
Two Different Isocitrate Dehydrogenases from Pseudomonas aeruginosa: Enzymology and Coenzyme-Evolutionary Implications


Abstract
:1. Introduction
2. Results
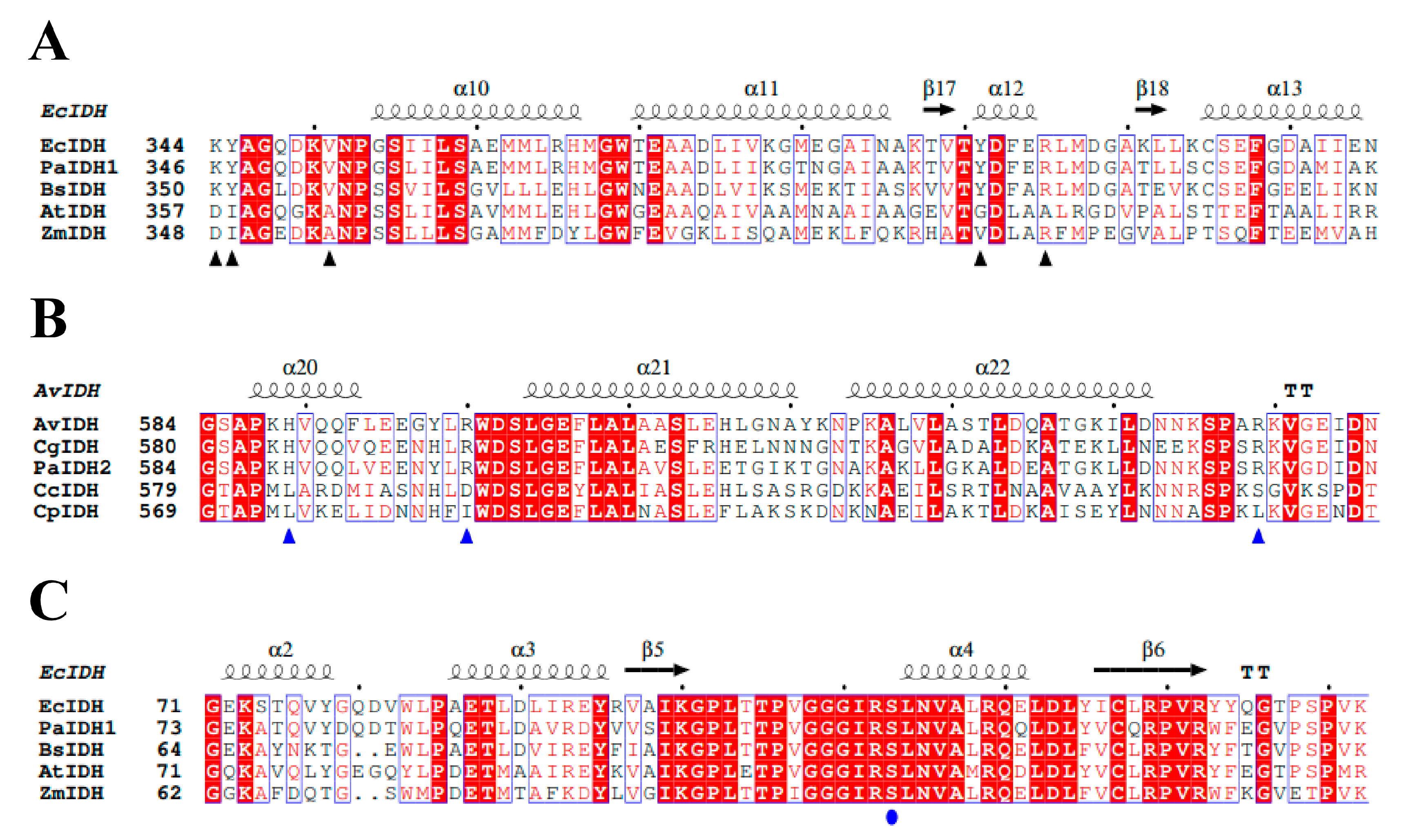
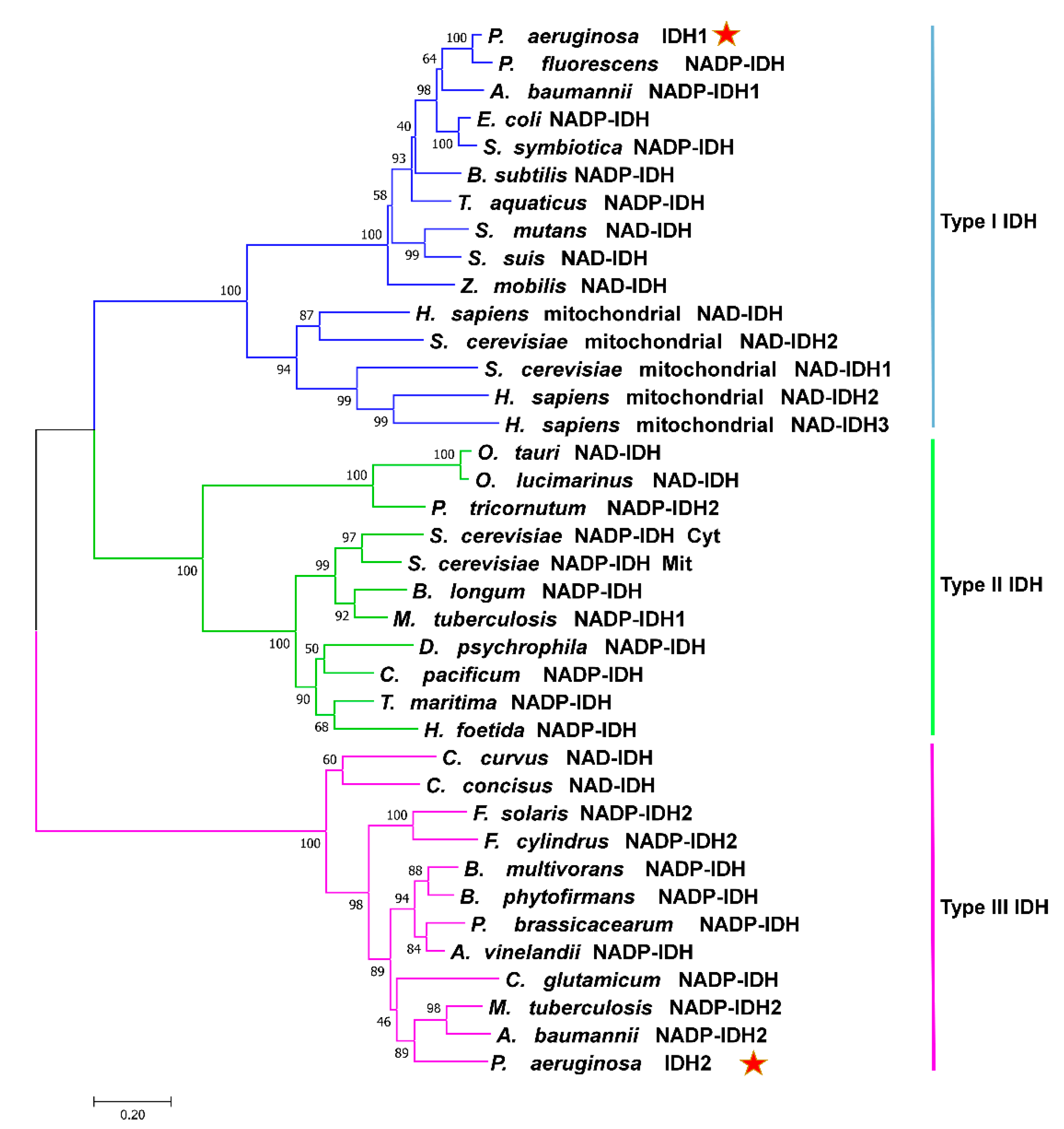
2.1. Sequence and Structure Analysis
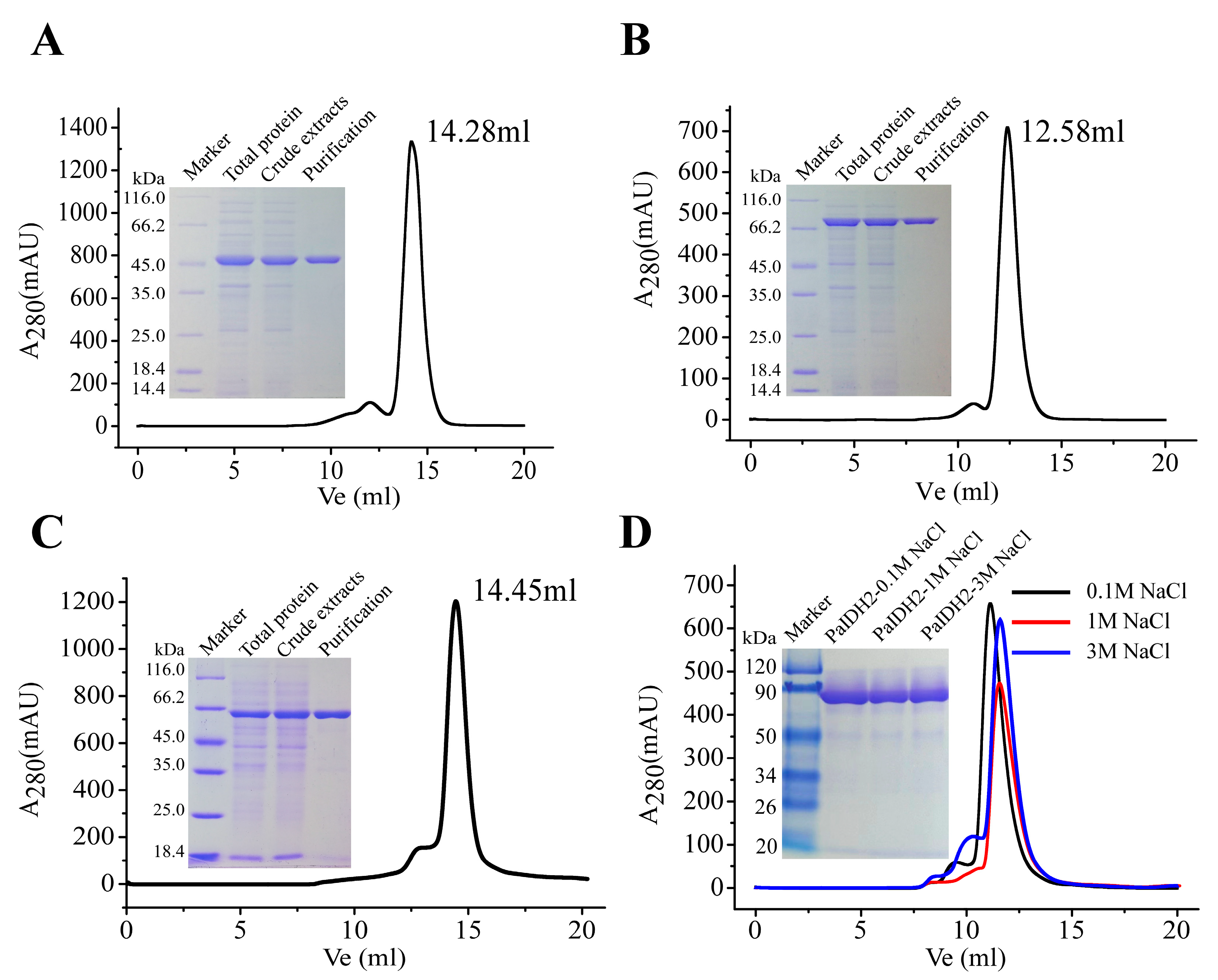
2.2. Overexpression and Purification of Recombinant PaIDH1, PaIDH2 and PaIDH K/P
2.3. Effects of pH, Temperature and Various Compounds on the Activity of PaIDH1 and PaIDH2
2.4. Kinetics Analysis
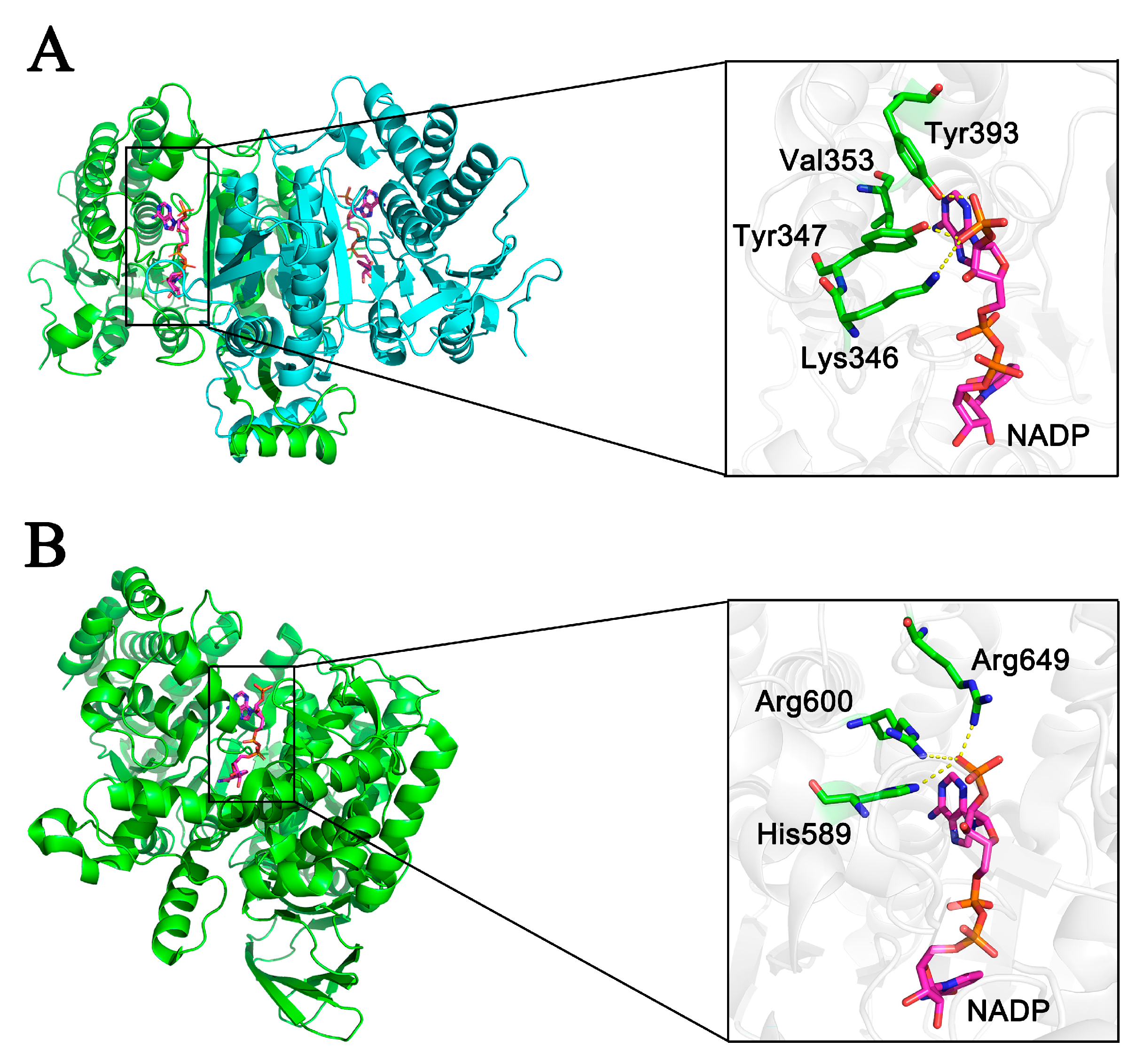
2.5. Evaluation of Coenzyme Specificity Determinants
2.6. In Vitro Phosphorylation and Identification of Phosphorylation Site
3. Discussion
4. Materials and Methods
4.1. Strains and Reagents
4.2. Plasmid Construction
4.3. Protein Expression and Purification
4.4. Enzyme Assay and Kinetic Studies
4.5. Effects of pH, Temperature and Compounds
4.6. Gel Filtration Chromatography
4.7. In Vitro Phosphorylation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Iversen, B.G.; Jacobsen, T.; Eriksen, H.M.; Bukholm, G.; Melby, K.K.; Nygard, K.; Aavitsland, P. An outbreak of Pseudomonas aeruginosa infection caused by contaminated mouth swabs. Clin. Infect. Dis. 2007, 44, 794–801. [Google Scholar] [CrossRef]
- Spiers, A.J.; Buckling, A.; Rainey, P.B. The causes of Pseudomonas diversity. Microbiology 2000, 146 Pt 10, 2345–2350. [Google Scholar] [CrossRef]
- Gilligan, P.H. Microbiology of airway disease in patients with cystic fibrosis. Clin. Microbiol. Rev. 1991, 4, 35–51. [Google Scholar] [CrossRef]
- Williams, P.; Camara, M. Quorum sensing and environmental adaptation in Pseudomonas aeruginosa: A tale of regulatory networks and multifunctional signal molecules. Curr. Opin. Microbiol. 2009, 12, 182–191. [Google Scholar] [CrossRef]
- Luckett, J.C.; Darch, O.; Watters, C.; Abuoun, M.; Wright, V.; Paredes-Osses, E.; Ward, J.; Goto, H.; Heeb, S.; Pommier, S.; et al. A novel virulence strategy for Pseudomonas aeruginosa mediated by an autotransporter with arginine-specific aminopeptidase activity. PLoS Pathog. 2012, 8, e1002854. [Google Scholar] [CrossRef]
- Gellatly, S.L.; Hancock, R.E. Pseudomonas aeruginosa: New insights into pathogenesis and host defenses. Pathog. Dis. 2013, 67, 159–173. [Google Scholar] [CrossRef]
- Zhu, G.; Golding, G.B.; Dean, A.M. The selective cause of an ancient adaptation. Science 2005, 307, 1279–1282. [Google Scholar] [CrossRef]
- Chen, R.; Greer, A.; Dean, A.M. A highly active decarboxylating dehydrogenase with rationally inverted coenzyme specificity. Proc. Natl. Acad. Sci. USA 1995, 92, 11666–11670. [Google Scholar] [CrossRef]
- Steen, I.H.; Lien, T.; Madsen, M.S.; Birkeland, N.K. Identification of cofactor discrimination sites in NAD-isocitrate dehydrogenase from Pyrococcus furiosus. Arch. Microbiol. 2002, 178, 297–300. [Google Scholar] [CrossRef]
- Wang, P.; Jin, M.; Zhu, G. Biochemical and molecular characterization of NAD(+)-dependent isocitrate dehydrogenase from the ethanologenic bacterium Zymomonas mobilis. FEMS Microbiol. Lett. 2011, 327, 134–141. [Google Scholar] [CrossRef]
- McAlister-Henn, L. Ligand binding and structural changes associated with allostery in yeast NAD(+)-specific isocitrate dehydrogenase. Arch. Biochem. Biophys. 2012, 519, 112–117. [Google Scholar] [CrossRef]
- Yang, F.; Zhang, S.; Zhou, Y.J.; Zhu, Z.; Lin, X.; Zhao, Z.K. Characterization of the mitochondrial NAD+ -dependent isocitrate dehydrogenase of the oleaginous yeast Rhodosporidium toruloides. Appl. Microbiol. Biotechnol. 2012, 94, 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- Lancien, M.; Gadal, P.; Hodges, M. Molecular characterization of higher plant NAD-dependent isocitrate dehydrogenase: Evidence for a heteromeric structure by the complementation of yeast mutants. Plant J. 1998, 16, 325–333. [Google Scholar] [CrossRef]
- Soundar, S.; Park, J.H.; Huh, T.L.; Colman, R.F. Evaluation by mutagenesis of the importance of 3 arginines in alpha, beta, and gamma subunits of human NAD-dependent isocitrate dehydrogenase. J. Biol. Chem. 2003, 278, 52146–52153. [Google Scholar] [CrossRef]
- Bzymek, K.P.; Colman, R.F. Role of alpha-Asp181; beta-Asp192, and gamma-Asp190 in the distinctive subunits of human NAD-specific isocitrate dehydrogenase. Biochemistry 2007, 46, 5391–5397. [Google Scholar] [CrossRef] [PubMed]
- Lemaitre, T.; Urbanczyk-Wochniak, E.; Flesch, V.; Bismuth, E.; Fernie, A.R.; Hodges, M. NAD-dependent isocitrate dehydrogenase mutants of Arabidopsis suggest the enzyme is not limiting for nitrogen assimilation. Plant Physiol. 2007, 144, 1546–1558. [Google Scholar] [CrossRef]
- Qi, F.; Chen, X.; Beard, D.A. Detailed kinetics and regulation of mammalian NAD-linked isocitrate dehydrogenase. Biochim. Biophys. Acta 2008, 1784, 1641–1651. [Google Scholar] [CrossRef]
- Liu, Y.; Hu, L.; Ma, T.; Yang, J.; Ding, J. Insights into the inhibitory mechanisms of NADH on the αγ heterodimer of human NAD-dependent isocitrate dehydrogenase. Sci. Rep. 2018, 8, 3146. [Google Scholar] [CrossRef]
- Cohen, P.F.; Colman, R.F. Purification of NAD-specific isocitrate dehydrogenase from porcine heart. Biochim. Biophys. Acta 1971, 242, 325–330. [Google Scholar] [CrossRef]
- Kim, S.Y.; Park, J.W. Cellular defense against singlet oxygen-induced oxidative damage by cytosolic NADP+-dependent isocitrate dehydrogenase. Free. Radic. Res. 2003, 37, 309–316. [Google Scholar] [CrossRef]
- Jo, S.H.; Son, M.K.; Koh, H.J.; Lee, S.M.; Song, I.H.; Kim, Y.O.; Lee, Y.S.; Jeong, K.S.; Kim, W.B.; Park, J.W.; et al. Control of mitochondrial redox balance and cellular defense against oxidative damage by mitochondrial NADP+-dependent isocitrate dehydrogenase. J. Biol. Chem. 2001, 276, 16168–16176. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Lee, D.C.; Jia, Z. Purification, crystallization and preliminary X-ray analysis of isocitrate dehydrogenase kinase/phosphatase from Escherichia coli. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2009, 65, 536–539. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Greer, A.F.; Dean, A.M. Structural constraints in protein engineering--the coenzyme specificity of Escherichia coli isocitrate dehydrogenase. Eur. J. Biochem. 1997, 250, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Nimmo, H.G. Kinetic mechanism of Escherichia coli isocitrate dehydrogenase and its inhibition by glyoxylate and oxaloacetate. Biochem. J. 1986, 234, 317–323. [Google Scholar] [CrossRef]
- Reeves, H.C.; Malloy, P.J. The phosphorylation of isocitrate dehydrogenase in Escherichia coli. Biochem. Soc. Trans. 1982, 10, 321–322. [Google Scholar] [CrossRef]
- Reeves, H.C.; Daumy, G.O.; Lin, C.C.; Houston, M. NADP + -specific isocitrate dehydrogenase of Escherichia coli. I. Purification and characterization. Biochim. Biophys. Acta 1972, 258, 27–39. [Google Scholar] [CrossRef]
- Wang, P.; Lv, C.; Zhu, G. Novel type II and monomeric NAD+ specific isocitrate dehydrogenases: Phylogenetic affinity, enzymatic characterization, and evolutionary implication. Sci. Rep. 2015, 5, 9150. [Google Scholar] [CrossRef]
- Wu, M.C.; Tian, C.Q.; Cheng, H.M.; Xu, L.; Wang, P.; Zhu, G.P. A Novel Type II NAD+-Specific Isocitrate Dehydrogenase from the Marine Bacterium Congregibacter litoralis KT71. PLoS ONE 2015, 10, e0125229. [Google Scholar] [CrossRef]
- Tang, W.G.; Song, P.; Cao, Z.Y.; Wang, P.; Zhu, G.P. A unique homodimeric NAD+-linked isocitrate dehydrogenase from the smallest autotrophic eukaryote Ostreococcus tauri. FASEB J. 2015, 29, 2462–2472. [Google Scholar] [CrossRef]
- Wang, P.; Wu, Y.; Liu, J.; Song, P.; Li, S.; Zhou, X.; Zhu, G. Crystal Structure of the Isocitrate Dehydrogenase 2 from Acinetobacter baumannii (AbIDH2) Reveals a Novel Dimeric Structure with Two Monomeric-IDH-Like Subunits. Int. J. Mol. Sci. 2018, 19, 1131. [Google Scholar] [CrossRef]
- Yasutake, Y.; Watanabe, S.; Yao, M.; Takada, Y.; Fukunaga, N.; Tanaka, I. Structure of the monomeric isocitrate dehydrogenase: Evidence of a protein monomerization by a domain duplication. Structure 2002, 10, 1637–1648. [Google Scholar] [CrossRef] [PubMed]
- Sidhu, N.S.; Delbaere, L.T.; Sheldrick, G.M. Structure of a highly NADP+-specific isocitrate dehydrogenase. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 856–869. [Google Scholar] [CrossRef]
- Zhang, B.; Wang, B.; Wang, P.; Cao, Z.; Huang, E.; Hao, J.; Dean, A.M.; Zhu, G. Enzymatic characterization of a monomeric isocitrate dehydrogenase from Streptomyces lividans TK54. Biochimie 2009, 91, 1405–1410. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Nandyala, A.; Podili, R.; Katoch, V.M.; Hasnain, S.E. Comparison of Mycobacterium tuberculosis isocitrate dehydrogenases (ICD-1 and ICD-2) reveals differences in coenzyme affinity, oligomeric state, pH tolerance and phylogenetic affiliation. BMC Biochem. 2005, 6, 20. [Google Scholar] [CrossRef]
- Crousilles, A.; Dolan, S.K.; Brear, P.; Chirgadze, D.Y.; Welch, M. Gluconeogenic precursor availability regulates flux through the glyoxylate shunt in Pseudomonas aeruginosa. J. Biol. Chem. 2018, 293, 14260–14269. [Google Scholar] [CrossRef]
- Zheng, J.; Jia, Z. Structure of the bifunctional isocitrate dehydrogenase kinase/phosphatase. Nature 2010, 465, 961–965. [Google Scholar] [CrossRef]
- Song, P.; Wang, M.L.; Zheng, Q.Y.; Wang, P.; Zhu, G.P. Isocitrate dehydrogenase 1 from Acinetobacter baummanii (AbIDH1) enzymatic characterization and its regulation by phosphorylation. Biochimie 2020, 181, 77–85. [Google Scholar] [CrossRef]
- Wang, P.; Jin, M.; Su, R.; Song, P.; Wang, M.; Zhu, G. Enzymatic characterization of isocitrate dehydrogenase from an emerging zoonotic pathogen Streptococcus suis. Biochimie 2011, 93, 1470–1475. [Google Scholar] [CrossRef]
- Wang, P.; Chen, X.; Yang, J.; Pei, Y.; Bian, M.; Zhu, G. Characterization of the nicotinamide adenine dinucleotides (NAD(+) and NADP(+)) binding sites of the monomeric isocitrate dehydrogenases from Campylobacter species. Biochimie 2019, 160, 148–155. [Google Scholar] [CrossRef]
- Hurley, J.H.; Thorsness, P.E.; Ramalingam, V.; Helmers, N.H.; Koshland, D.E., Jr.; Stroud, R.M. Structure of a bacterial enzyme regulated by phosphorylation, isocitrate dehydrogenase. Proc. Natl. Acad. Sci. USA 1989, 86, 8635–8639. [Google Scholar] [CrossRef]
- Vasquez, B.; Reeves, H.C. NADP-specific isocitrate dehydrogenase of Escherichia coli. IV. Purification by chromatography on Affi-Gel Blue. Biochim. Biophys. Acta 1979, 578, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Eikmanns, B.J.; Rittmann, D.; Sahm, H. Cloning; sequence analysis; expression, and inactivation of the Corynebacterium glutamicum icd gene encoding isocitrate dehydrogenase and biochemical characterization of the enzyme. J. Bacteriol. 1995, 177, 774–782. [Google Scholar] [CrossRef] [PubMed]
- Sahara, T.; Takada, Y.; Takeuchi, Y.; Yamaoka, N.; Fukunaga, N. Cloning sequencing, and expression of a gene encoding the monomeric isocitrate dehydrogenase of the nitrogen-fixing bacterium, Azotobacter vinelandii. Biosci. Biotechnol. Biochem. 2002, 66, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Cao, Z.Y.; Wang, P.; Liu, A.M.; Pan, W.; Wang, J.; Zhu, G.P. Heteroexpression and characterization of a monomeric isocitrate dehydrogenase from the multicellular prokaryote Streptomyces avermitilis MA-4680. Mol. Biol. Rep. 2010, 38, 3717–3724. [Google Scholar] [CrossRef] [PubMed]
- Lv, C.; Wang, P.; Wang, W.; Su, R.; Ge, Y.; Zhu, Y.; Zhu, G. Two isocitrate dehydrogenases from a plant pathogen Xanthomonas campestris pv. campestris 8004. Bioinformatic analysis, enzymatic characterization, and implication in virulence. J. Basic Microbiol. 2016, 56, 975–985. [Google Scholar] [CrossRef] [PubMed]
- Cozzone, A.J.; El-Mansi, M. Control of isocitrate dehydrogenase catalytic activity by protein phosphorylation in Escherichia coli. J. Mol. Microbiol. Biotechnol. 2005, 9, 132–146. [Google Scholar] [CrossRef]
- El-Mansi, M.; Cozzone, A.J.; Shiloach, J.; Eikmanns, B.J. Control of carbon flux through enzymes of central and intermediary metabolism during growth of Escherichia coli on acetate. Curr. Opin. Microbiol. 2006, 9, 173–179. [Google Scholar] [CrossRef]
- Murima, P.; Zimmermann, M.; Chopra, T.; Pojer, F.; Fonti, G.; Peraro, M.D.; Alonso, S.; Sauer, U.; Pethe, K.; McKinney, J.D. A rheostat mechanism governs the bifurcation of carbon flux in mycobacteria. Nat. Commun. 2016, 7, 12527. [Google Scholar] [CrossRef]
- Banerjee, S.; Nandyala, A.; Podili, R.; Katoch, V.M.; Murthy, K.J.; Hasnain, S.E. Mycobacterium tuberculosis (Mtb) isocitrate dehydrogenases show strong B cell response and distinguish vaccinated controls from TB patients. Proc. Natl. Acad. Sci. USA 2004, 101, 12652–12657. [Google Scholar] [CrossRef]
- Ochiai, T.; Fukunaga, N.; Sasaki, S. Purification and some properties of two NADP+-specific isocitrate dehydrogenases from an obligately psychrophilic marine bacterium, Vibrio sp. strain ABE-1. J. Biochem. 1979, 86, 377–384. [Google Scholar] [CrossRef]
- Dean, A.M.; Golding, G.B. Protein engineering reveals ancient adaptive replacements in isocitrate dehydrogenase. Proc. Natl. Acad. Sci. USA 1997, 94, 3104–3109. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolite | Relative Activity (%) | |
|---|---|---|
| PaIDH1 | PaIDH2 | |
| Control (none) | 100 ± 0.00 | 100 ± 0.00 |
| ATP | 89.16 ± 5.17 | 75.91 ± 5.92 |
| ADP | 81.79 ± 3.55 | 82.66 ± 3.37 |
| AMP | 87.30 ± 1.95 | 91.17 ± 6.95 |
| ATP + ADP | 79.04 ± 7.50 | 52.48 ± 4.76 |
| AMP + ADP + AMP | 68.07 ± 6.59 | 59.61 ± 5.56 |
| ATP + AMP | 83.89 ± 3.81 | 51.02 ± 5.04 |
| ADP + AMP | 81.15 ± 5.73 | 80.68 ± 7.31 |
| GTP | 85.13 ± 1.48 | 93.99 ± 1.3 |
| GDP | 86.69 ± 1.16 | 88.43 ± 2.80 |
| GMP | 83.32 ± 2.58 | 93.26 ± 3.09 |
| GTP + GDP | 85.97 ± 1.38 | 90.88 ± 2.47 |
| GTP + GMP | 85.74 ± 2.14 | 89.83 ± 4.12 |
| GDP + GMP | 86.79 ± 1.66 | 87.63 ± 3.59 |
| GTP + GDP + GMP | 84.43 ± 1.59 | 79.25 ± 4.96 |
| CIA | 86.97 ± 4.95 | 94.81 ± 4.82 |
| GLA | 93.05 ± 3.88 | 87.06 ± 3.73 |
| α-KG | 79.52 ± 4.64 | 82.79 ± 5.65 |
| OAA | 77.45 ± 4.56 | 88.32 ± 7.97 |
| α-KG + OAA | 71.54 ± 4.76 | 70.7 ± 2.97 |
| GLA + OAA | 75.71 ± 6.02 | 79.88 ± 7.08 |
| α-KG + OAA + GLA | 68.17 ± 4.42 | 16.23 ± 0.56 |
| α-KG + OAA + GLA + CIA | 68.53 ± 0.86 | 9.926 ± 1.22 |
| Enzymes | ICT | NADP+ | References | ||||
|---|---|---|---|---|---|---|---|
| Km (μM) | kcat (s−1) | kcat/Km (μM−1s−1) | Km (μM) | kcat (s−1) | kcat/Km (μM−1s−1) | ||
| PaIDH1 | 14.41 ± 1.82 | 29.26 ± 10.92 | 2.10 ± 0.93 | 20.00 ± 6.29 | 104.51 ± 19.91 | 5.42 ± 0.97 | This study |
| PaIDH2 | 23.61 ± 1.21 | 171.34 ± 6.77 | 7.26 ± 0.13 | 30.68 ± 2.73 | 133.98 ± 17.04 | 4.42 ± 1.13 | This study |
| EcIDH | 24.2 ± 6.6 | 51.8 ± 4.4 | 2.1 | 17 | 80.5 | 4.7 | [8] |
| MtIDH1 | 10 ± 5 | 3.8 | 0.38 | 125 ± 5 | 4 | 0.032 | [34] |
| MtIDH2 | 20 ± 1 | 37.13 | 1.857 | 19.6 ± 6 | 37.4 | 1.908 | [34] |
| AbIDH1 | 50.5 ± 3.2 | 50.4 | 1.0 | 46.6 ± 6.1 | 60.1 ± 3.3 | 1.3 | [37] |
| AbIDH2 | 21 ± 3 | 39.2 ± 2.1 | 1.9 | 94 ± 6 | 36.9 ± 1.2 | 0.39 | [37] |
| Enzymes | NADP+ | NAD+ | ||||
|---|---|---|---|---|---|---|
| Km (μM) | kcat (s−1) | kcat/Km (A) (μM−1s−1) | Km (μM) | kcat (s−1) | kcat/Km (B) (μM−1s−1) | |
| PaIDH1 PaIDH1-D346 PaIDH1-D346I347 PaIDH1-D346I347A353 PaIDH1-D346I347A353K393 PaIDH1-K393 | 20.00 ± 6.29 1715.67 ± 133.5 - - - 74.36 ± 5.28 | 104.51 ± 19.91 39.56 ± 2.43 - - - 41.32 ± 1.95 | 5.42 ± 0.97 0.02 ± 0.003 - - - 0.56 ± 0.04 | - 15,368.79 ± 2818 - 4500 ± 157.36 925.1 ± 83.81 - | - 0.003 ± 0.0003 - 4.73 ± 0.76 7.04 ± 0.71 - | - 0.02 × 10−6 - 0.001 ± 0.0002 0.008 ± 0.001 - |
| Enzymes | NADP+ | NAD+ | ||||
|---|---|---|---|---|---|---|
| Km (μM) | kcat (s−1) | kcat/Km (A) (μM−1s−1) | Km (μM) | kcat (s−1) | kcat/Km (B) (μM−1s−1) | |
| PaIDH2 | 30.68 ± 2.73 | 13 3.98 ± 17.04 | 4.42 ± 1.13 | - | - | - |
| PaIDH2-L589 | 135.33 ± 14.4 | 52.36 ± 9.08 | 0.39 ± 0.1 | 5927.3 ± 205.25 | 10.89 ± 0.15 | 0.002 ± 0.00008 |
| PaIDH2-L589 D600 | 849.33 ± 71.92 | 1.7 ± 0.35 | 0.002 ± 0.0004 | 8949.33 ± 748.06 | 4.5 ± 0.41 | 0.0005 ± 0.000003 |
| PaIDH2-L589 D600 S649 | - | - | - | 12242.33 ± 497.22 | 6.63 ± 0.12 | 0.0005 ± 0.00001 |
| PaIDH2-L589 I600 | 7770.67 ± 270.55 | 16.03 ± 0.79 | 0.002 ± 0.00005 | 5824.33 ± 346.01 | 20.74 ± 0.43 | 0.004 ± 0.0002 |
| PaIDH2-L589 I600 L649 | - | - | - | 15625.33 ± 1751 | 28.92 ± 2.73 | 0.002 ± 0.0001 |
| PaIDH2-L589 I600 D649 | - | - | - | 9968.67 ± 1051.5 | 48.19 ± 4.15 | 0.005 ± 0.0002 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.; Wei, W.; Xiong, W.; Wu, S.; Wu, Q.; Wang, P.; Zhu, G. Two Different Isocitrate Dehydrogenases from Pseudomonas aeruginosa: Enzymology and Coenzyme-Evolutionary Implications. Int. J. Mol. Sci. 2023, 24, 14985. https://doi.org/10.3390/ijms241914985
Chen X, Wei W, Xiong W, Wu S, Wu Q, Wang P, Zhu G. Two Different Isocitrate Dehydrogenases from Pseudomonas aeruginosa: Enzymology and Coenzyme-Evolutionary Implications. International Journal of Molecular Sciences. 2023; 24(19):14985. https://doi.org/10.3390/ijms241914985
Chicago/Turabian StyleChen, Xuefei, Wei Wei, Wei Xiong, Shen Wu, Quanchao Wu, Peng Wang, and Guoping Zhu. 2023. "Two Different Isocitrate Dehydrogenases from Pseudomonas aeruginosa: Enzymology and Coenzyme-Evolutionary Implications" International Journal of Molecular Sciences 24, no. 19: 14985. https://doi.org/10.3390/ijms241914985
APA StyleChen, X., Wei, W., Xiong, W., Wu, S., Wu, Q., Wang, P., & Zhu, G. (2023). Two Different Isocitrate Dehydrogenases from Pseudomonas aeruginosa: Enzymology and Coenzyme-Evolutionary Implications. International Journal of Molecular Sciences, 24(19), 14985. https://doi.org/10.3390/ijms241914985