
Interaction of Substrates with γ-Secretase at the Level of Individual Transmembrane Helices—A Methodological Approach
 , , , , , and
, , , , , and 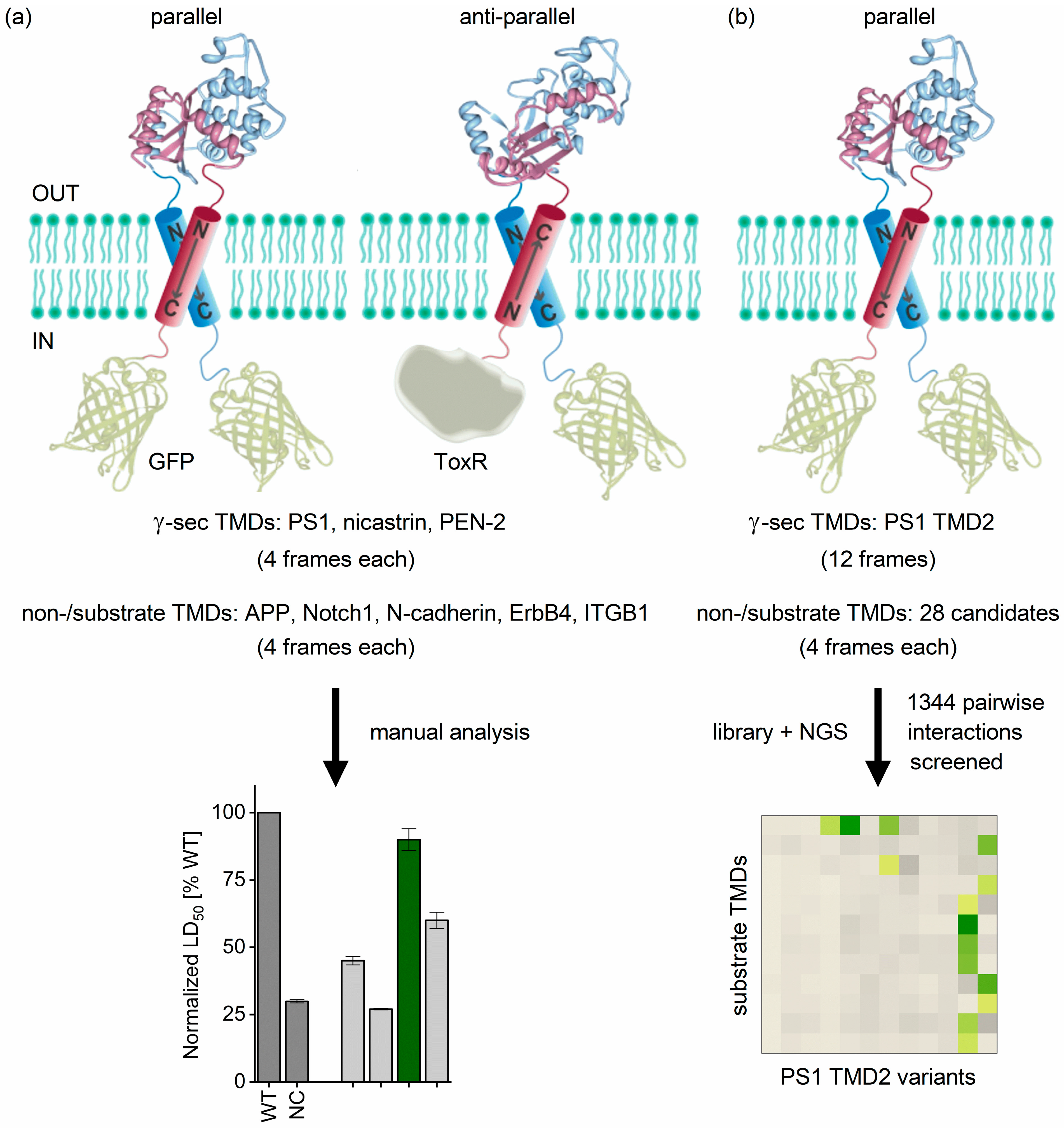{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
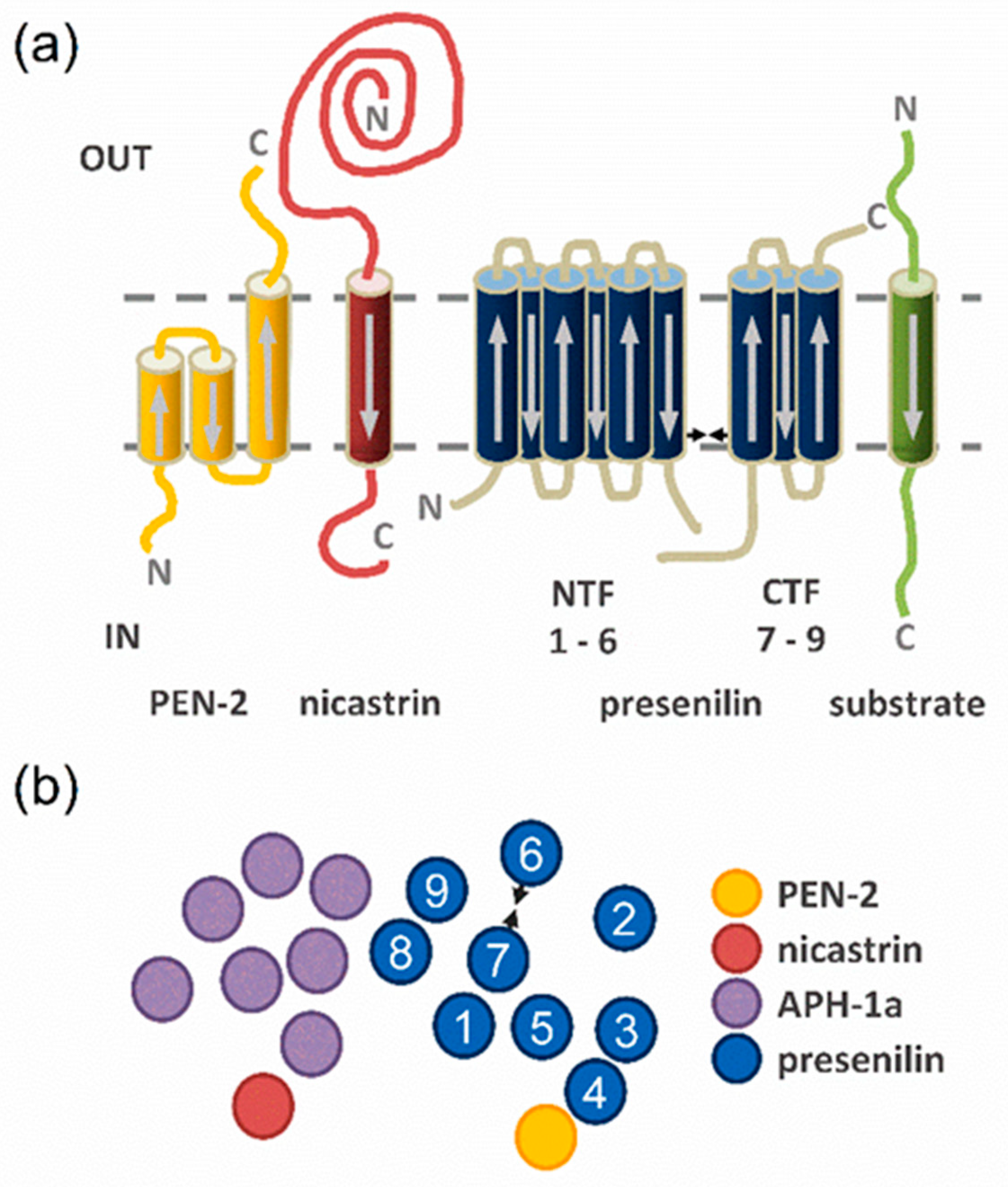
2. Results
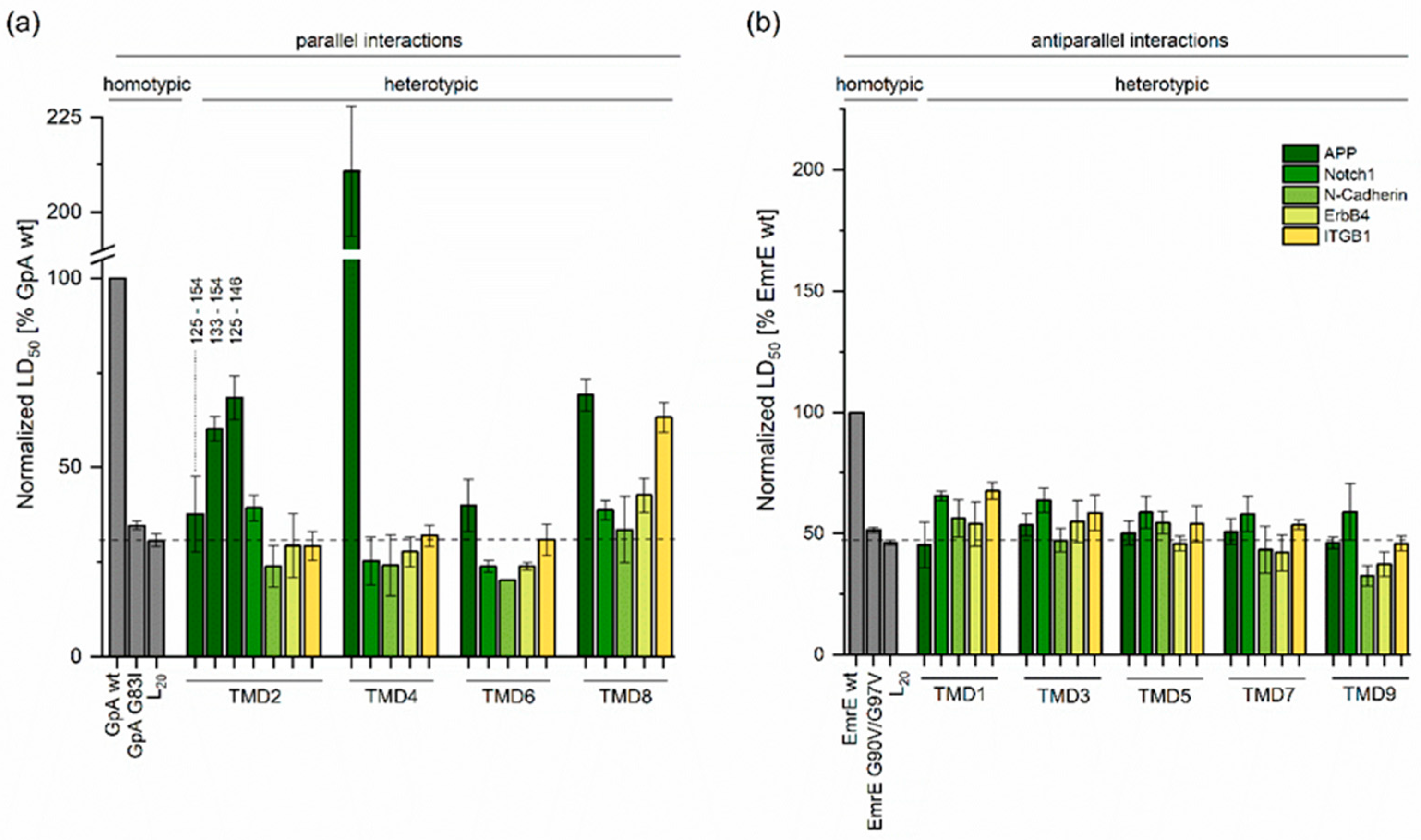
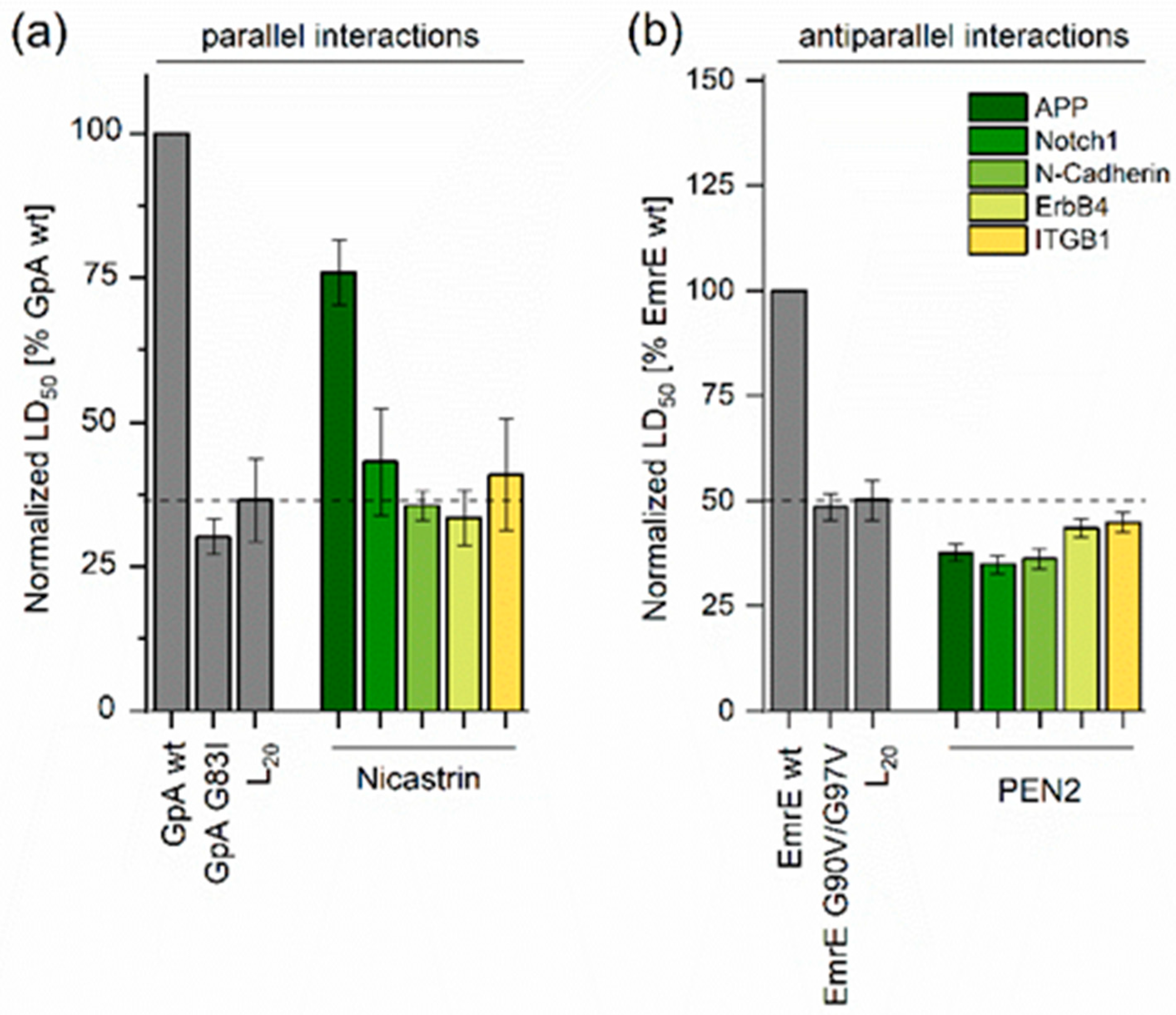
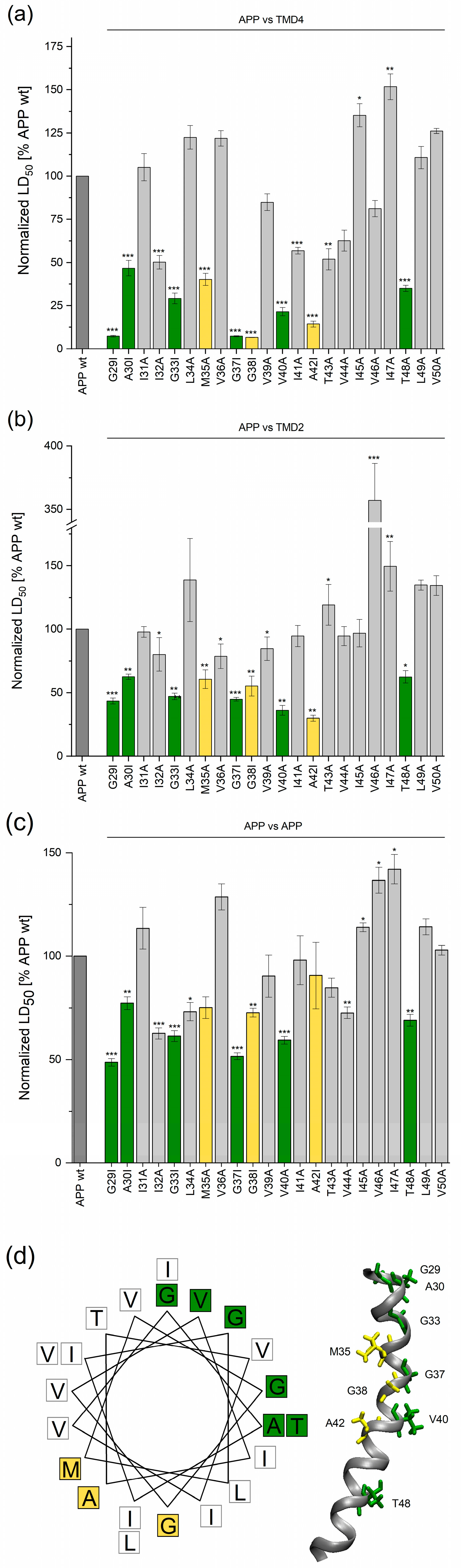
2.1. Determining TMD−TMD Interactions by Manual Testing of Candidate Pairs
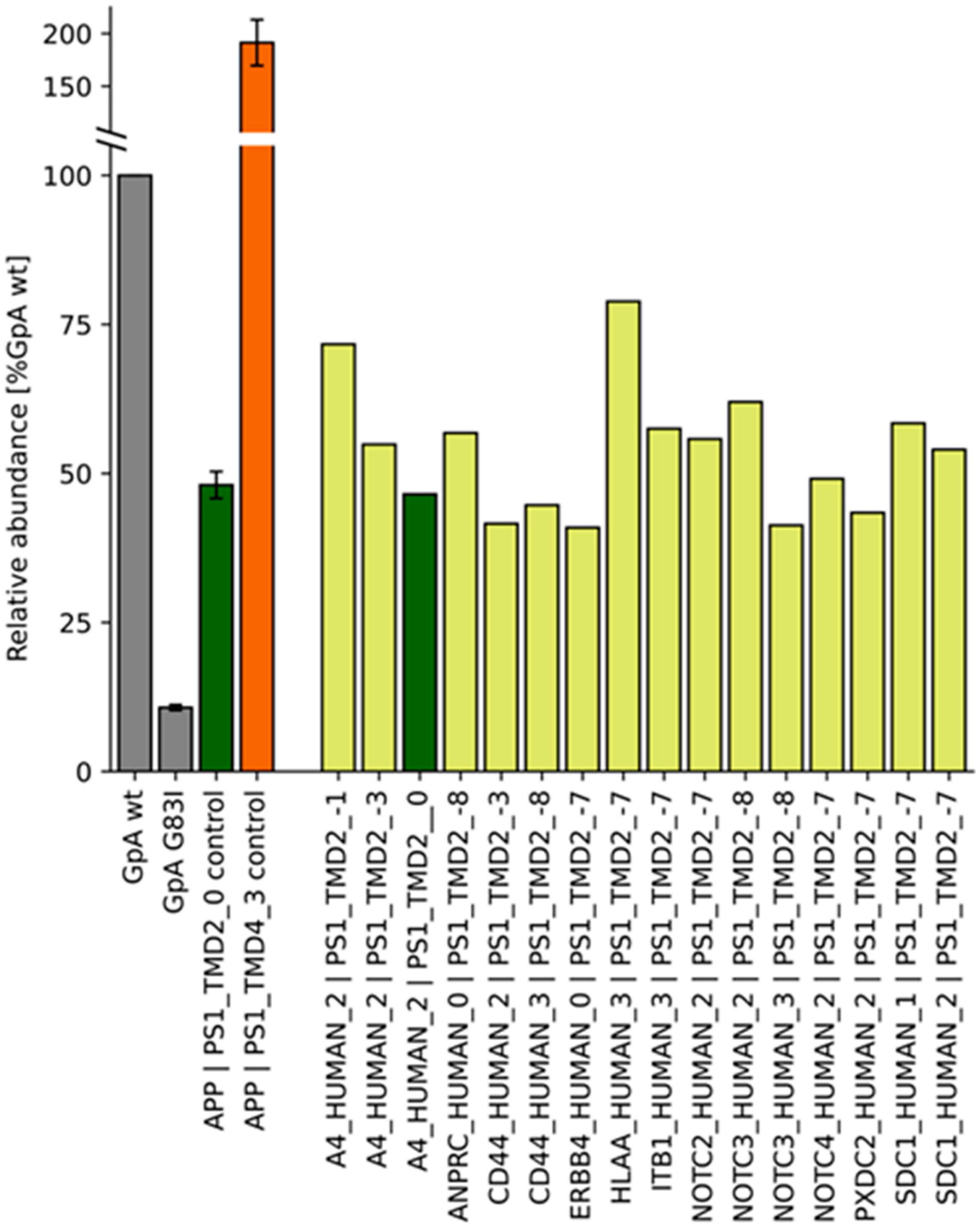
2.2. TMD−TMD Interactions Determined by Library Screening
3. Discussion
4. Materials and Methods
4.1. Plasmid Design and Construction
4.2. Determining Ampicillin LD50 Values
4.3. Determining GFP Expression
4.4. Design and Screening of Combinatorial TMD Libraries
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Langosch, D.; Arkin, I.T. Interaction and Conformational Dynamics of Membrane-Spanning Protein Helices. Protein Sci. 2009, 18, 1343–1358. [Google Scholar] [CrossRef]
- Neumann, J.; Klein, N.; Otzen, D.E.; Schneider, D. Folding energetics and oligomerization of polytopic alpha-helical transmembrane proteins. Arch. Biochem. Biophys. 2014, 564, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Schneider, D.; Engelman, D.M. GALLEX: A measurement of heterologous association of transmembrane helices in a biological membrane. J. Biol. Chem. 2003, 278, 3105–3111. [Google Scholar] [CrossRef] [PubMed]
- Petschnigg, J.; Groisman, B.; Kotlyar, M.; Taipale, M.; Zheng, Y.; Kurat, C.F.; Sayad, A.; Sierra, J.R.; Usaj, M.M.; Snider, J.; et al. The mammalian-membrane two-hybrid assay (MaMTH) for probing membrane-protein interactions in human cells. Nat. Methods 2014, 11, 585–592. [Google Scholar] [CrossRef]
- Karimova, G.; Dautin, N.; Ladant, D. Interaction network among Escherichia coli membrane proteins involved in cell division as revealed by bacterial two-hybrid analysis. J. Bacteriol. 2005, 187, 2233–2243. [Google Scholar] [CrossRef] [PubMed]
- Sawma, P.; Roth, L.; Blanchard, C.; Bagnard, D.; Cremel, G.; Bouveret, E.; Duneau, J.P.; Sturgis, J.N.; Hubert, P. Evidence for new homotypic and heterotypic interactions between transmembrane helices of proteins involved in receptor tyrosine kinase and neuropilin signaling. J. Mol. Biol. 2014, 426, 4099–4111. [Google Scholar] [CrossRef]
- Steindorf, D.; Schneider, D. In vivo selection of heterotypically interacting transmembrane helices: Complementary helix surfaces, rather than conserved interaction motifs, drive formation of transmembrane hetero-dimers. Biochim. Biophys. Acta 2017, 1859, 245–256. [Google Scholar] [CrossRef]
- Duart, G.; Grau, B.; Mingarro, I.; Martinez-Gil, L. Methodological approaches for the analysis of transmembrane domain interactions: A systematic review. Biochim. Biophys. Acta-Biomembr. 2021, 1863, 183712. [Google Scholar] [CrossRef]
- Schanzenbach, C.; Schmidt, F.C.; Breckner, P.; Teese, M.G.; Langosch, D. BLaTM—A genetic tool to measure heterotypic interactions of transmembrane helices. Sci. Rep. 2017, 7, 43476. [Google Scholar] [CrossRef]
- Julius, A.; Laur, L.; Schanzenbach, C.; Langosch, D. BLaTM 2.0, a Genetic Tool Revealing Preferred Antiparallel Interaction of Transmembrane Helix 4 of the Dual-Topology Protein EmrE. J. Mol. Biol. 2017, 11, 1630–1637. [Google Scholar] [CrossRef]
- Steiner, H.; Fukumori, A.; Tagami, S.; Okochi, M. Making the final cut: Pathogenic amyloid-β peptide generation by γ-secretase. Cell Stress 2018, 2, 292–310. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, M.S. Processive proteolysis by gamma-secretase and the mechanism of Alzheimer’s disease. Biol. Chem. 2012, 393, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Guner, G.; Lichtenthaler, S.F. The substrate repertoire of gamma-secretase/presenilin. Semin. Cell Dev. Biol. 2020, 105, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Lichtenthaler, S.F.; Haass, C.; Steiner, H. Regulated intramembrane proteolysis—Lessons from amyloid precursor protein processing. J. Neurochem. 2011, 117, 779–796. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Bai, X.C.; Ma, D.; Xie, T.; Yan, C.; Sun, L.; Yang, G.; Zhao, Y.; Zhou, R.; Scheres, S.H.W.; et al. Three-dimensional structure of human γ-secretase. Nature 2014, 512, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Bolduc, D.M.; Montagna, D.R.; Gu, Y.; Selkoe, D.J.; Wolfe, M.S. Nicastrin functions to sterically hinder gamma-secretase-substrate interactions driven by substrate transmembrane domain. Proc. Natl. Acad. Sci. USA 2016, 113, E509–E518. [Google Scholar] [CrossRef]
- Zhou, R.; Yang, G.; Guo, X.; Zhou, Q.; Lei, J.; Shi, Y. Recognition of the amyloid precursor protein by human gamma-secretase. Science 2019, 363, eaaw0930. [Google Scholar] [CrossRef]
- Yang, G.; Zhou, R.; Zhou, Q.; Guo, X.; Yan, C.; Ke, M.; Lei, J.; Shi, Y. Structural basis of Notch recognition by human γ-secretase. Nature 2019, 565, 192–197. [Google Scholar] [CrossRef]
- Petit, D.; Hitzenberger, M.; Lismont, S.; Zoltowska, K.M.; Ryan, N.S.; Mercken, M.; Bischoff, F.; Zacharias, M.; Chavez-Gutierrez, L. Extracellular interface between APP and Nicastrin regulates A beta length and response to gamma-secretase modulators. EMBO J. 2019, 38, e101494. [Google Scholar] [CrossRef]
- Kornilova, A.Y.; Bihel, F.; Das, C.; Wolfe, M.S. The initial substrate-binding site of γ-secretase is located on presenilin near the active site. Proc. Natl. Acad. Sci. USA 2005, 102, 3230–3235. [Google Scholar] [CrossRef] [PubMed]
- Fukumori, A.; Steiner, H. Substrate recruitment of gamma-secretase and mechanism of clinical presenilin mutations revealed by photoaffinity mapping. EMBO J. 2016, 35, 1628–1643. [Google Scholar] [CrossRef]
- Tomita, T.; Iwatsubo, T. Structural Biology of Presenilins and Signal Peptide Peptidases. J. Biol. Chem. 2013, 288, 14673–14680. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Y.; Zhao, J.; Zhang, Y.K.; Ubarretxena-Belandia, I.; Forth, S.; Lieberman, R.L.; Wang, C.Y. Substrate-Enzyme Interactions in Intramembrane Proteolysis: Gamma-Secretase as the Prototype. Front. Mol. Neurosci. 2020, 13, 65. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.C.; Yan, C.; Yang, G.; Lu, P.; Ma, D.; Sun, L.; Zhou, R.; Scheres, S.H.; Shi, Y. An atomic structure of human gamma-secretase. Nature 2015, 525, 212–217. [Google Scholar] [CrossRef]
- Aguayo-Ortiz, R.; Dominguez, L. Simulating the gamma-secretase enzyme: Recent advances and future directions. Biochimie 2018, 147, 130–135. [Google Scholar] [CrossRef]
- Hitzenberger, M.; Gotz, A.; Menig, S.; Brunschweiger, B.; Zacharias, M.; Scharnagl, C. The dynamics of gamma-secretase and its substrates. Semin. Cell Dev. Biol. 2020, 105, 86–101. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Flanagan, J.M.; Treutlein, H.R.; Zhang, J.; Engelman, D.M. Sequence specificity in the dimerization of transmembrane alpha-helices. Biochemistry 1992, 31, 12719–12725. [Google Scholar] [CrossRef]
- Bai, X.C.; Rajendra, E.; Yang, G.; Shi, Y.; Scheres, S.H. Sampling the conformational space of the catalytic subunit of human gamma-secretase. eLife 2015, 4, e1182. [Google Scholar] [CrossRef]
- Gurezka, R.; Laage, R.; Brosig, B.; Langosch, D. A Heptad Motif of Leucine Residues Found in Membrane Proteins Can Drive Self-Assembly of Artificial Transmembrane Segments. J. Biol. Chem. 1999, 274, 9265–9270. [Google Scholar] [CrossRef]
- Chavez-Gutierrez, L.; Bammens, L.; Benilova, I.; Vandersteen, A.; Benurwar, M.; Borgers, M.; Lismont, S.; Zhou, L.; Van Cleynenbreugel, S.; Esselmann, H.; et al. The mechanism of gamma-Secretase dysfunction in familial Alzheimer disease. EMBO J. 2012, 31, 2261–2274. [Google Scholar] [CrossRef] [PubMed]
- Hemming, M.L.; Elias, J.E.; Gygi, S.P.; Selkoe, D.J. Proteomic profiling of gamma-secretase substrates and mapping of substrate requirements. PLoS Biol. 2008, 6, e257. [Google Scholar] [CrossRef] [PubMed]
- Silber, M.; Hitzenberger, M.; Zacharias, M.; Muhle-Goll, C. Altered Hinge Conformations in APP Transmembrane Helix Mutants May Affect Enzyme-Substrate Interactions of gamma-Secretase. ACS Chem. Neurosci. 2020, 11, 4426–4433. [Google Scholar] [CrossRef]
- Barrett, P.J.; Song, Y.; Van Horn, W.D.; Hustedt, E.J.; Schafer, J.M.; Hadziselimovic, A.; Beel, A.J.; Sanders, C.R. The amyloid precursor protein has a flexible transmembrane domain and binds cholesterol. Science 2012, 336, 1168–1171. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, N.; Image, I., II; Takagi, S.; Tominaga, A.; Image Image, I.; Tomita, T.; Iwatsubo, T. Functional analysis of the transmembrane domains of presenilin 1: Participation of transmembrane domains 2 and 6 in the formation of initial substrate-binding site of γ-secretase. J. Biol. Chem. 2010, 285, 19738–19746. [Google Scholar] [CrossRef]
- Takagi-Niidome, S.; Sasaki, T.; Osawa, S.; Sato, T.; Morishima, K.; Cai, T.; Iwatsubo, T.; Tomita, T. Cooperative roles of hydrophilic loop 1 and the C-terminus of presenilin 1 in the substrate-gating mechanism of gamma-secretase. J. Neurosci. 2015, 35, 2646–2656. [Google Scholar] [CrossRef]
- Beel, A.J.; Sanders, C.R. Substrate specificity of gamma-secretase and other intramembrane proteases. Cell. Mol. Life Sci. 2008, 65, 1311–1334. [Google Scholar] [CrossRef]
- Sato, C.; Takagi, S.; Tomita, T.; Iwatsubo, T. The C-terminal PAL motif and transmembrane domain 9 of presenilin 1 are involved in the formation of the catalytic pore of the gamma-secretase. J. Neurosci. 2008, 28, 6264–6271. [Google Scholar] [CrossRef]
- Kong, R.; Chang, S.; Xia, W.; Wong, S.T. Molecular dynamics simulation study reveals potential substrate entry path into gamma-secretase/presenilin-1. J. Struct. Biol. 2015, 191, 120–129. [Google Scholar] [CrossRef]
- Somavarapu, A.K.; Kepp, K.P. The dynamic mechanism of presenilin-1 function: Sensitive gate dynamics and loop unplugging control protein access. Neurobiol. Dis. 2016, 89, 147–156. [Google Scholar] [CrossRef]
- Li, S.; Zhang, W.; Han, W. Initial Substrate Binding of gamma-Secretase: The Role of Substrate Flexibility. ACS Chem. Neurosci. 2017, 8, 1279–1290. [Google Scholar] [CrossRef]
- Aguayo-Ortiz, R.; Chavez-Garcia, C.; Straub, J.E.; Dominguez, L. Characterizing the structural ensemble of gamma-secretase using a multiscale molecular dynamics approach. Chem. Sci. 2017, 8, 5576–5584. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, A.; Cai, T.; Takagi-Niidome, S.; Iwatsubo, T.; Tomita, T. Conformational Changes in Transmembrane Domain 4 of Presenilin 1 Are Associated with Altered Amyloid-beta 42 Production. J. Neurosci. 2016, 36, 1362–1372. [Google Scholar] [CrossRef]
- Hogel, P.; Gotz, A.; Kuhne, F.; Ebert, M.; Stelzer, W.; Rand, K.D.; Scharnagl, C.; Langosch, D. Glycine Perturbs Local and Global Conformational Flexibility of a Transmembrane Helix. Biochemistry 2018, 57, 1326–1337. [Google Scholar] [CrossRef] [PubMed]
- Teese, M.G.; Langosch, D. Role of GxxxG Motifs in Transmembrane Domain Interactions. Biochemistry 2015, 54, 5125–5135. [Google Scholar] [CrossRef] [PubMed]
- Munter, L.M.; Voigt, P.; Harmeier, A.; Kaden, D.; Gottschalk, K.E.; Weise, C.; Pipkorn, R.; Schaefer, M.; Langosch, D.; Multhaup, G. GxxxG motifs within the amyloid precursor protein transmembrane sequence are critical for the etiology of Aβ42. EMBO J. 2007, 26, 1702–1712. [Google Scholar] [CrossRef]
- Sato, T.; Tang, T.C.; Reubins, G.; Fei, J.Z.; Fujimoto, T.; Kienlen-Campard, P.; Constantinescu, S.N.; Octave, J.N.; Aimoto, S.; Smith, S.O. A helix-to-coil transition at the epsilon-cut site in the transmembrane dimer of the amyloid precursor protein is required for proteolysis. Proc. Natl. Acad. Sci. USA 2009, 106, 1421–1426. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Gamache, E.; Rosenman, D.J.; Xie, J.; Lopez, M.M.; Li, Y.M.; Wang, C. Familial Alzheimer’s mutations within APPTM increase Abeta42 production by enhancing accessibility of epsilon-cleavage site. Nat. Commun. 2014, 5, 3037. [Google Scholar] [CrossRef]
- Nadezhdin, K.D.; Bocharova, O.V.; Bocharov, E.V.; Arseniev, A.S. Dimeric Structure of Transmembrane Domain of Amyloid Precursor Protein in Micellar Environment. FEBS Lett. 2012, 586, 1687–1692. [Google Scholar] [CrossRef]
- Dominguez, L.; Meredith, S.C.; Straub, J.E.; Thirumalai, D. Transmembrane fragment structures of amyloid precursor protein depend on membrane surface curvature. J. Am. Chem. Soc. 2014, 136, 854–857. [Google Scholar] [CrossRef]
- Winkler, E.; Julius, A.; Steiner, H.; Langosch, D. Homodimerization Protects the Amyloid Precursor Protein C99 Fragment from Cleavage by gamma-Secretase. Biochemistry 2015, 54, 6149–6152. [Google Scholar] [CrossRef]
- Mall, S.; Broadbridge, R.; Sharma, R.P.; Lee, A.G.; East, J.M. Effects of aromatic residues at the ends of transmembrane alpha-helices on helix interactions with lipid bilayers. Biochemistry 2000, 39, 2071–2078. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Murria, M.J.; Duart, G.; Grau, B.; Diaz-Beneitez, E.; Rodriguez, D.; Mingarro, I.; Martinez-Gil, L. Viral Bcl2s’ transmembrane domain interact with host Bcl2 proteins to control cellular apoptosis. Nat. Commun. 2020, 11, 6056. [Google Scholar] [CrossRef] [PubMed]
- Duart, G.; Elazar, A.; Weinstein, J.Y.; Gadea-Salom, L.; Ortiz-Mateu, J.; Fleishman, S.J.; Mingarro, I.; Martinez-Gil, L. Computational design of BclxL inhibitors that target transmembrane domain interactions. Proc. Natl. Acad. Sci. USA 2023, 120, e2219648120. [Google Scholar] [CrossRef]
- Vincent, M.S.; Comas Hervada, C.; Sebban-Kreuzer, C.; Le Guenno, H.; Chabalier, M.; Kosta, A.; Guerlesquin, F.; Mignot, T.; McBride, M.J.; Cascales, E.; et al. Dynamic proton-dependent motors power type IX secretion and gliding motility in Flavobacterium. PLoS Biol. 2022, 20, e3001443. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pauli, T.M.; Julius, A.; Costa, F.; Eschrig, S.; Moosmüller, J.; Fischer, L.; Schanzenbach, C.; Schmidt, F.C.; Ortner, M.; Langosch, D. Interaction of Substrates with γ-Secretase at the Level of Individual Transmembrane Helices—A Methodological Approach. Int. J. Mol. Sci. 2023, 24, 14396. https://doi.org/10.3390/ijms241814396
Pauli TM, Julius A, Costa F, Eschrig S, Moosmüller J, Fischer L, Schanzenbach C, Schmidt FC, Ortner M, Langosch D. Interaction of Substrates with γ-Secretase at the Level of Individual Transmembrane Helices—A Methodological Approach. International Journal of Molecular Sciences. 2023; 24(18):14396. https://doi.org/10.3390/ijms241814396
Chicago/Turabian StylePauli, Theresa M., Ayse Julius, Francesco Costa, Sabine Eschrig, Judith Moosmüller, Lea Fischer, Christoph Schanzenbach, Fabian C. Schmidt, Martin Ortner, and Dieter Langosch. 2023. "Interaction of Substrates with γ-Secretase at the Level of Individual Transmembrane Helices—A Methodological Approach" International Journal of Molecular Sciences 24, no. 18: 14396. https://doi.org/10.3390/ijms241814396
APA StylePauli, T. M., Julius, A., Costa, F., Eschrig, S., Moosmüller, J., Fischer, L., Schanzenbach, C., Schmidt, F. C., Ortner, M., & Langosch, D. (2023). Interaction of Substrates with γ-Secretase at the Level of Individual Transmembrane Helices—A Methodological Approach. International Journal of Molecular Sciences, 24(18), 14396. https://doi.org/10.3390/ijms241814396