
The ABA/LANCL Hormone/Receptor System in the Control of Glycemia, of Cardiomyocyte Energy Metabolism, and in Neuroprotection: A New Ally in the Treatment of Diabetes Mellitus?



Abstract
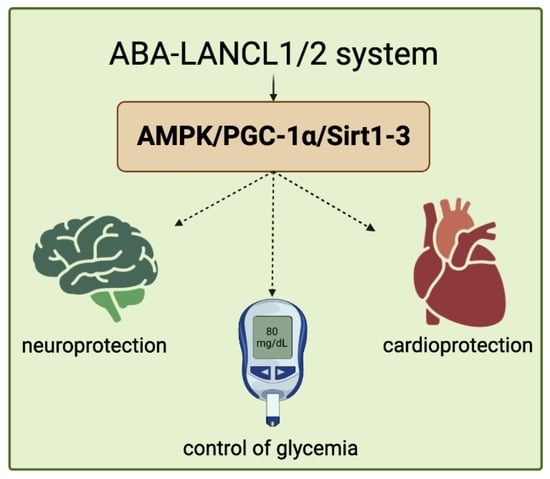
:
{kind=link}
{kind=link}
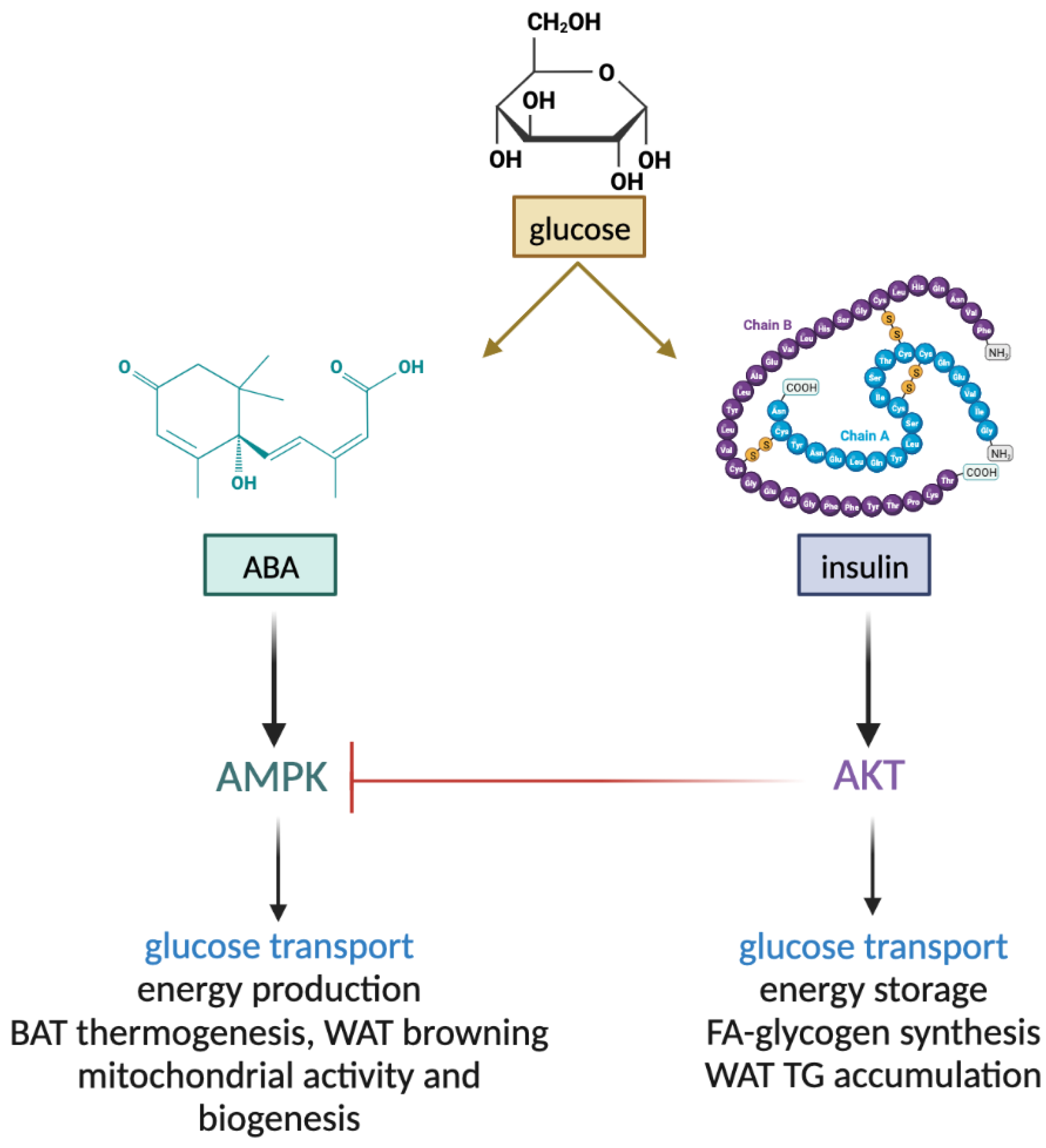{kind=link}
{kind=link}
1. The ABA/LANCL1/2 Hormone/Receptor System
1.1. Abscisic Acid, an Ancient Stress Signal
1.2. Mammalian LANCL Proteins
1.2.1. The Membrane-Bound LANCL2 Receptor
1.2.2. LANCL1 Is Also an ABA Receptor
1.2.3. AMPK Activation Downstream of LANCL1/2
1.2.4. ABA Signaling in Skeletal Muscle
1.2.5. The ABA/LANCL System in the Adipose Tissue
1.2.6. Mitochondrial Effects of the ABA/LANCL System
1.2.7. Non-Overlapping Roles of ABA and Insulin in Energy Metabolism
2. The ABA/LANCL System in Diabetes
2.1. Plasma ABA in Diabetic Subjects
2.2. Clinical Studies on Borderline and Prediabetic Subjects
2.3. Oral ABA Ameliorates Glycemia in Insulin-Deficient Mice
3. The ABA/LANCL System Protects Cardiomyocytes from Hypoxia
3.1. The ABA/LANCL System Regulates NO Production in Rodent Cardiomyocytes
3.1.1. ABA Is Released by H9c2 under Hypoxia and Stimulates NO Production
3.1.2. ABA Stimulates eNOS Transcription, Expression, and Phosphorylation
3.1.3. The ABA-Induced Increase in NO Improves Survival of H9c2 under Hypoxia
3.1.4. Transcriptional Control of LANCL1/2 on eNOS Transcription, Expression, and Function under Normoxia and Hypoxia
3.1.5. Signaling Downstream of LANCL1/2 Involves the AMPK/PGC-1α/Sirt1 Axis
4. Neuroprotective Effects of the ABA/LANCL System
4.1. LANCL1 Protects Neurons from Oxidative Stress In Vivo
4.2. LANCL1 Protects Neurons against Death from Oxygen and Glucose Deprivation
4.3. ABA Improves Cognitive Impairment in Animal Models of Alzheimer’s Disease (AD)
4.4. Transcriptional Activity of LANCL1/2
5. Conclusions and Future Perspectives
5.1. Conclusions
5.2. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bruzzone, S.; Moreschi, I.; Usai, C.; Guida, L.; Damonte, G.; Salis, A.; Scarfì, S.; Millo, E.; De Flora, A.; Zocchi, E. Abscisic acid is an endogenous cytokine in human granulocytes with cyclic ADP-ribose as second messenger. Proc. Natl. Acad. Sci. USA 2007, 104, 5759–5764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desikan, R.; Cheung, M.K.; Bright, J.; Henson, D.; Hancock, J.T.; Neill, S.J. ABA, hydrogen peroxide and nitric oxide signalling in stomatal guard cells. J. Exp. Bot. 2004, 55, 205–212. [Google Scholar] [CrossRef] [Green Version]
- Bruzzone, S.; Basile, G.; Mannino, E.; Sturla, L.; Magnone, M.; Grozio, A.; Salis, A.; Fresia, C.; Vigliarolo, T.; Guida, L.; et al. Autocrine Abscisic Acid Mediates the UV-B-Induced Inflammatory Response in Human Granulocytes and Keratinocytes. J. Cell Physiol. 2012, 227, 2502–2510. [Google Scholar] [CrossRef] [PubMed]
- Tossi, V.; Cassia, R.; Bruzzone, S.; Zocchi, E.; Lamattina, L. ABA Says NO to UV-B: A Universal Response? Trends Plant Sci. 2012, 17, 510–517. [Google Scholar] [CrossRef]
- Sun, Y.; Pri-Tal, O.; Michaeli, D.; Mosquna, A. Evolution of Abscisic Acid Signaling Module and Its Perception. Front. Plant Sci. 2020, 11, 934. [Google Scholar] [CrossRef] [PubMed]
- Bauer, H.; Mayer, H.; Marchler-Bauer, A.; Salzer, U.; Prohaska, R. Characterization of P40/GPR69A as a Peripheral Membrane Protein Related to the Lantibiotic Synthetase Component C. Biochem. Biophys. Res. Commun. 2000, 275, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Landlinger, C.; Salzer, U.; Prohaska, R. Myristoylation of Human LanC-like Protein 2 (LANCL2) Is Essential for the Interaction with the Plasma Membrane and the Increase in Cellular Sensitivity to Adriamycin. Biochim. Biophys. Acta Biomembr. 2006, 1758, 1759–1767. [Google Scholar] [CrossRef] [Green Version]
- He, C.; Zeng, M.; Dutta, D.; Koh, T.H.; Chen, J.; van der Donk, W.A. LanCL Proteins Are Not Involved in Lanthionine Synthesis in Mammals. Sci. Rep. 2017, 7, 40980. [Google Scholar] [CrossRef] [Green Version]
- Lai, K.-Y.; Galan, S.R.G.; Zeng, Y.; Zhou, T.H.; He, C.; Raj, R.; Riedl, J.; Liu, S.; Chooi, K.P.; Garg, N.; et al. LanCLs Add Glutathione to Dehydroamino Acids Generated at Phosphorylated Sites in the Proteome. Cell 2021, 184, 2680–2695.e26. [Google Scholar] [CrossRef]
- Liu, X.; Yue, Y.; Li, B.; Nie, Y.; Li, W.; Wu, W.H.; Ma, L. A G Protein-Coupled Receptor Is a Plasma Membrane Receptor for the Plant Hormone Abscisic Acid. Science 2007, 315, 1712–1716. [Google Scholar] [CrossRef]
- Pandey, S.; Nelson, D.C.; Assmann, S.M. Two Novel GPCR-Type G Proteins Are Abscisic Acid Receptors in Arabidopsis. Cell 2009, 136, 136–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kharenko, O.A.; Choudhary, P.; Loewen, M.C. Abscisic Acid Binds to Recombinant Arabidopsis Thaliana G-Protein Coupled Receptor-Type G-Protein 1 in Sacaromycese Cerevisiae and in Vitro. Plant Physiol. Biochem. 2013, 68, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Cichero, E.; Fresia, C.; Guida, L.; Booz, V.; Millo, E.; Scotti, C.; Iamele, L.; de Jonge, H.; Galante, D.; De Flora, A.; et al. Identification of a High Affinity Binding Site for Abscisic Acid on Human Lanthionine Synthetase Component C-like Protein 2. Int. J. Biochem. Cell Biol. 2018, 97, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Sturla, L.; Fresia, C.; Guida, L.; Bruzzone, S.; Scarfì, S.; Usai, C.; Fruscione, F.; Magnone, M.; Millo, E.; Basile, G.; et al. LANCL2 Is Necessary for Abscisic Acid Binding and Signaling in Human Granulocytes and in Rat Insulinoma Cells. J. Biol. Chem. 2009, 284, 28045–28057. [Google Scholar] [CrossRef] [Green Version]
- Fresia, C.; Vigliarolo, T.; Guida, L.; Booz, V.; Bruzzone, S.; Sturla, L.; Di Bona, M.; Pesce, M.; Usai, C.; De Flora, A.; et al. G-Protein Coupling and Nuclear Translocation of the Human Abscisic Acid Receptor LANCL2. Sci. Rep. 2016, 6, 26658. [Google Scholar] [CrossRef] [Green Version]
- Vigliarolo, T.; Zocchi, E.; Fresia, C.; Booz, V.; Guida, L. Abscisic Acid Influx into Human Nucleated Cells Occurs through the Anion Exchanger AE2. Int. J. Biochem. Cell Biol. 2016, 75, 99–103. [Google Scholar] [CrossRef]
- Magnone, M.; Ameri, P.; Salis, A.; Andraghetti, G.; Emionite, L.; Murialdo, G.; De Flora, A.; Zocchi, E. Microgram Amounts of Abscisic Acid in Fruit Extracts Improve Glucose Tolerance and Reduce Insulinemia in Rats and in Humans. FASEB J. 2015, 29, 4783–4793. [Google Scholar] [CrossRef] [Green Version]
- Sturla, L.; Mannino, E.; Scarfì, S.; Bruzzone, S.; Magnone, M.; Sociali, G.; Booz, V.; Guida, L.; Vigliarolo, T.; Fresia, C.; et al. Abscisic Acid Enhances Glucose Disposal and Induces Brown Fat Activity in Adipocytes in Vitro and in Vivo. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 131–144. [Google Scholar] [CrossRef]
- Spinelli, S.; Begani, G.; Guida, L.; Magnone, M.; Galante, D.; D’Arrigo, C.; Scotti, C.; Iamele, L.; De Jonge, H.; Zocchi, E.; et al. LANCL1 Binds Abscisic Acid and Stimulates Glucose Transport and Mitochondrial Respiration in Muscle Cells via the AMPK/PGC-1α/Sirt1 Pathway. Mol. Metab. 2021, 53, 101263. [Google Scholar] [CrossRef]
- Magnone, M.; Emionite, L.; Guida, L.; Vigliarolo, T.; Sturla, L.; Spinelli, S.; Buschiazzo, A.; Marini, C.; Sambuceti, G.; De Flora, A.; et al. Insulin-Independent Stimulation of Skeletal Muscle Glucose Uptake by Low-Dose Abscisic Acid via AMPK Activation. Sci. Rep. 2020, 10, 1454. [Google Scholar] [CrossRef]
- Horman, S.; Vertommen, D.; Heath, R.; Neumann, D.; Mouton, V.; Woods, A.; Schlattner, U.; Wallimann, T.; Carling, D.; Hue, L.; et al. Insulin Antagonizes Ischemia-Induced Thr172 Phosphorylation of AMP-Activated Protein Kinase α-Subunits in Heart via Hierarchical Phosphorylation of Ser485/491. J. Biol. Chem. 2006, 281, 5335–5340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valentine, R.J.; Coughlan, K.A.; Ruderman, N.B.; Saha, A.K. Insulin Inhibits AMPK Activity and Phosphorylates AMPK Ser485/491 through Akt in Hepatocytes, Myotubes and Incubated Rat Skeletal Muscle. Arch. Biochem. Biophys. 2014, 562, 62–69. [Google Scholar] [CrossRef] [Green Version]
- Guri, A.J.; Hontecillas, R.; Si, H.; Liu, D.; Bassaganya-Riera, J. Dietary Abscisic Acid Ameliorates Glucose Tolerance and Obesity-Related Inflammation in Db/Db Mice Fed High-Fat Diets. Clin. Nutr. 2007, 26, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Derosa, G.; Maffioli, P.; D’Angelo, A.; Preti, P.S.; Tenore, G.; Novellino, E. Abscisic Acid Treatment in Patients with Prediabetes. Nutrients 2020, 12, 2931. [Google Scholar] [CrossRef] [PubMed]
- Pan, R.; Zhu, X.; Maretich, P.; Chen, Y. Metabolic Improvement via Enhancing Thermogenic Fat-Mediated Non-Shivering Thermogenesis: From Rodents to Humans. Front. Endocrinol. 2020, 11, 633. [Google Scholar] [CrossRef] [PubMed]
- Ameri, P.; Bruzzone, S.; Mannino, E.; Sociali, G.; Andraghetti, G.; Salis, A.; Ponta, M.L.; Briatore, L.; Adami, G.F.; Ferraiolo, A.; et al. Impaired Increase of Plasma Abscisic Acid in Response to Oral Glucose Load in Type 2 Diabetes and in Gestational Diabetes. PLoS ONE 2015, 10, e0115992. [Google Scholar] [CrossRef] [Green Version]
- Hunter, K.; Rainbow, D.; Plagnol, V.; Todd, J.A.; Peterson, L.B.; Wicker, L.S. Interactions between Idd5.1/Ctla4 and Other Type 1 Diabetes Genes. J. Immunol. 2007, 179, 8341–8349. [Google Scholar] [CrossRef] [Green Version]
- Westphal, D.S.; Andres, S.; Makowski, C.; Meitinger, T.; Hoefele, J. MAP2—A Candidate Gene for Epilepsy, Developmental Delay and Behavioral Abnormalities in a Patient with Microdeletion 2q34. Front. Genet. 2018, 9, 99. [Google Scholar] [CrossRef]
- Magnone, M.; Leoncini, G.; Vigliarolo, T.; Emionite, L.; Sturla, L.; Zocchi, E.; Murialdo, G. Chronic Intake of Micrograms of Abscisic Acid Improves Glycemia and Lipidemia in a Human Study and in High-Glucose Fed Mice. Nutrients 2018, 10, 1495. [Google Scholar] [CrossRef] [Green Version]
- Leber, A.; Hontecillas, R.; Tubau-Juni, N.; Zoccoli-Rodriguez, V.; Goodpaster, B.; Bassaganya-Riera, J. Abscisic Acid Enriched Fig Extract Promotes Insulin Sensitivity by Decreasing Systemic Inflammation and Activating LANCL2 in Skeletal Muscle. Sci. Rep. 2020, 10, 10463. [Google Scholar] [CrossRef]
- Uchida, K.; Dezaki, K.; Damdindorj, B.; Inada, H.; Shiuchi, T.; Mori, Y.; Yada, T.; Minokoshi, Y.; Tominaga, M. Lack of TRPM2 Impaired Insulin Secretion and Glucose Metabolisms in Mice. Diabetes 2011, 60, 119–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magnone, M.; Spinelli, S.; Begani, G.; Guida, L.; Sturla, L.; Emionite, L.; Zocchi, E. Abscisic Acid Improves Insulin Action on Glycemia in Insulin-Deficient Mouse Models of Type 1 Diabetes. Metabolites 2022, 12, 523. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.; Zhang, W.; Zhao, Z.; Ma, C.; Zhang, T.; Meng, Q.; Yan, P.; Zhang, L.; Zhao, Y. Regulation of Mitochondrial Quality Control by Natural Drugs in the Treatment of Cardiovascular Diseases: Potential and Advantages. Front. Cell Dev. Biol. 2020, 8, 616139. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, S.; Guida, L.; Vigliarolo, T.; Passalacqua, M.; Begani, G.; Magnone, M.; Sturla, L.; Benzi, A.; Ameri, P.; Lazzarini, E.; et al. The ABA-LANCL1/2 Hormone-Receptors System Protects H9c2 Cardiomyocytes from Hypoxia-Induced Mitochondrial Injury via an AMPK- and NO-Mediated Mechanism. Cells 2022, 11, 2888. [Google Scholar] [CrossRef]
- Carlström, M. Nitric Oxide Signalling in Kidney Regulation and Cardiometabolic Health. Nat. Rev. Nephrol. 2021, 17, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Hong, F.F.; Liang, X.Y.; Liu, W.; Lv, S.; He, S.J.; Kuang, H.B.; Yang, S.L. Roles of eNOS in atherosclerosis treatment. Inflamm. Res. 2019, 68, 429–441. [Google Scholar] [CrossRef]
- Pisarenko, O.; Studneva, I. Modulating the bioactivity of nitric oxide as a therapeutic strategy in cardiac surgery. J. Surg. Res. 2021, 257, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Eid, R.A.; Bin-Meferij, M.M.; El-Kott, A.F.; Eleawa, S.M.; Zaki, M.S.A.; Al-Shraim, M.; El-Sayed, F.; Eldeen, M.A.; Alkhateeb, M.A.; Alharbi, S.A.; et al. Exendin-4 protects against myocardial ischemia-reperfusion injury by upregulation of SIRT1 and SIRT3 and activation of AMPK. J. Cardiovasc. Transl. Res. 2020, 14, 619–635. [Google Scholar] [CrossRef]
- Hsu, C.P.; Oka, S.; Shao, D.; Hariharan, N.; Sadoshima, J. Nicotinamide phosphoribosyltransferase regulates cell survival through NAD+ synthesis in cardiac myocytes. Circ. Res. 2009, 105, 481–491. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.M.; Tsai, J.Y.; Chen, Y.C.; Huang, C.Y.; Hsu, H.L.; Weng, C.F.; Shih, C.C.; Hsu, C.P. Downregulation of Sirt1 as aging change in advanced heart failure. J. Biomed. Sci. 2014, 21, 57. [Google Scholar] [CrossRef]
- Zhang, J.; He, Z.; Fedorova, J.; Logan, C.; Bates, L.; Davitt, K.; Le, V.; Murphy, J.; Li, M.; Wang, M.; et al. Alterations in mitochondrial dynamics with age-related Sirtuin1/Sirtuin3 deficiency impair cardiomyocyte contractility. Aging Cell 2021, 20, 13419. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Zhou, L.; Wu, X.; Liu, C.; Fan, Y.; Li, Q. Hypoxic preconditioning protects cardiomyocytes against hypoxia/reoxygenation injury through AMPK/eNOS/PGC-1α signaling pathway. Int. J. Clin. Exp. Pathol. 2014, 7, 7378–7388. [Google Scholar] [PubMed]
- Gan, L.; Xie, D.; Liu, J.; Bond Lau, W.; Christopher, T.A.; Lopez, B.; Zhang, L.; Gao, E.; Koch, W.; Ma, X.L.; et al. Small Extracellular microvesicles mediated pathological communications between dysfunctional adipocytes and cardiomyocytes as a novel mechanism exacerbating ischemia/reperfusion injury in diabetic mice. Circulation 2020, 141, 968–983. [Google Scholar] [CrossRef]
- Luo, G.; Jian, Z.; Zhu, Y.; Zhu, Y.; Chen, B.; Ma, R.; Tang, F.; Xiao, Y. Sirt1 promotes autophagy and inhibits apoptosis to protect cardiomyocytes from hypoxic stress. Int. J. Mol. Med. 2019, 43, 2033–2043. [Google Scholar] [CrossRef] [Green Version]
- Tian, L.; Cao, W.; Yue, R.; Yuan, Y.; Guo, X.; Qin, D.; Xing, J.; Wang, X. Pretreatment with Tilianin improves mitochondrial energy metabolism and oxidative stress in rats with myocardial ischemia/reperfusion injury via AMPK/SIRT1/PGC-1 alpha signaling pathway. J. Pharmacol. Sci. 2019, 139, 352–360. [Google Scholar] [CrossRef]
- Fang, Y.; Wang, X.; Yang, D.; Lu, Y.; Wei, G.; Yu, W.; Liu, X.; Zheng, Q.; Ying, J.; Hua, F. Relieving cellular energy stress in aging, neurodegenerative, and metabolic diseases, SIRT1 as a therapeutic and promising node. Front. Aging Neurosci. 2021, 13, 738686. [Google Scholar] [CrossRef] [PubMed]
- Biessels, G.J.; Whitmer, R.A. Cognitive dysfunction in diabetes: How to implement emerging guidelines. Diabetologia 2020, 63, 3–9. [Google Scholar] [CrossRef] [Green Version]
- van Praag, H.; Christie, B.R.; Sejnowski, T.J.; Gage, F.H. Running enhances neurogenesis, learning, and long-term potentiation in mice. Proc. Natl. Acad. Sci. USA 1999, 96, 13427–13431. [Google Scholar] [CrossRef] [Green Version]
- Mahalakshmi, B.; Maurya, N.; Lee, S.D.; Bharath Kumar, V. Possible Neuroprotective Mechanisms of Physical Exercise in Neurodegeneration. Int. J. Mol. Sci. 2020, 21, 5895. [Google Scholar] [CrossRef]
- Koepsell, H. Glucose transporters in brain in health and disease. Pflug. Arch. 2020, 472, 1299–1343. [Google Scholar] [CrossRef]
- Ruze, R.; Xu, Q.; Liu, G.; Li, Y.; Chen, W.; Cheng, Z.; Xiong, Y.; Liu, S.; Zhang, G.; Hu, S.; et al. Central GLP-1 contributes to improved cognitive function and brain glucose uptake after duodenum-jejunum bypass on obese and diabetic rats. Am. J. Physiol. Endocrinol. Metab. 2021, 321, E392–E409. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, L.; Liu, Y.; Xu, J.; Zhu, G.; Cang, H.; Li, X.; Bartlam, M.; Hensley, K.; Li, G.; et al. Structure of human lanthionine synthetase C-like protein 1 and its interaction with Eps8 and glutathione. Genes Dev. 2009, 23, 1387–1392. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Chen, M.; Pang, D.; Bi, D.; Zou, Y.; Xia, X.; Yang, W.; Luo, L.; Deng, R.; Tan, H.; et al. Developmental and activity-dependent expression of LanCL1 confers antioxidant activity required for neuronal survival. Dev. Cell 2014, 30, 479–487. [Google Scholar] [CrossRef] [Green Version]
- Tan, H.; Chen, M.; Pang, D.; Xia, X.; Du, C.; Yang, W.; Cui, Y.; Huang, C.; Jiang, W.; Bi, D.; et al. LanCL1 promotes motor neuron survival and extends the lifespan of amyotrophic lateral sclerosis mice. Cell Death Differ. 2020, 27, 1369–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.; Cao, B.Q.; Wang, T.; Lei, Q.; Kang, T.; Ge, C.Y.; Gao, W.J.; Hui, H. LanCL1 attenuates ischemia-induced oxidative stress by Sirt3-mediated preservation of mitochondrial function. Brain Res. Bull. 2018, 142, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Perez, A.M. Abscisic acid, a promising therapeutic molecule to prevent Alzheimer’s and neurodegenerative diseases. Neural. Regen. Res. 2020, 15, 1035–1036. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.H.; Kim, N.; Ju, Y.J.; Gee, M.S.; Lee, D.; Lee, J.K. Phytohormone Abscisic Acid Improves Memory Impairment and Reduces Neuroinflammation in 5xFAD Mice by Upregulation of LanC-Like Protein 2. Int. J. Mol. Sci. 2020, 21, 8425. [Google Scholar] [CrossRef]
- Chen, Y.; Xie, X.; Wu, A. A synthetic cell-penetrating peptide derived from nuclear localization signal of EPS8 exerts anticancer activity against acute myeloid leukemia. J. Exp. Clin. Cancer Res. 2018, 37, 12. [Google Scholar] [CrossRef] [Green Version]
- Daliu, P.; Annunziata, G.; Tenore, G.C.; Santini, A. Abscisic acid identification in Okra, Abelmoschus esculentus L. (Moench): Perspective nutraceutical use for the treatment of diabetes. Nat. Prod. Res. 2020, 34, 3–9. [Google Scholar] [CrossRef]
- Neumann, N.R.; Thompson, D.C.; Vasiliou, V. AMPK Activators for the Prevention and Treatment of Neurodegenerative Diseases. Expert. Opin. Drug Metab. Toxicol. 2021, 17, 1199–1210. [Google Scholar] [CrossRef]
- Le Page-Degivry, M.T.; Bidard, J.N.; Rouvier, E.; Bulard, C.; Lazdunski, M. Presence of Abscisic Acid, a Phytohormone, in the Mammalian Brain. Proc. Natl. Acad. Sci. USA 1986, 83, 1155–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khorasani, A.; Abbasnejad, M.; Esmaeili-Mahani, S. Phytohormone Abscisic Acid Ameliorates Cognitive Impairments in Streptozotocin-Induced Rat Model of Alzheimer’s Disease through PPARβ/δ and PKA Signaling. Int. J. Neurosci. 2019, 129, 1053–1065. [Google Scholar] [CrossRef]
- Ribes-Navarro, A.; Atef, M.; Sánchez-Sarasúa, S.; Beltrán-Bretones, M.T.; Olucha-Bordonau, F.; Sánchez-Pérez, A.M. Abscisic Acid Supplementation Rescues High Fat Diet-Induced Alterations in Hippocampal Inflammation and IRSs Expression. Mol. Neurobiol. 2019, 56, 454–464. [Google Scholar] [CrossRef]
- Shabani, M.; Naderi, R. Phytohormone Abscisic Acid Elicits Positive Effects on Harmaline-induced Cognitive and Motor Disturbances in a Rat Model of Essential Tremor. Brain Behav. 2022, 12, e2564. [Google Scholar] [CrossRef] [PubMed]
- Kooshki, R.; Anaeigoudari, A.; Abbasnejad, M.; Askari-Zahabi, K.; Esmaeili-Mahani, S. Abscisic Acid Interplays with PPARγ Receptors and Ameliorates Diabetes-Induced Cognitive Deficits in Rats. Avicenna J. Phytomed. 2021, 11, 247–257. [Google Scholar]
- Naderi, R.; Esmaeili-Mahani, S.; Abbasnejad, M. Extracellular Calcium Influx through L-Type Calcium Channels, Intracellular Calcium Currents and Extracellular Signal-Regulated Kinase Signaling Are Involved in the Abscisic Acid-Induced Precognitive and Anti-Anxiety Effects. Biomed. Pharmacother. 2019, 109, 582–588. [Google Scholar] [CrossRef]
- Espinosa-Fernández, V.; Mañas-Ojeda, A.; Pacheco-Herrero, M.; Castro-Salazar, E.; Ros-Bernal, F.; Sánchez-Pérez, A.M. Early Intervention with ABA Prevents Neuroinflammation and Memory Impairment in a Triple Transgenic Mice Model of Alzheimer’s Disease. Behav. Brain Res. 2019, 374, 112106. [Google Scholar] [CrossRef]
- Xue, M.; Xu, W.; Ou, Y.-N.; Cao, X.-P.; Tan, M.-S.; Tan, L.; Yu, J.-T. Diabetes Mellitus and Risks of Cognitive Impairment and Dementia: A Systematic Review and Meta-Analysis of 144 Prospective Studies. Ageing Res. Rev. 2019, 55, 100944. [Google Scholar] [CrossRef] [PubMed]
- Hanyu, H. Diabetes-Related Dementia. Adv. Exp. Med. Biol. 2019, 1128, 147–160. [Google Scholar] [CrossRef]
- Srikanth, V.; Sinclair, A.J.; Hill-Briggs, F.; Moran, C.; Biessels, G.J. Type 2 Diabetes and Cognitive Dysfunction—Towards Effective Management of Both Comorbidities. Lancet Diabetes Endocrinol. 2020, 8, 535–545. [Google Scholar] [CrossRef]
- Tang, S.W.; Leonard, B.E.; Helmeste, D.M. Long COVID, neuropsychiatric disorders, psychotropics, present and future. Acta Neuropsychiatr. 2022, 34, 109–126. [Google Scholar] [CrossRef] [PubMed]
- Song, W.J.; Hui, C.K.M.; Hull, J.H.; Birring, S.S.; McGarvey, L.; Mazzone, S.B.; Chung, K.F. Confronting COVID-19-associated cough and the post-COVID syndrome: Role of viral neurotropism, neuroinflammation, and neuroimmune responses. Lancet Respir. Med. 2021, 9, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Lyra e Silva, N.M.; Barros-Aragão, F.G.Q.; De Felice, F.G.; Ferreira, S.T. Inflammation at the crossroads of COVID-19, cognitive deficits and depression. Neuropharmacology 2022, 209, 109023. [Google Scholar] [CrossRef] [PubMed]
- Toogood, P.L.; Clauw, D.J.; Phadke, S.; Hoffman, D. Myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS): Where will the drugs come from? Pharmacol. Res. 2021, 165, 105465. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spinelli, S.; Magnone, M.; Guida, L.; Sturla, L.; Zocchi, E. The ABA/LANCL Hormone/Receptor System in the Control of Glycemia, of Cardiomyocyte Energy Metabolism, and in Neuroprotection: A New Ally in the Treatment of Diabetes Mellitus? Int. J. Mol. Sci. 2023, 24, 1199. https://doi.org/10.3390/ijms24021199
Spinelli S, Magnone M, Guida L, Sturla L, Zocchi E. The ABA/LANCL Hormone/Receptor System in the Control of Glycemia, of Cardiomyocyte Energy Metabolism, and in Neuroprotection: A New Ally in the Treatment of Diabetes Mellitus? International Journal of Molecular Sciences. 2023; 24(2):1199. https://doi.org/10.3390/ijms24021199
Chicago/Turabian StyleSpinelli, Sonia, Mirko Magnone, Lucrezia Guida, Laura Sturla, and Elena Zocchi. 2023. "The ABA/LANCL Hormone/Receptor System in the Control of Glycemia, of Cardiomyocyte Energy Metabolism, and in Neuroprotection: A New Ally in the Treatment of Diabetes Mellitus?" International Journal of Molecular Sciences 24, no. 2: 1199. https://doi.org/10.3390/ijms24021199
APA StyleSpinelli, S., Magnone, M., Guida, L., Sturla, L., & Zocchi, E. (2023). The ABA/LANCL Hormone/Receptor System in the Control of Glycemia, of Cardiomyocyte Energy Metabolism, and in Neuroprotection: A New Ally in the Treatment of Diabetes Mellitus? International Journal of Molecular Sciences, 24(2), 1199. https://doi.org/10.3390/ijms24021199