
Toll-like Receptor 4 in Acute Kidney Injury

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. TLRS Structure and Ligands
3. TLR4 Expression
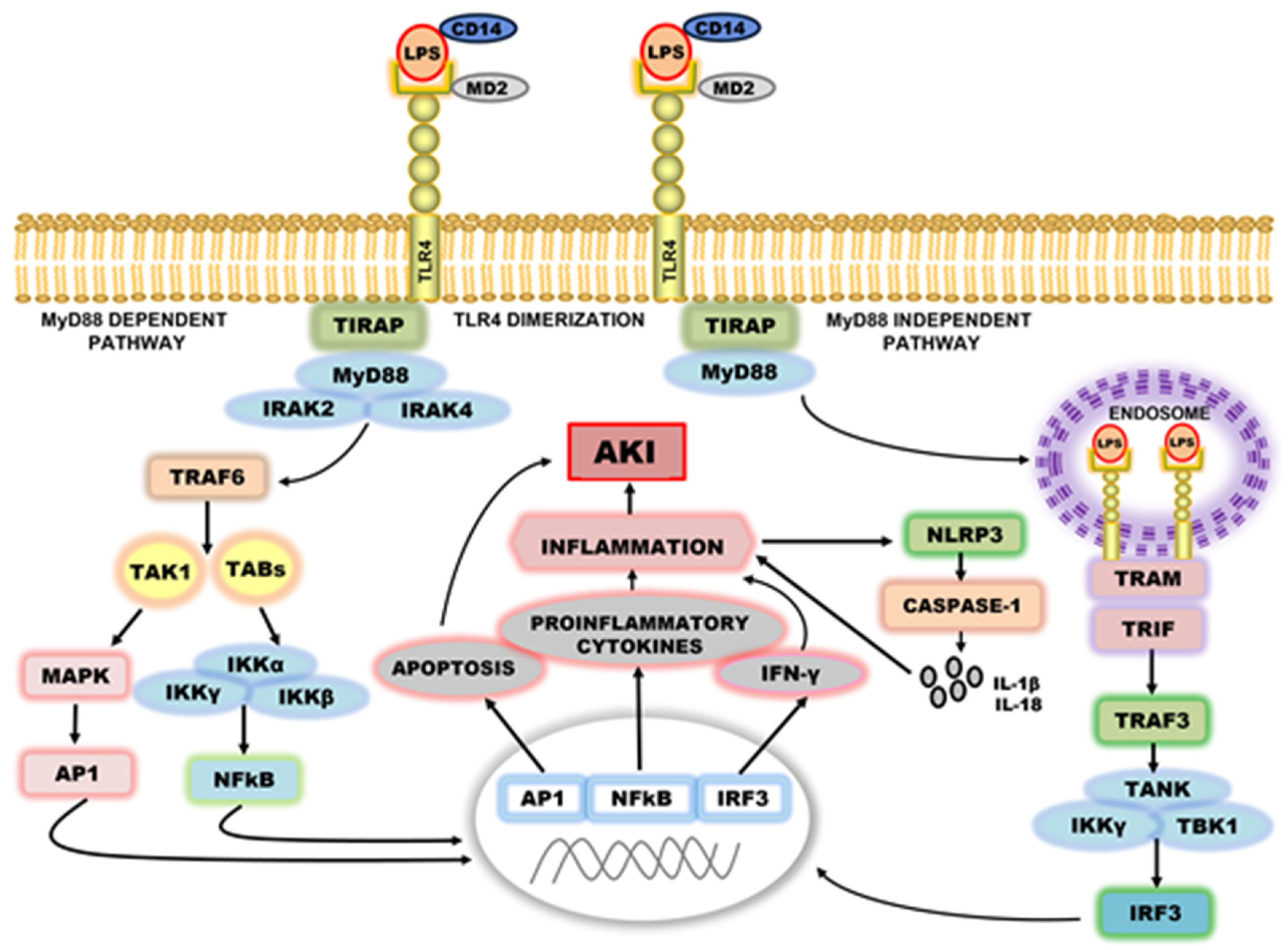
4. The Intracellular Signal Transduction of TLR4
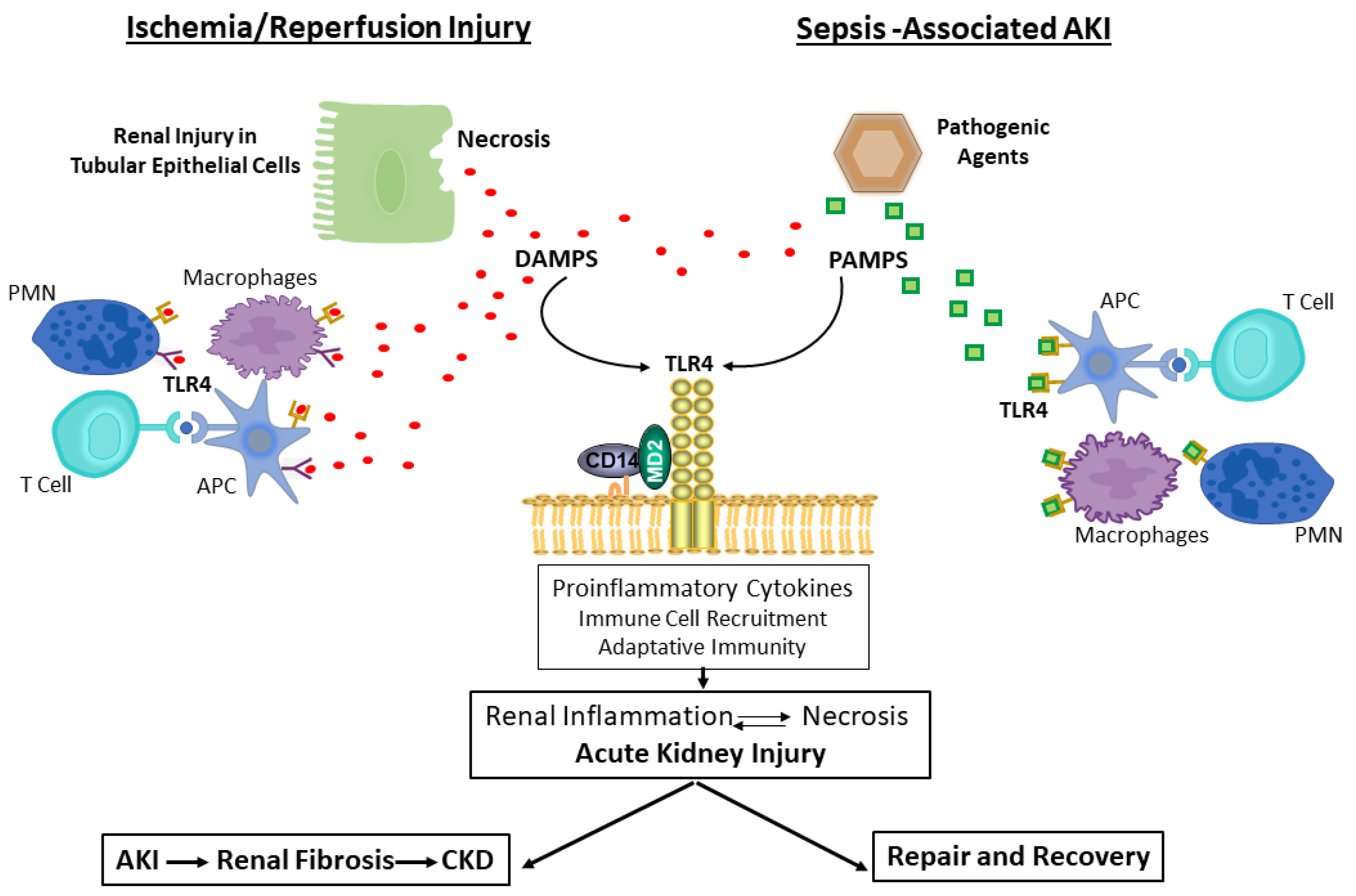
4.1. TLR4 Signaling in Ischemia/Reperfusion-Induced AKI
4.2. TLR4 Signaling in Sepsis-Associated AKI
4.3. TLR4 Inhibition in Acute Kidney Injury
4.4. TLR4 and Cell Regeneration
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bellomo, R.; Ronco, C.; Kellum, J.A.; Mehta, R.L.; Palevsky, P.; Acute Dialysis Quality Initiative Workgroup. Acute renal failure—Definition, outcome measures, animal models, fluid therapy and information technology needs: The Second International Consensus Conference of the Acute Dialysis Quality Initiative (ADQI) Group. Crit. Care 2004, 8, R204–R212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostermann, M.; Chang, R.W.S. Acute kidney injury in the intensive care unit according to RIFLE. Crit. Care Med. 2007, 35, 1837–1843. [Google Scholar] [CrossRef] [PubMed]
- Singbartl, K.; Kellum, J.A. AKI in the ICU: Definition, epidemiology, risk stratification, and outcomes. Kidney Int. 2012, 81, 819–825. [Google Scholar] [CrossRef] [Green Version]
- Hoste, E.A.J.; Schurgers, M. Epidemiology of acute kidney injury: How big is the problem? Crit. Care Med. 2008, 36, S146–S151. [Google Scholar] [CrossRef] [PubMed]
- Ostermann, M.; Liu, K. Pathophysiology of AKI. Best Pract. Res. Clin. Anaesthesiol. 2017, 31, 305–314. [Google Scholar] [CrossRef]
- Basile, D.P.; Anderson, M.D.; Sutton, T.A. Pathophysiology of Acute Kidney Injury. Compr. Physiol. 2012, 2, 1303–1353. [Google Scholar] [CrossRef] [Green Version]
- Hanif, M.O.; Bali, A.; Ramphul, K. Acute Renal Tubular Necrosis; StatPearls Publishing: Tampa, FL, USA, 2022. [Google Scholar] [PubMed]
- Kurts, C.; Panzer, U.; Anders, H.-J.; Rees, A.J. The immune system and kidney disease: Basic concepts and clinical implications. Nat. Rev. Immunol. 2013, 13, 738–753. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Chen, G.; Wyburn, K.R.; Yin, J.; Bertolino, P.; Eris, J.M.; Alexander, S.I.; Sharland, A.F.; Chadban, S.J. TLR4 activation mediates kidney ischemia/reperfusion injury. J. Clin. Investig. 2007, 117, 2847–2859. [Google Scholar] [CrossRef] [Green Version]
- Anders, H.-J.; Schaefer, L. Beyond Tissue Injury—Damage-Associated Molecular Patterns, Toll-Like Receptors, and Inflammasomes Also Drive Regeneration and Fibrosis. J. Am. Soc. Nephrol. 2014, 25, 1387–1400. [Google Scholar] [CrossRef]
- Yang, D.; Han, Z.; Oppenheim, J.J. Alarmins and immunity. Immunol. Rev. 2017, 280, 41–56. [Google Scholar] [CrossRef]
- Eleftheriadis, T.; Pissas, G.; Liakopoulos, V.; Stefanidis, I.; Lawson, B.R. Toll-like receptors and their role in renal pathologies. Inflamm. Allergy-Drug Targets 2012, 11, 464–477. [Google Scholar] [CrossRef] [PubMed]
- Bowie, A.; O’Neill, L. The interleukin-1 receptor/Toll-like receptor superfamily: Signal generators for pro-inflammatory interleukins and microbial products. J. Leukoc. Biol. 2000, 67, 508–514. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [Green Version]
- Jezierska, A.; Kolosova, I.A.; Verin, A.D. Toll Like Receptors Signaling Pathways as a Target for Therapeutic Interventions. Curr. Signal Transduct. Ther. 2011, 6, 428–440. [Google Scholar] [CrossRef] [PubMed]
- Gay, N.J.; Symmons, M.F.; Gangloff, M.; Bryant, C.E. Assembly and localization of Toll-like receptor signalling complexes. Nat. Rev. Immunol. 2014, 14, 546–558. [Google Scholar] [CrossRef] [PubMed]
- Sellge, G.; Kufer, T.A. PRR-signaling pathways: Learning from microbial tactics. Semin. Immunol. 2015, 27, 75–84. [Google Scholar] [CrossRef]
- Chen, G.Y.; Nuñez, G. Sterile inflammation: Sensing and reacting to damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef] [Green Version]
- Matzinger, P. Tolerance, Danger, and the Extended Family. Annu. Rev. Immunol. 1994, 12, 991–1045. [Google Scholar] [CrossRef]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef]
- Quintana, F.J.; Cohen, I.R. Heat Shock Proteins as Endogenous Adjuvants in Sterile and Septic Inflammation. J. Immunol. 2005, 175, 2777–2782. [Google Scholar] [CrossRef] [Green Version]
- Bours, M.; Swennen, E.; Di Virgilio, F.; Cronstein, B.; Dagnelie, P. Adenosine 5′-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol. Ther. 2006, 112, 358–404. [Google Scholar] [CrossRef]
- Kono, H.; Chen, C.-J.; Ontiveros, F.; Rock, K.L. Uric acid promotes an acute inflammatory response to sterile cell death in mice. J. Clin. Investig. 2010, 120, 1939–1949. [Google Scholar] [CrossRef] [PubMed]
- Babelova, A.; Moreth, K.; Tsalastra-Greul, W.; Zeng-Brouwers, J.; Eickelberg, O.; Young, M.F.; Bruckner, P.; Pfeilschifter, J.; Schaefer, R.M.; Gröne, H.-J.; et al. Biglycan, a Danger Signal That Activates the NLRP3 Inflammasome via Toll-like and P2X Receptors. J. Biol. Chem. 2009, 284, 24035–24048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eigenbrod, T.; Park, J.-H.; Harder, J.; Iwakura, Y.; Núñez, G. Cutting Edge: Critical Role for Mesothelial Cells in Necrosis-Induced Inflammation through the Recognition of IL-1α Released from Dying Cells. J. Immunol. 2008, 181, 8194–8198. [Google Scholar] [CrossRef] [Green Version]
- Moussion, C.; Ortega, N.; Girard, J.-P. The IL-1-Like Cytokine IL-33 Is Constitutively Expressed in the Nucleus of Endothelial Cells and Epithelial Cells In Vivo: A Novel ‘Alarmin’? PLoS ONE 2008, 3, e3331. [Google Scholar] [CrossRef] [Green Version]
- El-Achkar, T.M.; Dagher, P.C. Renal Toll-like receptors: Recent advances and implications for disease. Nat. Clin. Pract. Nephrol. 2006, 2, 568–581. [Google Scholar] [CrossRef]
- Takeda, K.; Kaisho, T.; Akira, S. Toll-Like Receptors. Annu. Rev. Immunol. 2003, 21, 335–376. [Google Scholar] [CrossRef]
- Bethany, M.T.; Tesar, B.M.; Goldstein, D.R. Toll-like receptors and their role in transplantation. Front. Biosci. 2007, 12, 4221–4238. [Google Scholar] [CrossRef] [Green Version]
- Tsuboi, N.; Yoshikai, Y.; Matsuo, S.; Kikuchi, T.; Iwami, K.-I.; Nagai, Y.; Takeuchi, O.; Akira, S.; Matsuguchi, T. Roles of Toll-Like Receptors in C-C Chemokine Production by Renal Tubular Epithelial Cells. J. Immunol. 2002, 169, 2026–2033. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, P.N.; Wang, Y.; Guo, R.; He, G.; Quigg, R.J. Role of Toll-Like Receptor 4 in Endotoxin-Induced Acute Renal Failure. J. Immunol. 2004, 172, 2629–2635. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Akira, S. TLR signaling. Cell Death Differ. 2006, 13, 816–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, T.; Akira, S. Toll-like Receptors and Their Crosstalk with Other Innate Receptors in Infection and Immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagai, Y.; Akashi, S.; Nagafuku, M.; Ogata, M.; Iwakura, Y.; Akira, S.; Kitamura, T.; Kosugi, A.; Kimoto, M.; Miyake, K. Essential role of MD-2 in LPS responsiveness and TLR4 distribution. Nat. Immunol. 2002, 3, 667–672. [Google Scholar] [CrossRef]
- Park, B.S.; Song, D.H.; Kim, H.M.; Choi, B.-S.; Lee, H.; Lee, J.-O. The structural basis of lipopolysaccharide recognition by the TLR4–MD-2 complex. Nature 2009, 458, 1191–1195. [Google Scholar] [CrossRef]
- Zanoni, I.; Ostuni, R.; Marek, L.R.; Barresi, S.; Barbalat, R.; Barton, G.M.; Granucci, F.; Kagan, J.C. CD14 Controls the LPS-Induced Endocytosis of Toll-like Receptor 4. Cell 2011, 147, 868–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajaiah, R.; Perkins, D.J.; Ireland, D.D.C.; Vogel, S.N.; Kagan, J.C. CD14 dependence of TLR4 endocytosis and TRIF signaling displays ligand specificity and is dissociable in endotoxin tolerance. Proc. Natl. Acad. Sci. USA 2015, 112, 8391–8396. [Google Scholar] [CrossRef] [Green Version]
- Kagan, J.C.; Su, T.; Horng, T.; Chow, A.; Akira, S.; Medzhitov, R. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-β. Nat. Immunol. 2008, 9, 361–368. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Kagan, J.C.; Medzhitov, R. Phosphoinositide-Mediated Adaptor Recruitment Controls Toll-like Receptor Signaling. Cell 2006, 125, 943–955. [Google Scholar] [CrossRef]
- Jha, A.K.; Gairola, S.; Kundu, S.; Doye, P.; Syed, A.M.; Ram, C.; Murty, U.S.; Naidu, V.; Sahu, B.D. Toll-like receptor 4: An attractive therapeutic target for acute kidney injury. Life Sci. 2021, 271, 119155. [Google Scholar] [CrossRef]
- Sato, S.; Sanjo, H.; Takeda, K.; Ninomiya-Tsuji, J.; Yamamoto, M.; Kawai, T.; Matsumoto, K.; Takeuchi, O.; Akira, S. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat. Immunol. 2005, 6, 1087–1095. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, L.; Karin, M. Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Toll-like Receptor and RIG-1-like Receptor Signaling. Ann. N. Y. Acad. Sci. 2008, 1143, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Ullah, M.O.; Sweet, M.J.; Mansell, A.; Kellie, S.; Kobe, B. TRIF-dependent TLR signaling, its functions in host defense and inflammation, and its potential as a therapeutic target. J. Leukoc. Biol. 2016, 100, 27–45. [Google Scholar] [CrossRef] [Green Version]
- Okun, E.; Griffioen, K.J.; Lathia, J.D.; Tang, S.-C.; Mattson, M.P.; Arumugam, T.V. Toll-like receptors in neurodegeneration. Brain Res. Rev. 2009, 59, 278–292. [Google Scholar] [CrossRef] [Green Version]
- Arslan, F.; Keogh, B.; McGuirk, P.; Parker, A.E. TLR2 and TLR4 in Ischemia Reperfusion Injury. Mediat. Inflamm. 2010, 2010, 704202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaczorowski, D.J.; Nakao, A.; Vallabhaneni, R.; Mollen, K.; Sugimoto, R.; Kohmoto, J.; Zuckerbraun, B.S.; McCurry, K.R.; Billiar, T.R. Mechanisms of Toll-Like Receptor 4 (TLR4)-Mediated Inflammation After Cold Ischemia/Reperfusion in the Heart. Transplantation 2009, 87, 1455–1463. [Google Scholar] [CrossRef] [Green Version]
- Leaf, I.A.; Nakagawa, S.; Johnson, B.G.; Cha, J.J.; Mittelsteadt, K.; Guckian, K.M.; Gomez, I.G.; Altemeier, W.A.; Duffield, J.S. Pericyte MyD88 and IRAK4 control inflammatory and fibrotic responses to tissue injury. J. Clin. Investig. 2016, 127, 321–334. [Google Scholar] [CrossRef]
- Mulay, S.R.; Linkermann, A.; Anders, H.-J. Necroinflammation in Kidney Disease. J. Am. Soc. Nephrol. 2015, 27, 27–39. [Google Scholar] [CrossRef] [Green Version]
- Wallach, D.; Kang, T.-B.; Kovalenko, A. Concepts of tissue injury and cell death in inflammation: A historical perspective. Nat. Rev. Immunol. 2013, 14, 51–59. [Google Scholar] [CrossRef]
- Farrar, C.A.; Keogh, B.; McCormack, W.; O’Shaughnessy, A.; Parker, A.; Reilly, M.; Sacks, S.H. Inhibition of TLR2 promotes graft function in a murine model of renal transplant ischemia—Reperfusion injury. FASEB J. 2011, 26, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Fung, A.; Zhao, H.; Yang, B.; Lian, Q.; Ma, D. Ischaemic and inflammatory injury in renal graft from brain death donation: An update review. J. Anesth. 2016, 30, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Perez, J.S.; Lu, K.; George, A.; Ma, D. Role of Toll-like receptor-4 in renal graft ischemia-reperfusion injury. Am. J. Physiol. Physiol. 2014, 306, F801–F811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, K.P.; Kasimsetty, S.G.; McKay, D.B. Innate immunity in donor procurement. Curr. Opin. Organ Transplant. 2013, 18, 154–160. [Google Scholar] [CrossRef] [Green Version]
- Andrade-Oliveira, V.; Campos, E.F.; Goncalves-Primo, A.; Grenzi, P.; Pestana, J.M.; Tedesco-Silva, H.; Gerbase-DeLima, M. TLR4 mRNA Levels as Tools to Estimate Risk for Early Posttransplantation Kidney Graft Dysfunction. Transplantation 2012, 94, 589–595. [Google Scholar] [CrossRef]
- Jang, H.R.; Rabb, H. The innate immune response in ischemic acute kidney injury. Clin. Immunol. 2009, 130, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Yang, C.W. The pathogenesis and treatment of chronic allograft nephropathy. Nat. Rev. Nephrol. 2009, 5, 513–519. [Google Scholar] [CrossRef]
- Kim, B.S.; Lim, S.W.; Li, C.; Kim, J.S.; Sun, B.K.; Ahn, K.O.; Han, S.W.; Kim, J.; Yang, C.W. Ischemia-Reperfusion Injury Activates Innate Immunity in Rat Kidneys. Transplantation 2005, 79, 1370–1377. [Google Scholar] [CrossRef]
- Wolfs, T.; Buurman, W.A.; Van Schadewijk, A.; De Vries, B.; Daemen, M.A.R.C.; Hiemstra, P.S.; Veer, C.V. In Vivo Expression of Toll-Like Receptor 2 and 4 by Renal Epithelial Cells: IFN-γ and TNF-α Mediated Up-Regulation During Inflammation. J. Immunol. 2002, 168, 1286–1293. [Google Scholar] [CrossRef] [Green Version]
- Pulskens, W.P.; Teske, G.J.; Butter, L.M.; Roelofs, J.J.; Van Der Poll, T.; Florquin, S.; Leemans, J.C. Toll-Like Receptor-4 Coordinates the Innate Immune Response of the Kidney to Renal Ischemia/Reperfusion Injury. PLoS ONE 2008, 3, e3596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shigeoka, A.A.; Holscher, T.D.; King, A.J.; Hall, F.W.; Kiosses, W.B.; Tobias, P.S.; Mackman, N.; McKay, D.B. TLR2 is constitutively expressed within the kidney and participates in ischemic renal injury through both MyD88-dependent and -independent pathways. J. Immunol. 2007, 178, 6252–6258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Hartono, J.R.; John, R.; Bennett, M.; Zhou, X.J.; Wang, Y.; Wu, Q.; Winterberg, P.D.; Nagami, G.T.; Lu, C.Y. Early interleukin 6 production by leukocytes during ischemic acute kidney injury is regulated by TLR4. Kidney Int. 2011, 80, 504–515. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Ma, J.; Wang, P.; Corpuz, T.M.; Panchapakesan, U.; Wyburn, K.R.; Chadban, S.J. HMGB1 Contributes to Kidney Ischemia Reperfusion Injury. J. Am. Soc. Nephrol. 2010, 21, 1878–1890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rusai, K.; Sollinger, D.; Baumann, M.; Wagner, B.; Strobl, M.; Schmaderer, C.; Roos, M.; Kirschning, C.; Heemann, U.; Lutz, J. Toll-like receptors 2 and 4 in renal ischemia/reperfusion injury. Pediatr. Nephrol. 2010, 25, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Dagher, P.C.; Hato, T.; Mang, H.E.; Plotkin, Z.; Richardson, Q.V.; Massad, M.; Mai, E.; Kuehl, S.E.; Graham, P.; Kumar, R.; et al. Inhibition of Toll-Like Receptor 4 Signaling Mitigates Microvascular Loss but Not Fibrosis in a Model of Ischemic Acute Kidney Injury. Int. J. Mol. Sci. 2016, 17, 647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wang, D.; Jin, Z. miR-27a suppresses TLR4-induced renal ischemia-reperfusion injury. Mol. Med. Rep. 2019, 20, 967–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergler, T.; Hoffmann, U.; Bergler, E.; Jung, B.; Banas, M.C.; Reinhold, S.W.; Krämer, B.K.; Banas, B. Toll-Like Receptor 4 in Experimental Kidney Transplantation: Early Mediator of Endogenous Danger Signals. Nephron Exp. Nephrol. 2012, 121, e59–e70. [Google Scholar] [CrossRef] [Green Version]
- Leventhal, J.S.; Schröppel, B. Toll-like receptors in transplantation: Sensing and reacting to injury. Kidney Int. 2012, 81, 826–832. [Google Scholar] [CrossRef]
- Kaczorowski, D.J.; Nakao, A.; Mollen, K.P.; Vallabhaneni, R.; Sugimoto, R.; Kohmoto, J.; Tobita, K.; Zuckerbraun, B.S.; McCurry, K.R.; Murase, N.; et al. Toll-Like Receptor 4 Mediates the Early Inflammatory Response After Cold Ischemia/Reperfusion. Transplantation 2007, 84, 1279–1287. [Google Scholar] [CrossRef]
- Palmer, S.M.; Burch, L.H.; Mir, S.; Smith, S.R.; Kuo, P.C.; Herczyk, W.F.; Reinsmoen, N.L.; Schwartz, D.A. Donor polymorphisms in Toll-like receptor-4 influence the development of rejection after renal transplantation. Clin. Transplant. 2005, 20, 30–36. [Google Scholar] [CrossRef]
- Krüger, B.; Krick, S.; Dhillon, N.; Lerner, S.M.; Ames, S.; Bromberg, J.S.; Lin, M.; Walsh, L.; Vella, J.; Fischereder, M.; et al. Donor Toll-like receptor 4 contributes to ischemia and reperfusion injury following human kidney transplantation. Proc. Natl. Acad. Sci. USA 2009, 106, 3390–3395. [Google Scholar] [CrossRef] [Green Version]
- Gomez, H.; Ince, C.; De Backer, D.; Pickkers, P.; Payen, D.; Hotchkiss, J.; Kellum, J.A. A Unified Theory of Sepsis-Induced Acute Kidney Injury: Inflammation, Microcirculatory Dysfunction, Bioenergetics, and the Tubular Cell Adaptation to Injury. Shock 2014, 41, 3–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabb, H.; Griffin, M.D.; McKay, D.B.; Swaminathan, S.; Pickkers, P.; Rosner, M.H.; Kellum, J.A.; Ronco, C. Inflammation in AKI: Current Understanding, Key Questions, and Knowledge Gaps. J. Am. Soc. Nephrol. 2016, 27, 371–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Okusa, M.D. Macrophages, Dendritic Cells, and Kidney Ischemia-Reperfusion Injury. Semin. Nephrol. 2010, 30, 268–277. [Google Scholar] [CrossRef] [Green Version]
- Kinsey, G.R.; Li, L.; Okusa, M.D. Inflammation in Acute Kidney Injury. Nephron Exp. Nephrol. 2008, 109, e102–e107. [Google Scholar] [CrossRef] [Green Version]
- Creagh, E.M.; O’Neill, L.A. TLRs, NLRs and RLRs: A trinity of pathogen sensors that co-operate in innate immunity. Trends Immunol. 2006, 27, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, S.T.; Larivière, L.; Leveque, G.; Clermont, S.; Moore, K.J.; Gros, P.; Malo, D. Endotoxin-tolerant Mice Have Mutations in Toll-like Receptor 4 (Tlr4). J. Exp. Med. 1999, 189, 615–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Achkar, T.M.; Hosein, M.; Dagher, P.C. Pathways of renal injury in systemic gram-negative sepsis. Eur. J. Clin. Investig. 2008, 38, 39–44. [Google Scholar] [CrossRef]
- Roger, T.; Froidevaux, C.; Le Roy, D.; Reymond, M.K.; Chanson, A.-L.; Mauri, D.; Burns, K.; Riederer, B.M.; Akira, S.; Calandra, T. Protection from lethal Gram-negative bacterial sepsis by targeting Toll-like receptor 4. Proc. Natl. Acad. Sci. USA 2009, 106, 2348–2352. [Google Scholar] [CrossRef] [Green Version]
- Daubeuf, B.; Mathison, J.; Spiller, S.; Hugues, S.; Herren, S.; Ferlin, W.; Kosco-Vilbois, M.; Wagner, H.; Kirschning, C.J.; Ulevitch, R.; et al. TLR4/MD-2 Monoclonal Antibody Therapy Affords Protection in Experimental Models of Septic Shock. J. Immunol. 2007, 179, 6107–6114. [Google Scholar] [CrossRef] [Green Version]
- Tulic, M.; Hurrelbrink, R.J.; Prêle, C.M.; Laing, I.; Upham, J.; Le Souef, P.; Sly, P.; Holt, P. TLR4 Polymorphisms Mediate Impaired Responses to Respiratory Syncytial Virus and Lipopolysaccharide. J. Immunol. 2007, 179, 132–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matzinger, P. The Danger Model: A Renewed Sense of Self. Science 2002, 296, 301–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keating, S.E.; Maloney, G.M.; Moran, E.M.; Bowie, A.G. IRAK-2 Participates in Multiple Toll-like Receptor Signaling Pathways to NFκB via Activation of TRAF6 Ubiquitination. J. Biol. Chem. 2007, 282, 33435–33443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalakeche, R.; Hato, T.; Rhodes, G.; Dunn, K.W.; El-Achkar, T.M.; Plotkin, Z.; Sandoval, R.M.; Dagher, P.C. Endotoxin Uptake by S1 Proximal Tubular Segment Causes Oxidative Stress in the Downstream S2 Segment. J. Am. Soc. Nephrol. 2011, 22, 1505–1516. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.; Yiu, W.H.; Wu, H.J.; Chan, L.Y.; Leung, J.C.; Au, W.S.; Chan, K.W.; Lai, K.N.; Tang, S.C. Toll-Like Receptor 4 Promotes Tubular Inflammation in Diabetic Nephropathy. J. Am. Soc. Nephrol. 2011, 23, 86–102. [Google Scholar] [CrossRef] [Green Version]
- Castoldi, A.; Braga, T.; Correa-Costa, M.; De Aguiar, C.F.; Bassi, Ê.J.; Correa-Silva, R.; Elias, R.M.; Salvador, F.; Vieira, P.; Cenedeze, M.A.; et al. TLR2, TLR4 and the MYD88 Signaling Pathway Are Crucial for Neutrophil Migration in Acute Kidney Injury Induced by Sepsis. PLoS ONE 2012, 7, e37584. [Google Scholar] [CrossRef] [Green Version]
- El-Achkar, T.M.; Huang, X.; Plotkin, Z.; Sandoval, R.M.; Rhodes, G.J.; Dagher, P.C. Sepsis induces changes in the expression and distribution of Toll-like receptor 4 in the rat kidney. Am. J. Physiol. Physiol. 2006, 290, F1034–F1043. [Google Scholar] [CrossRef] [Green Version]
- Good, D.W.; George, T.; Watts, B.A., III. Lipopolysaccharide directly alters renal tubule transport through distinct TLR4-dependent pathways in basolateral and apical membranes. Am. J. Physiol. Ren. Physiol. 2009, 297, F866–F874. [Google Scholar] [CrossRef]
- Watts, B.A.; George, T.; Sherwood, E.R.; Good, D.W. Basolateral LPS inhibits NHE3 and HCO3− absorption through TLR4/MyD88-dependent ERK activation in medullary thick ascending limb. Am. J. Physiol. Physiol. 2011, 301, C1296–C1306. [Google Scholar] [CrossRef] [Green Version]
- Watts, B.A., III; George, T.; Good, D.W. Lumen LPS inhibits HCO3− absorption in the medullary thick ascending limb through TLR4-PI3K-Akt-mTOR-dependent inhibition of basolateral Na+/H+ exchange. Am. J. Physiol. Ren. Physiol. 2013, 305, F451–F462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Good, D.W.; George, T.; Watts, B.A. Toll-like receptor 2 mediates inhibition of HCO3− absorption by bacterial lipoprotein in medullary thick ascending limb. Am. J. Physiol. Physiol. 2010, 299, F536–F544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bussolati, B.; David, S.; Cambi, V.; Tobias, P.S.; Camussi, G. Urinary soluble CD14 mediates human proximal tubular epithelial cell injury induced by LPS. Int. J. Mol. Med. 2002, 10, 441–449. [Google Scholar] [CrossRef]
- Roh, J.S.; Sohn, D.H. Damage-Associated Molecular Patterns in Inflammatory Diseases. Immune Netw. 2018, 18, e27. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, G.M.; Zamboni, D.; Camara, N. The role of innate immunity in septic acute kidney injuries. Shock 2010, 34, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Zamyatina, A.; Heine, H. Lipopolysaccharide Recognition in the Crossroads of TLR4 and Caspase-4/11 Mediated Inflammatory Pathways. Front. Immunol. 2020, 11, 585146. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Fei, D.; Chen, M.; Sun, M.; Xu, J.; Kang, K.; Jiang, L.; Zhao, M. Role of the nucleotide-binding domain-like receptor protein 3 inflammasome in acute kidney injury. FEBS J. 2015, 282, 3799–3807. [Google Scholar] [CrossRef] [Green Version]
- Huang, G.; Bao, J.; Shao, X.; Zhou, W.; Wu, B.; Ni, Z.; Wang, L. Inhibiting pannexin-1 alleviates sepsis-induced acute kidney injury via decreasing NLRP3 inflammasome activation and cell apoptosis. Life Sci. 2020, 254, 117791. [Google Scholar] [CrossRef]
- Arulkumaran, N.; Sixma, M.L.; Pollen, S.; Ceravola, E.; Jentho, E.; Prendecki, M.; Bass, P.S.; Tam, F.W.K.; Unwin, R.J.; Singer, M. P2X7 receptor antagonism ameliorates renal dysfunction in a rat model of sepsis. Physiol. Rep. 2018, 6, e13622. [Google Scholar] [CrossRef]
- Molema, G.; Zijlstra, J.G.; van Meurs, M.; Kamps, J.A.A.M. Renal microvascular endothelial cell responses in sepsis-induced acute kidney injury. Nat. Rev. Nephrol. 2021, 18, 95–112. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Tiwari, M.M.; Messer, K.J.; Holthoff, J.H.; Gokden, N.; Brock, R.W.; Mayeux, P.R. Peritubular capillary dysfunction and renal tubular epithelial cell stress following lipopolysaccharide administration in mice. Am. J. Physiol. Physiol. 2007, 292, F261–F268. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Zheng, S.; Rosin, D.L.; Poudel, N.; Yao, J.; Perry, H.M.; Cao, R.; Okusa, M.D.; Hu, S. Development of a photoacoustic microscopy technique to assess peritubular capillary function and oxygen metabolism in the mouse kidney. Kidney Int. 2021, 100, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Zafrani, L.; Payen, D.; Azoulay, E.; Ince, C. The Microcirculation of the Septic Kidney. Semin. Nephrol. 2015, 35, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Kuwabara, S.; Goggins, E.; Okusa, M.D. The Pathophysiology of Sepsis-Associated AKI. Clin. J. Am. Soc. Nephrol. 2022, 17, 1050–1069. [Google Scholar] [CrossRef] [PubMed]
- Anderberg, S.B.; Luther, T.; Frithiof, R. Physiological aspects of Toll-like receptor 4 activation in sepsis-induced acute kidney injury. Acta Physiol. 2016, 219, 575–590. [Google Scholar] [CrossRef]
- Fenhammar, J.; Rundgren, M.; Hultenby, K.; Forestier, J.; Taavo, M.; Kenne, E.; Weitzberg, E.; Eriksson, S.; Ozenci, V.; Wernerson, A.; et al. Renal effects of treatment with a TLR4 inhibitor in conscious septic sheep. Crit. Care 2014, 18, 488. [Google Scholar] [CrossRef] [Green Version]
- Kramann, R.; Humphreys, B.D. Kidney Pericytes: Roles in Regeneration and Fibrosis. Semin. Nephrol. 2014, 34, 374–383. [Google Scholar] [CrossRef] [Green Version]
- Humphreys, B.D.; Lin, S.-L.; Kobayashi, A.; Hudson, T.E.; Nowlin, B.T.; Bonventre, J.V.; Valerius, M.T.; McMahon, A.P.; Duffield, J.S. Fate Tracing Reveals the Pericyte and Not Epithelial Origin of Myofibroblasts in Kidney Fibrosis. Am. J. Pathol. 2010, 176, 85–97. [Google Scholar] [CrossRef] [Green Version]
- Lemos, D.R.; Marsh, G.; Huang, A.; Campanholle, G.; Aburatani, T.; Dang, L.; Gomez, I.; Fisher, K.; Ligresti, G.; Peti-Peterdi, J.; et al. Maintenance of vascular integrity by pericytes is essential for normal kidney function. Am. J. Physiol. Physiol. 2016, 311, F1230–F1242. [Google Scholar] [CrossRef]
- Li, P.; Zhou, Y.; Goodwin, A.J.; Cook, J.A.; Halushka, P.V.; Zhang, X.K.; Wilson, C.L.; Schnapp, L.M.; Zingarelli, B.; Fan, H. Fli-1 Governs Pericyte Dysfunction in a Murine Model of Sepsis. J. Infect. Dis. 2018, 218, 1995–2005. [Google Scholar] [CrossRef]
- Castellano, G.; Stasi, A.; Franzin, R.; Sallustio, F.; Divella, C.; Spinelli, A.; Netti, G.S.; Fiaccadori, E.; Cantaluppi, V.; Crovace, A.; et al. LPS-Binding Protein Modulates Acute Renal Fibrosis by Inducing Pericyte-to-Myofibroblast Trans-Differentiation through TLR-4 Signaling. Int. J. Mol. Sci. 2019, 20, 3682. [Google Scholar] [CrossRef] [Green Version]
- Spizzirri, F.D.; Rahman, R.C.; Bibiloni, N.; Ruscasso, J.D.; Amoreo, O.R. Childhood hemolytic uremic syndrome in Argentina: Long-term follow-up and prognostic features. Pediatr. Nephrol. 1997, 11, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Petruzziello, T.N.; Mawji, I.A.; Khan, M.; Marsden, P.A. Verotoxin biology: Molecular events in vascular endothelial injury. Kidney Int. 2009, 75, S17–S19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoja, C.; Buelli, S.; Morigi, M. Shiga toxin-associated hemolytic uremic syndrome: Pathophysiology of endothelial dysfunction. Pediatr. Nephrol. 2010, 25, 2231–2240. [Google Scholar] [CrossRef]
- Clayton, F.; Pysher, T.J.; Lou, R.; Kohan, D.E.; Denkers, N.D.; Tesh, V.L.; Taylor, F.B., Jr.; Siegler, R.L. Lipopolysaccharide Upregulates Renal Shiga Toxin Receptors in a Primate Model of Hemolytic Uremic Syndrome. Am. J. Nephrol. 2005, 25, 536–540. [Google Scholar] [CrossRef]
- Exeni, R.A.; Fernandez-Brando, R.J.; Santiago, A.P.; Fiorentino, G.A.; Exeni, A.M.; Ramos, M.V.; Palermo, M.S. Pathogenic role of inflammatory response during Shiga toxin-associated hemolytic uremic syndrome (HUS). Pediatr. Nephrol. 2018, 33, 2057–2071. [Google Scholar] [CrossRef]
- Luna, M.; Kamariski, M.; Principi, I.; Bocanegra, V.; Vallés, P.G. Severely ill pediatric patients with Shiga toxin-associated hemolytic uremic syndrome (STEC-HUS) who suffered from multiple organ involvement in the early stage. Pediatr. Nephrol. 2020, 36, 1499–1509. [Google Scholar] [CrossRef] [PubMed]
- Vallés, P.G.; Melechuck, S.; Gonzalez, A.; Manucha, W.; Bocanegra, V.; Vallés, R. Toll-like receptor 4 expression on circulating leucocytes in hemolytic uremic syndrome. Pediatr. Nephrol. 2011, 27, 407–415. [Google Scholar] [CrossRef]
- Vázquez-Carballo, C.; Guerrero-Hue, M.; García-Caballero, C.; Rayego-Mateos, S.; Opazo-Ríos, L.; Morgado-Pascual, J.L.; Herencia-Bellido, C.; Vallejo-Mudarra, M.; Cortegano, I.; Gaspar, M.L.; et al. Toll-Like Receptors in Acute Kidney Injury. Int. J. Mol. Sci. 2021, 22, 816. [Google Scholar] [CrossRef]
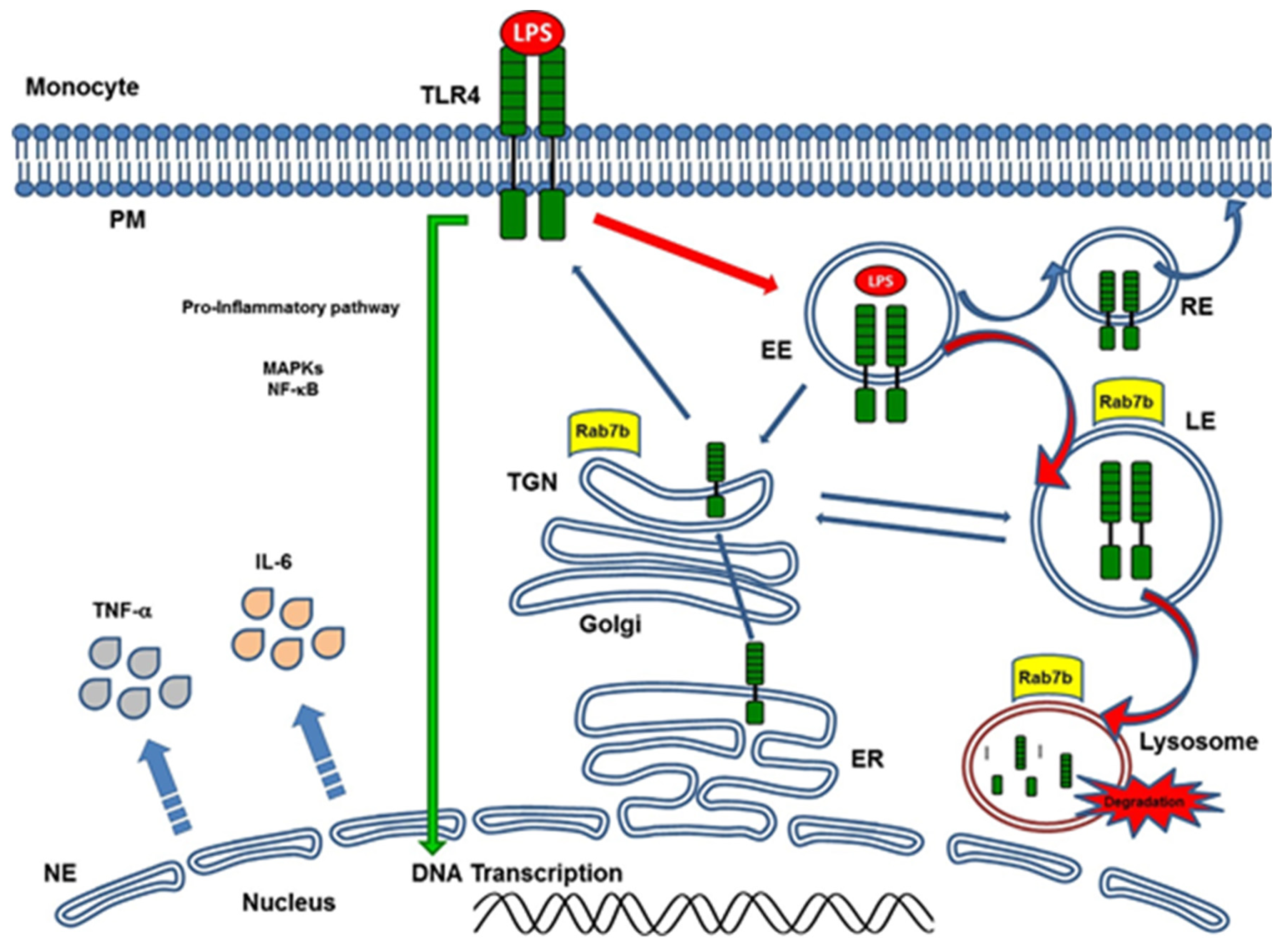
- Bucci, C.; Bakke, O.; Progida, C. Rab7b and receptors trafficking. Commun. Integr. Biol. 2010, 3, 401–404. [Google Scholar] [CrossRef]
- Manzano, A.F.L.; Gil Lorenzo, A.F.; Bocanegra, V.; Costantino, V.V.; Cacciamani, V.; Benardon, M.E.; Vallés, P.G. Rab7b participation on the TLR4 (Toll-like receptor) endocytic pathway in Shiga toxin-associated Hemolytic Uremic Syndrome (HUS). Cytokine 2019, 121, 154732. [Google Scholar] [CrossRef] [PubMed]
- Kuzmich, N.N.; Sivak, K.V.; Chubarev, V.N.; Porozov, Y.B.; Savateeva-Lyubimova, T.N.; Peri, F. TLR4 Signaling Pathway Modulators as Potential Therapeutics in Inflammation and Sepsis. Vaccines 2017, 5, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Fu, W.; Zheng, L.; Wang, Y.; Liang, G. Recent progress in the discovery of myeloid differentiation 2 (MD2) modulators for inflammatory diseases. Drug Discov. Today 2018, 23, 1187–1202. [Google Scholar] [CrossRef]
- Xu, Y.; Chen, S.; Cao, Y.; Zhou, P.; Chen, Z.; Cheng, K. Discovery of novel small molecule TLR4 inhibitors as potent anti-inflammatory agents. Eur. J. Med. Chem. 2018, 154, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Zaffaroni, L.; Peri, F. Recent advances on Toll-like receptor 4 modulation: New therapeutic perspectives. Futur. Med. Chem. 2018, 10, 461–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ain, Q.U.; Batool, M.; Choi, S. TLR4-Targeting Therapeutics: Structural Basis and Computer-Aided Drug Discovery Approaches. Molecules 2020, 25, 627. [Google Scholar] [CrossRef] [Green Version]
- Peri, F.; Calabrese, V. Toll-like Receptor 4 (TLR4) Modulation by Synthetic and Natural Compounds: An Update. J. Med. Chem. 2013, 57, 3612–3622. [Google Scholar] [CrossRef]
- Kim, H.M.; Park, B.S.; Kim, J.-I.; Kim, S.E.; Lee, J.; Oh, S.C.; Enkhbayar, P.; Matsushima, N.; Lee, H.; Yoo, O.J.; et al. Crystal Structure of the TLR4-MD-2 Complex with Bound Endotoxin Antagonist Eritoran. Cell 2007, 130, 906–917. [Google Scholar] [CrossRef] [Green Version]
- Bennett-Guerrero, E.; Grocott, H.P.; Levy, J.H.; Stierer, K.A.; Hogue, C.W.; Cheung, A.T.; Newman, M.F.; Carter, A.A.; Rossignol, D.P.; Collard, C.D. A Phase II, Double-Blind, Placebo-Controlled, Ascending-Dose Study of Eritoran (E5564), a Lipid A Antagonist, in Patients Undergoing Cardiac Surgery with Cardiopulmonary Bypass. Anesth. Analg. 2007, 104, 378–383. [Google Scholar] [CrossRef]
- Barochia, A.; Solomon, S.; Cui, X.; Natanson, C.; Eichacker, P.Q. Eritoran tetrasodium (E5564) treatment for sepsis: Review of preclinical and clinical studies. Expert Opin. Drug Metab. Toxicol. 2011, 7, 479–494. [Google Scholar] [CrossRef] [PubMed]
- Tidswell, M.; LaRosa, S.P. Toll-like receptor-4 antagonist eritoran tetrasodium for severe sepsis. Expert Rev. Anti-Infect. Ther. 2011, 9, 507–520. [Google Scholar] [CrossRef] [PubMed]
- Takashima, K.; Matsunaga, N.; Yoshimatsu, M.; Hazeki, K.; Kaisho, T.; Uekata, M.; Hazeki, O.; Akira, S.; Iizawa, Y.; Ii, M. Analysis of binding site for the novel small-molecule TLR4 signal transduction inhibitor TAK-242 and its therapeutic effect on mouse sepsis model. J. Cereb. Blood Flow Metab. 2009, 157, 1250–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habib, R. Multifaceted roles of Toll-like receptors in acute kidney injury. Heliyon 2021, 7, e06441. [Google Scholar] [CrossRef] [PubMed]
- Bonventre, J.V.; Zuk, A. Ischemic acute renal failure: An inflammatory disease? Kidney Int. 2004, 66, 480–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, H.-J.; Ryu, M. Renal microenvironments and macrophage phenotypes determine progression or resolution of renal inflammation and fibrosis. Kidney Int. 2011, 80, 915–925. [Google Scholar] [CrossRef] [Green Version]
- Lassen, S.; Lech, M.; Römmele, C.; Mittruecker, H.-W.; Mak, T.W.; Anders, H.-J. Ischemia Reperfusion Induces IFN Regulatory Factor 4 in Renal Dendritic Cells, which Suppresses Postischemic Inflammation and Prevents Acute Renal Failure. J. Immunol. 2010, 185, 1976–1983. [Google Scholar] [CrossRef] [Green Version]
- Humphreys, B.D.; Bonventre, J.V. Mesenchymal Stem Cells in Acute Kidney Injury. Annu. Rev. Med. 2008, 59, 311–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lech, M.; Gröbmayr, R.; Weidenbusch, M.; Anders, H.-J. Tissues Use Resident Dendritic Cells and Macrophages to Maintain Homeostasis and to Regain Homeostasis upon Tissue Injury: The Immunoregulatory Role of Changing Tissue Environments. Mediat. Inflamm. 2012, 2012, 951390. [Google Scholar] [CrossRef] [Green Version]
- Bonventre, J.V.; Yang, L. Cellular pathophysiology of ischemic acute kidney injury. J. Clin. Investig. 2011, 121, 4210–4221. [Google Scholar] [CrossRef]
- Sallustio, F.; Curci, C.; Aloisi, A.; Toma, C.C.; Marulli, E.; Serino, G.; Cox, S.N.; De Palma, G.; Stasi, A.; Divella, C.; et al. Inhibin-A and Decorin Secreted by Human Adult Renal Stem/Progenitor Cells Through the TLR2 Engagement Induce Renal Tubular Cell Regeneration. Sci. Rep. 2017, 7, 8225. [Google Scholar] [CrossRef]
- Kulkarni, O.P.; Hartter, I.; Mulay, S.R.; Hagemann, J.; Darisipudi, M.N.; Vr, S.K.; Romoli, S.; Thomasova, D.; Ryu, M.; Kobold, S.; et al. Toll-Like Receptor 4–Induced IL-22 Accelerates Kidney Regeneration. J. Am. Soc. Nephrol. 2014, 25, 978–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nechemia-Arbely, Y.; Barkan, D.; Pizov, G.; Shriki, A.; Rose-John, S.; Galun, E.; Axelrod, J.H. IL-6/IL-6R Axis Plays a Critical Role in Acute Kidney Injury. J. Am. Soc. Nephrol. 2008, 19, 1106–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flynn, C.M.; Garbers, Y.; Lokau, J.; Wesch, D.; Schulte, D.M.; Laudes, M.; Lieb, W.; Aparicio-Siegmund, S.; Garbers, C. Activation of Toll-like Receptor 2 (TLR2) induces Interleukin-6 trans-signaling. Sci. Rep. 2019, 9, 7306. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Huen, S.; Nishio, H.; Nishio, S.; Lee, H.K.; Choi, B.-S.; Ruhrberg, C.; Cantley, L.G. Distinct Macrophage Phenotypes Contribute to Kidney Injury and Repair. J. Am. Soc. Nephrol. 2011, 22, 317–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roedig, H.; Nastase, M.V.; Frey, H.; Moreth, K.; Zeng-Brouwers, J.; Poluzzi, C.; Hsieh, L.T.-H.; Brandts, C.; Fulda, S.; Wygrecka, M.; et al. Biglycan is a new high-affinity ligand for CD14 in macrophages. Matrix Biol. J. Int. Soc. Matrix Biol. 2019, 77, 4–22. [Google Scholar] [CrossRef]
- Poluzzi, C.; Nastase, M.-V.; Zeng-Brouwers, J.; Roedig, H.; Hsieh, L.T.-H.; Michaelis, J.B.; Buhl, E.M.; Rezende, F.; Manavski, Y.; Bleich, A.; et al. Biglycan evokes autophagy in macrophages via a novel CD44/Toll-like receptor 4 signaling axis in ischemia/reperfusion injury. Kidney Int. 2019, 95, 540–562. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vallés, P.G.; Gil Lorenzo, A.F.; Garcia, R.D.; Cacciamani, V.; Benardon, M.E.; Costantino, V.V. Toll-like Receptor 4 in Acute Kidney Injury. Int. J. Mol. Sci. 2023, 24, 1415. https://doi.org/10.3390/ijms24021415
Vallés PG, Gil Lorenzo AF, Garcia RD, Cacciamani V, Benardon ME, Costantino VV. Toll-like Receptor 4 in Acute Kidney Injury. International Journal of Molecular Sciences. 2023; 24(2):1415. https://doi.org/10.3390/ijms24021415
Chicago/Turabian StyleVallés, Patricia G., Andrea Fernanda Gil Lorenzo, Rodrigo D. Garcia, Valeria Cacciamani, María Eugenia Benardon, and Valeria Victoria Costantino. 2023. "Toll-like Receptor 4 in Acute Kidney Injury" International Journal of Molecular Sciences 24, no. 2: 1415. https://doi.org/10.3390/ijms24021415
APA StyleVallés, P. G., Gil Lorenzo, A. F., Garcia, R. D., Cacciamani, V., Benardon, M. E., & Costantino, V. V. (2023). Toll-like Receptor 4 in Acute Kidney Injury. International Journal of Molecular Sciences, 24(2), 1415. https://doi.org/10.3390/ijms24021415