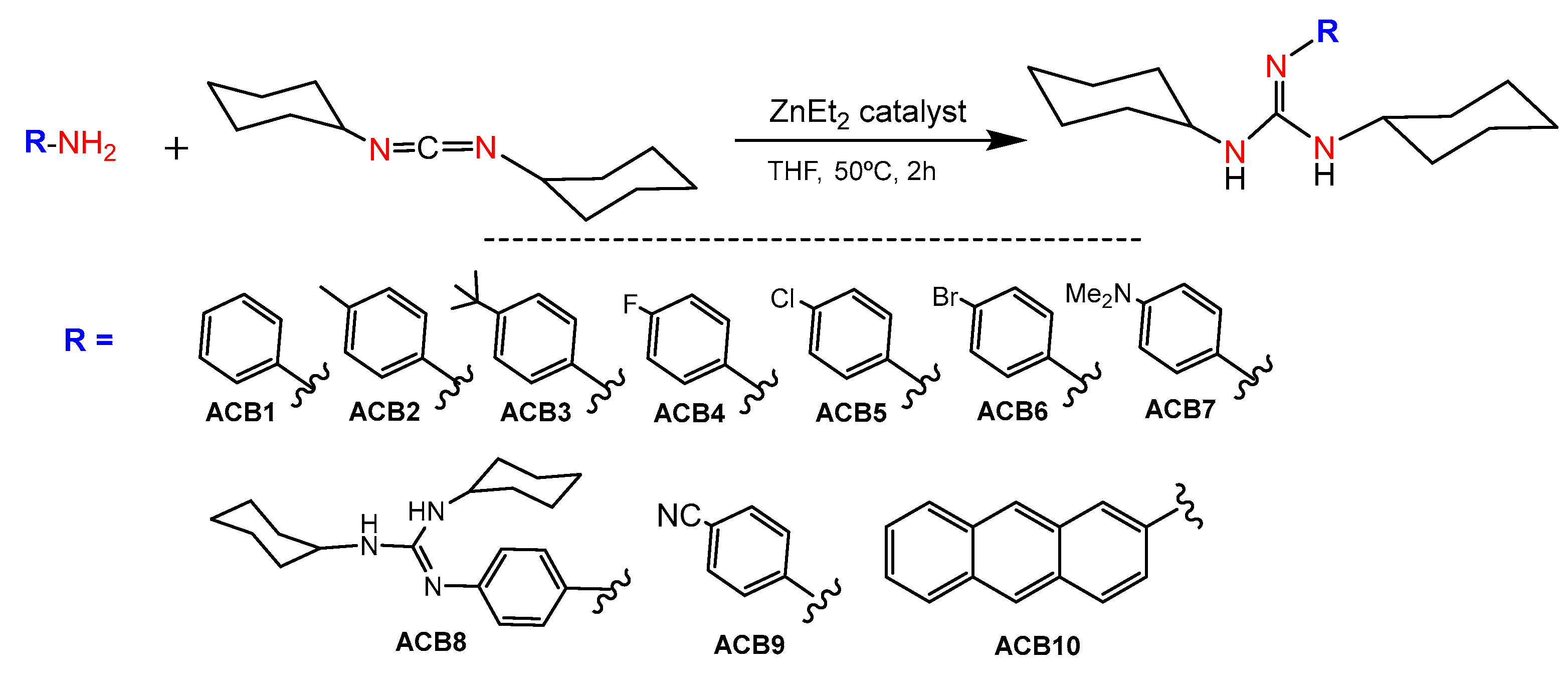
2.1. Synthesis of Guanidine-Based Agents
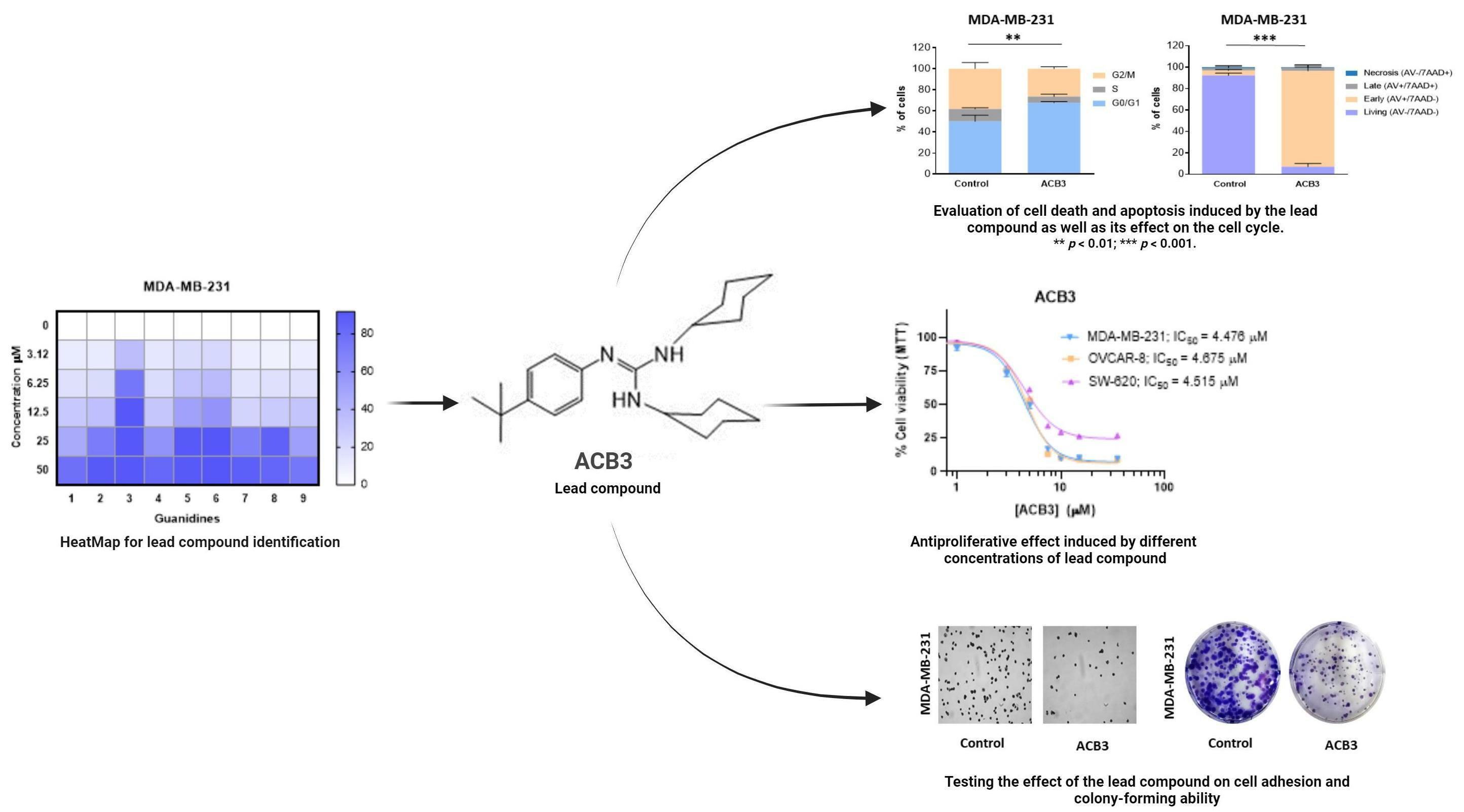
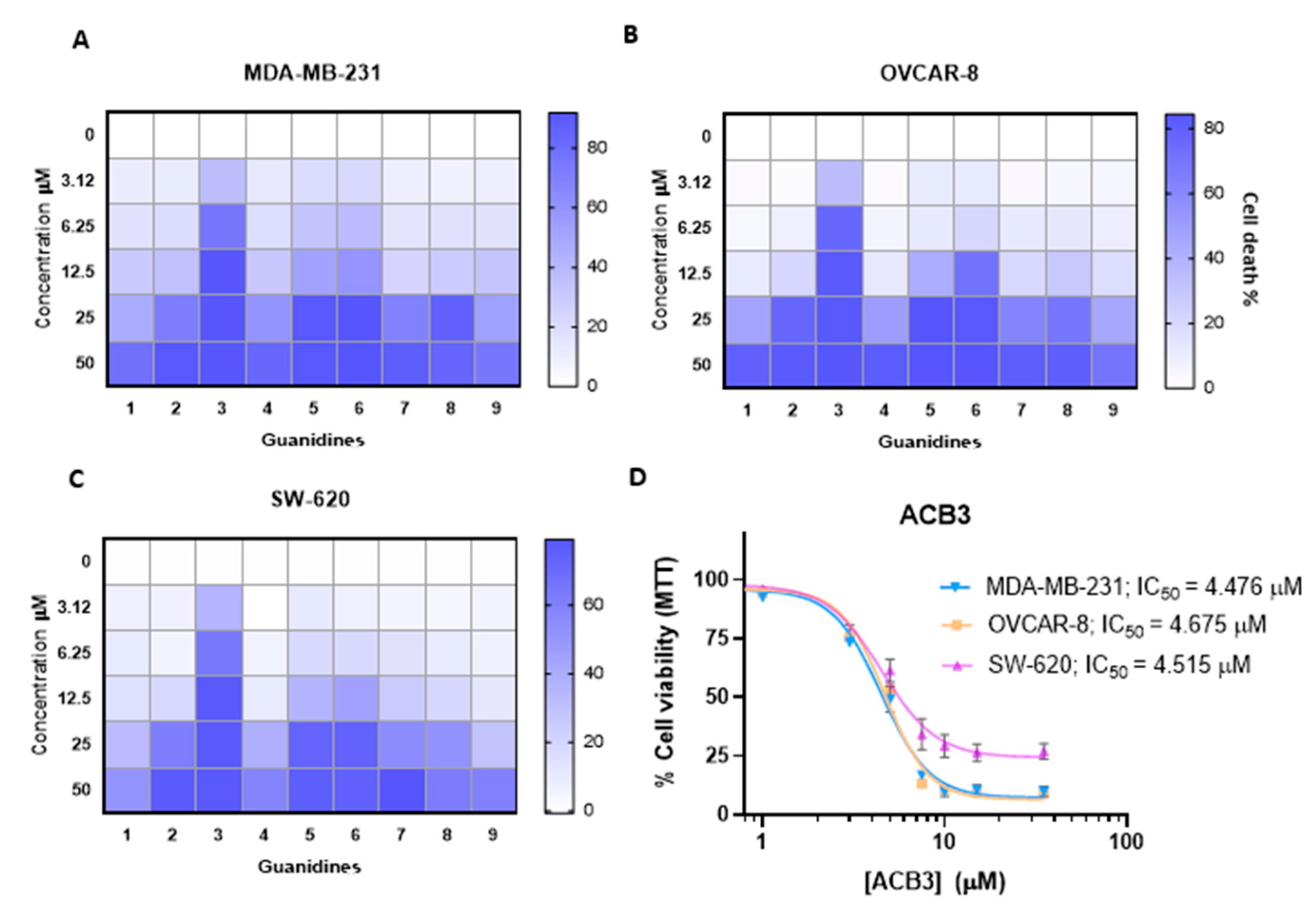
2.2. Antiproliferative Activity of Guanidines ACB0-ACB9 in Cancer Cell Lines
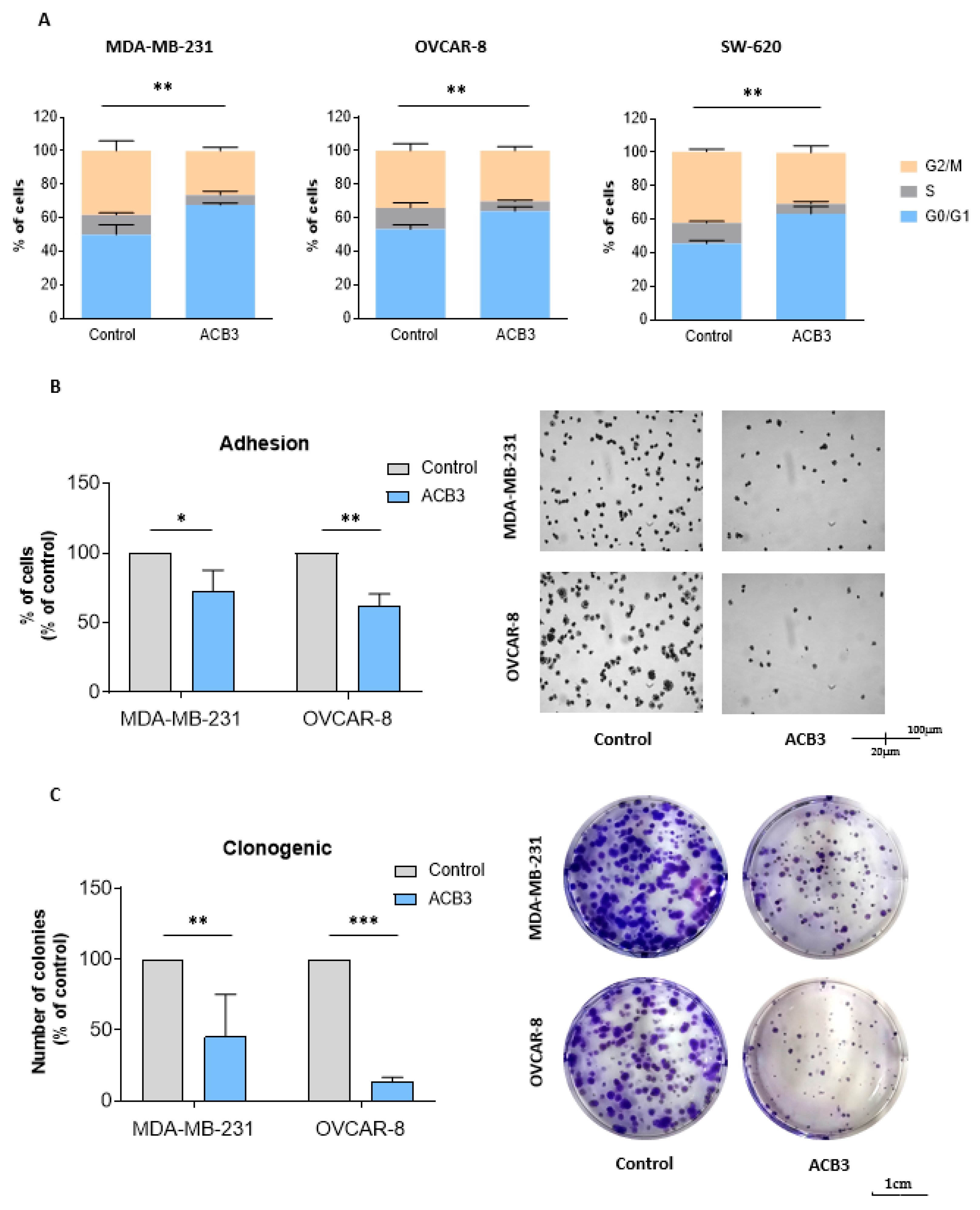
2.3. ACB3 Induces Cell Cycle Arrest and Reduces Cell Adhesion and Survival
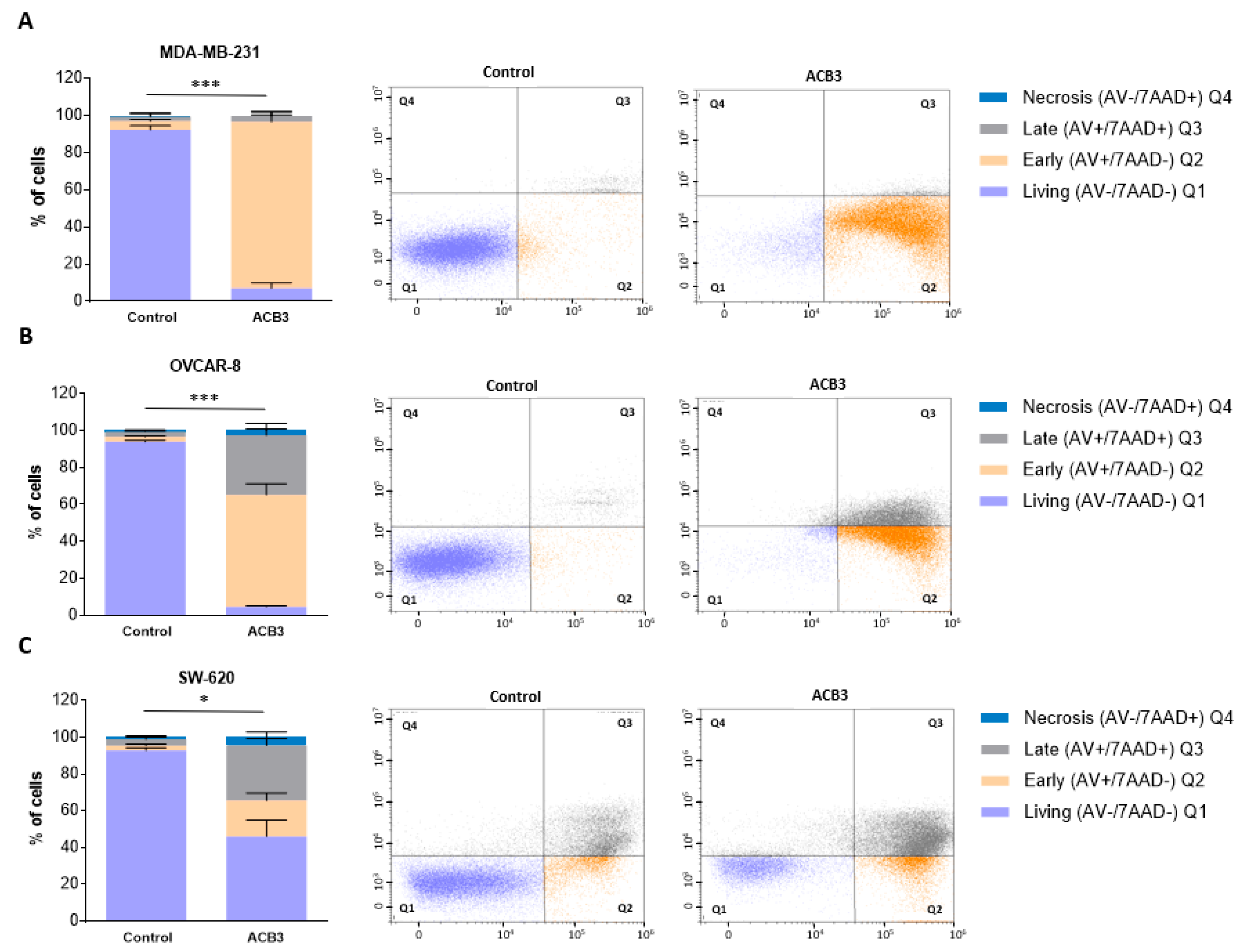
2.4. Treatment with ACB3 Leads to a Strong Induction of Apoptosis
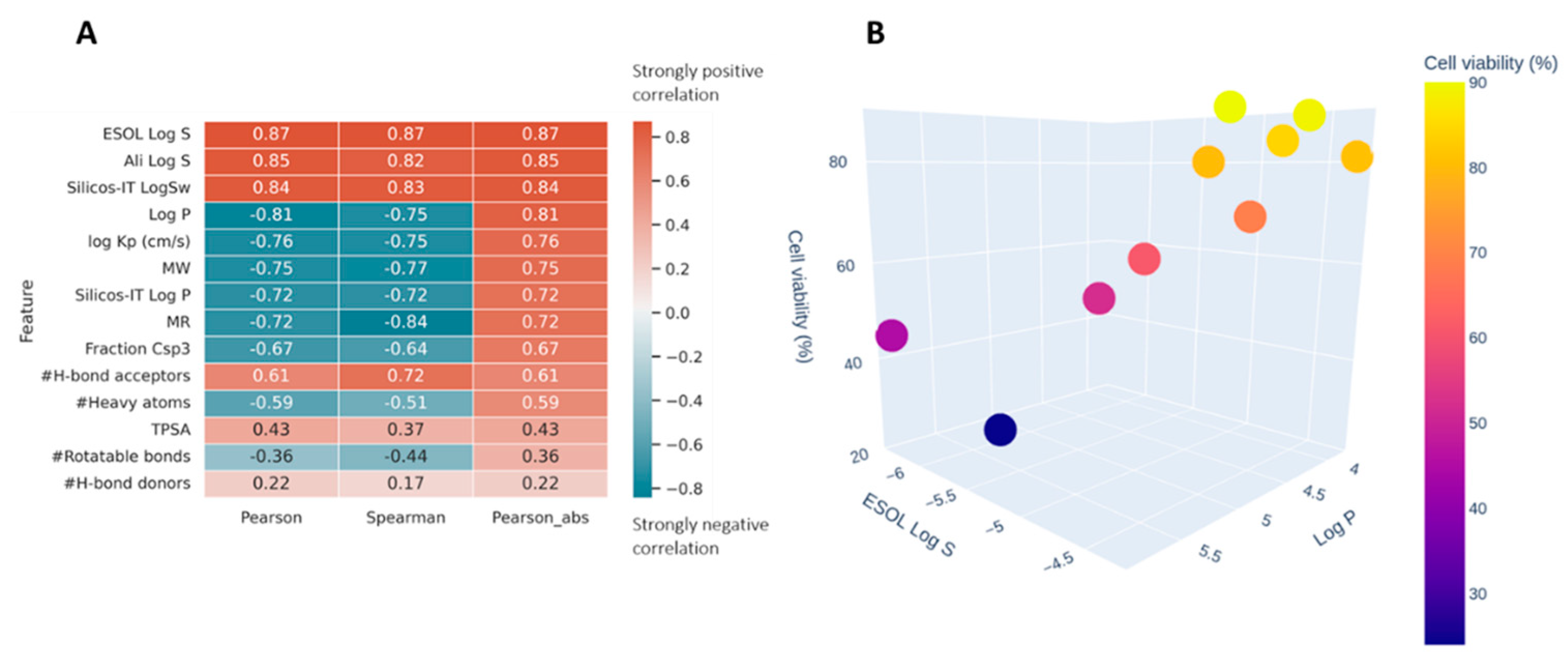
2.5. ADME Analysis for Providing Activity–Structure Relationships
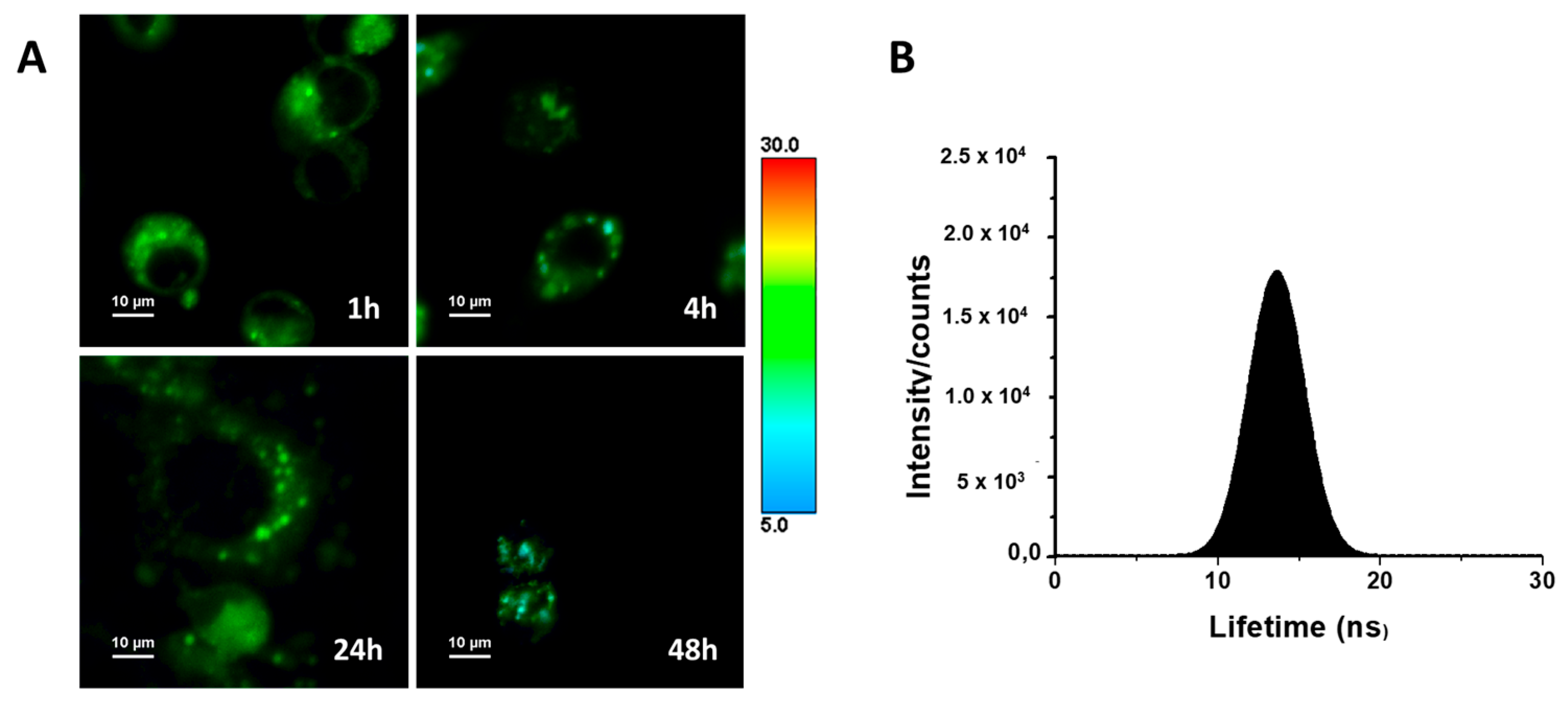
2.6. Uptake Studies for a Luminescent Guanidine Derivative
3. Discussion
4. Materials and Methods
4.1. General Procedure
4.2. Synthesis of Guanidine Derivatives ACB1-ACB9
4.3. Biological Assays
4.4. Fluorescence Lifetime Imaging of Cells
4.5. Estimation of ADME Parameters
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gomes, A.R.; Varela, C.L.; Pires, A.S.; Tavares-da-Silva, E.J.; Roleira, F.M.F. Synthetic and Natural Guanidine Derivatives as Antitumor and Antimicrobial Agents: A Review. Bioorganic Chem. 2023, 138, 106600. [Google Scholar] [CrossRef] [PubMed]
- Saczewski, F.; Balewski, Ł. Biological Activities of Guanidine Compounds. Expert Opin. Ther. Pat. 2009, 19, 1417–1448. [Google Scholar] [CrossRef] [PubMed]
- Oliver, D.W.; Dormehl, I.C.; Wikberg, J.; Dambrova, M. Guanidines: From molecule to primate. Med. Chem. Res. 2004, 13, 427–438. [Google Scholar] [CrossRef]
- Quirk, J.; Thornton, M.; Kirkpatrick, P. Rosuvastatin Calcium. Nat. Rev. Drug Discov. 2003, 2, 769–770. [Google Scholar] [CrossRef] [PubMed]
- Nasri, H.; Rafieian-Kopaei, M. Metformin: Current Knowledge. J. Res. Med. Sci. Off. J. Isfahan Univ. Med. Sci. 2014, 19, 658–664. [Google Scholar]
- Sica, D.A. Chapter 83-Centrally Acting Agents. In Comprehensive Hypertension; Lip, G.Y.H., Hall, J.E., Eds.; Mosby: Philadelphia, PA, USA, 2007; pp. 1027–1035. ISBN 978-0-323-03961-1. [Google Scholar]
- Shamburek, R.D.; Schubert, M.L. Control of Gastric Acid Secretion. Histamine H2-Receptor Antagonists and H+K(+)-ATPase Inhibitors. Gastroenterol. Clin. N. Am. 1992, 21, 527–550. [Google Scholar] [CrossRef]
- Weinblatt, M.E. Methotrexate in Rheumatoid Arthritis: A Quarter Century of Development. Trans. Am. Clin. Climatol. Assoc. 2013, 124, 16–25. [Google Scholar] [PubMed]
- Kim, S.-H.; Semenya, D.; Castagnolo, D. Antimicrobial Drugs Bearing Guanidine Moieties: A Review. Eur. J. Med. Chem. 2021, 216, 113293. [Google Scholar] [CrossRef] [PubMed]
- Von Itzstein, M. The War against Influenza: Discovery and Development of Sialidase Inhibitors. Nat. Rev. Drug Discov. 2007, 6, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Proguanil. In LiverTox: Clinical and Research Information on Drug-Induced Liver Injury; National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD, USA, 2012.
- Buchdunger, E.; Zimmermann, J.; Mett, H.; Meyer, T.; Müller, M.; Druker, B.J.; Lydon, N.B. Inhibition of the Abl Protein-Tyrosine Kinase in Vitro and in Vivo by a 2-Phenylaminopyrimidine Derivative. Cancer Res. 1996, 56, 100–104. [Google Scholar] [PubMed]
- Ostendorf, B.N.; le Coutre, P.; Kim, T.D.; Quintás-Cardama, A. Nilotinib. In Small Molecules in Oncology; Recent Results Cancer Res. Fortschritte Krebsforsch. Progres Dans Rech. Sur Cancer; Springer: Berlin/Heidelberg, Germany, 2014; Volume 201, pp. 67–80. [Google Scholar] [CrossRef]
- Dhillon, S. Gefitinib: A Review of Its Use in Adults with Advanced Non-Small Cell Lung Cancer. Target. Oncol. 2015, 10, 153–170. [Google Scholar] [CrossRef] [PubMed]
- Loesberg, C.; Van Rooij, H.; Romijn, J.C.; Smets, L.A. Mitochondrial Effects of the Guanidino Group-Containing Cytostatic Drugs, m-Iodobenzylguanidine and Methylglyoxal Bis (Guanylhydrazone). Biochem. Pharmacol. 1991, 42, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Ekelund, S.; Nygren, P.; Larsson, R. Guanidino-Containing Drugs in Cancer Chemotherapy: Biochemical and Clinical Pharmacology33Abbreviations: MIBG, m-Iodobenzylguanidine; MGBG, Methylglyoxal Bis(Guanylhydrazone); MIBA, m-Iodobenzylamine; BG, Benzylguanidine; NE, Norepinephrine; GBG, Glyoxal Bis(Guanylhydrazone); EGBG, Ethylglyoxal Bis(Guanylhydrazone); MGBCP, Methylglyoxal Bis(Cyclopentylamidinohydrazone); ODC, Ornithine Decarboxylase; and SAMDC, S-Adenosylmethionine Decarboxylase. Biochem. Pharmacol. 2001, 61, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Menna, P.L.; Comín, J.; Gómez, D.E.; Alonso, D.F. Phenyl-Guanidine Derivatives. US20140228388A1, 14 August 2014. [Google Scholar]
- Ohara, K.; Smietana, M.; Restouin, A.; Mollard, S.; Borg, J.-P.; Collette, Y.; Vasseur, J.-J. Amine-Guanidine Switch: A Promising Approach to Improve DNA Binding and Antiproliferative Activities. J. Med. Chem. 2007, 50, 6465–6475. [Google Scholar] [CrossRef]
- Nagle, P.S.; Rodriguez, F.; Kahvedzić, A.; Quinn, S.J.; Rozas, I. Asymmetrical Diaromatic Guanidinium/2-Aminoimidazolinium Derivatives: Synthesis and DNA Affinity. J. Med. Chem. 2009, 52, 7113–7121. [Google Scholar] [CrossRef] [PubMed]
- Nagle, P.S.; Rodriguez, F.; Nguyen, B.; Wilson, W.D.; Rozas, I. High DNA Affinity of a Series of Peptide Linked Diaromatic Guanidinium-like Derivatives. J. Med. Chem. 2012, 55, 4397–4406. [Google Scholar] [CrossRef] [PubMed]
- Doğan, N.; Yavuz, S.Ç.; Sahin, K.; Orhan, M.D.; Muhammed, H.K.; Calis, S.; Küp, F.Ö.; Avsar, T.; Akkoc, S.; Tapera, M.; et al. Synthesis, Characterization, Biological Activity and Molecular Modeling Studies of Novel Aminoguanidine Derivatives. ChemistrySelect 2022, 7, e202202819. [Google Scholar] [CrossRef]
- Franca, P.H.B.; Da Silva-Junior, E.F.; Aquino, P.G.V.; Santana, A.E.G.; Ferro, J.N.S.; De Oliveira Barreto, E.; Pessoa, C.D.O.; Meira, A.S.; De Aquino, T.M.; Alexandre-Moreira, M.S.; et al. Preliminary in Vitro Evaluation of the Anti-Proliferative Activity of Guanylhydrazone Derivatives. Acta Pharm. 2016, 66, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Rogovoy, A.R.K.B.V. Recent Developments in Guanylating Agents. ARKIVOC 2005, 4, 49–87. [Google Scholar] [CrossRef]
- Alonso-Moreno, C.; Antiñ, A.; Carrillo-Hermosilla, F.; Otero, A. Guanidines: From Classical Approaches to Efficient Catalytic Syntheses. Chem. Soc. Rev. Chem. Soc Rev. 2014, 3406, 3406–3425. [Google Scholar] [CrossRef]
- Zhang, W.-X.; Xu, L.; Xi, Z. Recent Development of Synthetic Preparation Methods for Guanidines via Transition Metal Catalysis. Chem. Commun. 2015, 51, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Moreno, C.; Carrillo-Hermosilla, F.; Garcés, A.; Otero, A.; López-Solera, I.; Rodríguez, A.M.; Antiñolo, A. Simple, Versatile, and Efficient Catalysts for Guanylation of Amines. Organometallics 2010, 29, 2789–2795. [Google Scholar] [CrossRef]
- Kocarnik, J.M.; Compton, K.; Dean, F.E.; Fu, W.; Gaw, B.L.; Harvey, J.D.; Henrikson, H.J.; Lu, D.; Pennini, A.; Xu, R.; et al. Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life Years for 29 Cancer Groups From 2010 to 2019. JAMA Oncol. 2022, 8, 420–444. [Google Scholar] [CrossRef] [PubMed]
- Burrell, R.A.; Swanton, C. Tumour Heterogeneity and the Evolution of Polyclonal Drug Resistance. Mol. Oncol. 2014, 8, 1095–1111. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, H.; Chen, X. Drug Resistance and Combating Drug Resistance in Cancer. Cancer Drug Resist. 2019, 2, 141–160. [Google Scholar] [CrossRef] [PubMed]
- Bravo, I.; Alonso-Moreno, C.; Posadas, I.; Albaladejo, J.; Carrillo-Hermosilla, F.; Ceña, V.; Garzón, A.; López-Solera, I.; Romero-Castillo, L. Phenyl-Guanidine Derivatives as Potential Therapeutic Agents for Glioblastoma Multiforme: Catalytic Syntheses, Cytotoxic Effects and DNA Affinity. RSC Adv. 2016, 6, 8267–8276. [Google Scholar] [CrossRef]
- Ong, T.-G.; O’Brien, J.S.; Korobkov, I.; Richeson, D.S. Facile and Atom-Efficient Amidolithium-Catalyzed C−C and C−N Formation for the Construction of Substituted Guanidines and Propiolamidines. Organometallics 2006, 25, 4728–4730. [Google Scholar] [CrossRef]
- Luo, F.; Li, J.; Wu, S.; Wu, X.; Chen, M.; Zhong, X.; Liu, K. Comparative Profiling between Primary Colorectal Carcinomas and Metastases Identifies Heterogeneity on Drug Resistance. Oncotarget 2016, 7, 63937–63949. [Google Scholar] [CrossRef] [PubMed]
- Balalaeva, I.V.; Zdobnova, T.A.; Krutova, I.V.; Brilkina, A.A.; Lebedenko, E.N.; Deyev, S.M. Passive and Active Targeting of Quantum Dots for Whole-Body Fluorescence Imaging of Breast Cancer Xenografts. J. Biophotonics 2012, 5, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, A.M.; Peter, D. Artificial Intelligence in Cancer Research: Trends, Challenges and Future Directions. Life Basel Switz. 2022, 12, 1991. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
del Campo-Balguerías, A.; Parra-Cadenas, B.; Nieto-Jimenez, C.; Bravo, I.; Ripoll, C.; Poyatos-Racionero, E.; Gancarski, P.; Carrillo-Hermosilla, F.; Alonso-Moreno, C.; Ocaña, A. Guanylation Reactions for the Rational Design of Cancer Therapeutic Agents. Int. J. Mol. Sci. 2023, 24, 13820. https://doi.org/10.3390/ijms241813820
del Campo-Balguerías A, Parra-Cadenas B, Nieto-Jimenez C, Bravo I, Ripoll C, Poyatos-Racionero E, Gancarski P, Carrillo-Hermosilla F, Alonso-Moreno C, Ocaña A. Guanylation Reactions for the Rational Design of Cancer Therapeutic Agents. International Journal of Molecular Sciences. 2023; 24(18):13820. https://doi.org/10.3390/ijms241813820
Chicago/Turabian Styledel Campo-Balguerías, Almudena, Blanca Parra-Cadenas, Cristina Nieto-Jimenez, Iván Bravo, Consuelo Ripoll, Elisa Poyatos-Racionero, Pawel Gancarski, Fernando Carrillo-Hermosilla, Carlos Alonso-Moreno, and Alberto Ocaña. 2023. "Guanylation Reactions for the Rational Design of Cancer Therapeutic Agents" International Journal of Molecular Sciences 24, no. 18: 13820. https://doi.org/10.3390/ijms241813820
APA Styledel Campo-Balguerías, A., Parra-Cadenas, B., Nieto-Jimenez, C., Bravo, I., Ripoll, C., Poyatos-Racionero, E., Gancarski, P., Carrillo-Hermosilla, F., Alonso-Moreno, C., & Ocaña, A. (2023). Guanylation Reactions for the Rational Design of Cancer Therapeutic Agents. International Journal of Molecular Sciences, 24(18), 13820. https://doi.org/10.3390/ijms241813820