
The Complex Histopathological and Immunohistochemical Spectrum of Neuroendocrine Tumors—An Overview of the Latest Classifications

 and
and
Abstract
:1. Introduction
2. Histopathological and Immunohistochemical Features in NENs
3. Histologic Grading of NENs
4. Differences between NET G3 and NEC
5. Pathological Features of Mixed Neuroendocrine–Non-Neuroendocrine Neoplasms
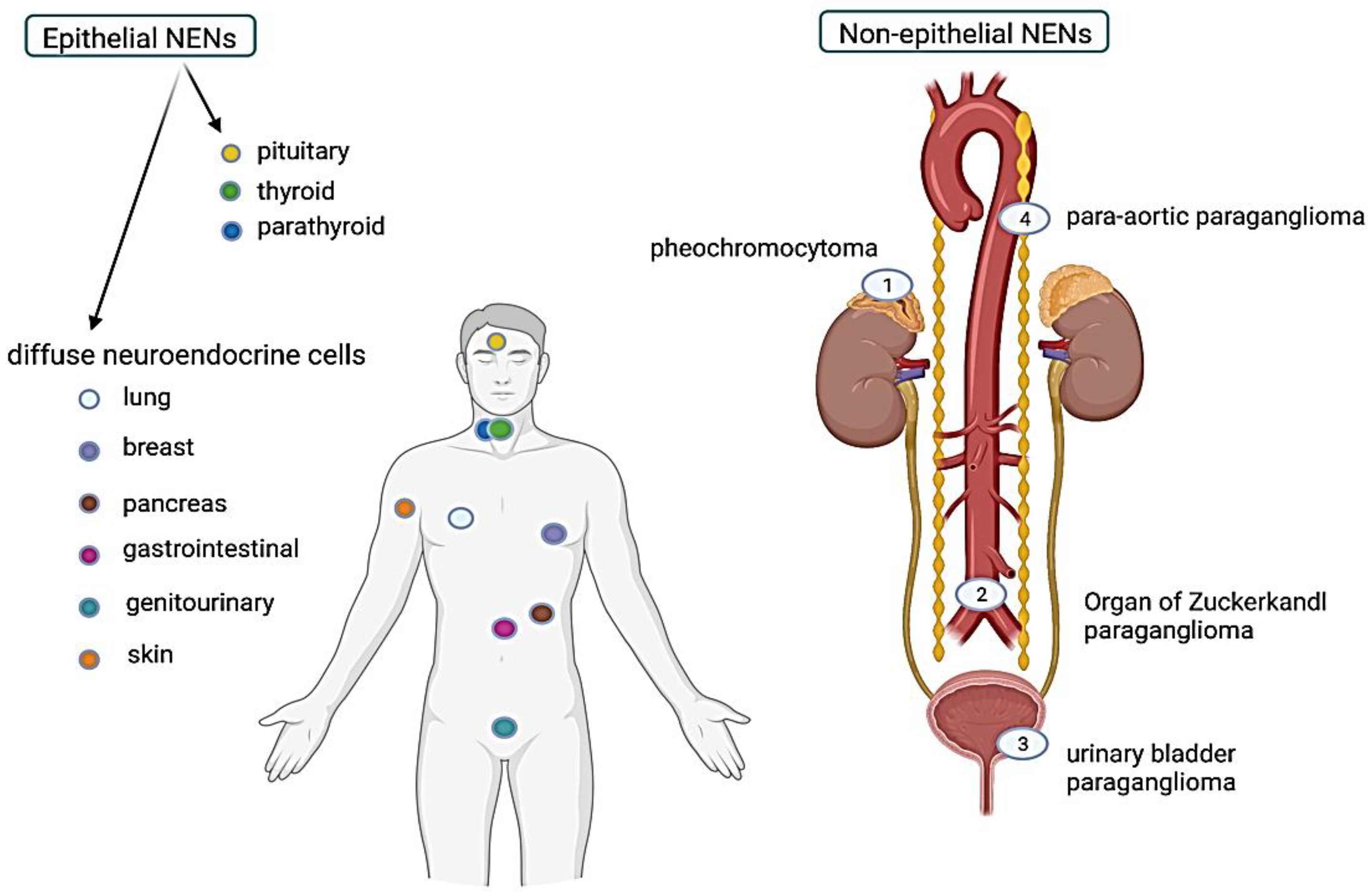
6. Characteristics of Epithelial NENs Based on the Organ of Origin
6.1. Lung NENs
6.2. Gastrointestinal and Pancreatobiliary NENs
6.3. Head and Neck NENs
6.4. Breast NENs
6.5. Femele Reproductive System NENs
6.6. Urinary and Male Genital System NENs
6.7. Skin NENs
6.8. Pituitary NENs
7. Non-Epithelial Neuroendocrine Neoplasms
7.1. Pheochromocytomas
7.2. Sympathetic Paragangliomas
7.3. Parasympathetic Paragangliomas
7.4. Composite Phaeochromocytoma and Paraganglioma
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kim, J.Y.; Hong, S.-M.; Ro, J.Y. Recent updates on grading and classification of neuroendocrine tumors. Ann. Diagn. Pathol. 2017, 29, 11–16. [Google Scholar] [CrossRef]
- Asa, S.L.; Mete, O.; Cusimano, M.D.; McCutcheon, I.E.; Perry, A.; Yamada, S.; Nishioka, H.; Casar-Borota, O.; Uccella, S.; La Rosa, S.; et al. Pituitary neuroendocrine tumors: A model for neuroendocrine tumor classification. Mod. Pathol. 2021, 34, 1634–1650. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, E.; Abo-Ahmed, A.I.; Latifi, F. Ultrastructure and histochemistry of the subepithelial glands of the nasal septal island in dromedaries with special reference to the possible functions. Saudi J. Biol. Sci. 2021, 28, 5325–5331. [Google Scholar] [CrossRef] [PubMed]
- Bussolati, G. C and APUD Cells and Endocrine Tumours. Pearse’s Laboratory in the Years 1965–1969: A Personal Recollection. Endocr. Pathol. 2014, 25, 133–140. [Google Scholar] [CrossRef]
- Pearse, A.G. The APUD cell concept and its implications in pathology. Pathol. Annu. 1974, 9, 27–41. [Google Scholar] [PubMed]
- Asa, S.L.; Ezzat, S.; Mete, O. The Diagnosis and Clinical Significance of Paragangliomas in Unusual Locations. J. Clin. Med. 2018, 7, 280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, W.; Ding, Z.Y.; Zhang, B.; Chen, L.; Li, G.X.; Wu, J.J.; Zhang, B.; Chen, X.P.; Zhu, P. Primary functioning hepatic para-ganglioma mimicking hepatocellular carcinoma: A case report and literature review. Medicine 2018, 97, e0293. [Google Scholar] [CrossRef]
- Rindi, G.; Klimstra, D.S.; Abedi-Ardekani, B.; Asa, S.L.; Bosman, F.T.; Brambilla, E.; Busam, K.J.; De Krijger, R.R.; Dietel, M.; El-Naggar, A.K.; et al. A common classification framework for neuroendocrine neoplasms: An International Agency for Research on Cancer (IARC) and World Health Organization (WHO) expert consensus proposal. Mod. Pathol. 2018, 31, 1770–1786. [Google Scholar] [CrossRef] [Green Version]
- Klöppel, G. Neuroendocrine Neoplasms: Dichotomy, Origin and Classifications. Visc. Med. 2017, 33, 324–330. [Google Scholar] [CrossRef]
- Maleki, Z.; Nadella, A.; Nadella, M.; Patel, G.; Patel, S.; Kholová, I. INSM1, a Novel Biomarker for Detection of Neuroendocrine Neoplasms: Cytopathologists’ View. Diagnostics 2021, 11, 2172. [Google Scholar] [CrossRef]
- Rindi, G.; Mete, O.; Uccella, S.; Basturk, O.; La Rosa, S.; Brosens, L.A.A.; Ezzat, S.; de Herder, W.W.; Klimstra, D.S.; Papotti, M.; et al. Overview of the 2022 WHO Classification of Neuroendocrine Neoplasms. Endocr. Pathol. 2022, 33, 115–154. [Google Scholar] [CrossRef]
- Oronsky, B.; Ma, P.C.; Morgensztern, D.; Carter, C.A. Nothing But NET: A Review of Neuroendocrine Tumors and Carci-nomas. Neoplasia 2017, 19, 991–1002. [Google Scholar] [CrossRef] [PubMed]
- Rekhtman, N. Lung neuroendocrine neoplasms: Recent progress and persistent challenges. Mod. Pathol. 2021, 35 (Suppl. S1), 36–50. [Google Scholar] [CrossRef]
- Yuan, C.; Jiao, F.; Zhai, C.; Zhang, J.; Wang, S.; Zhu, L. Application of INSM1 in Diagnosis and Grading of Laryngeal Neu-roendocrine Carcinoma. Laryngoscope 2021, 131, E2662–E2668. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Cen, H.B.; Wei, J.G.; Qin, L.Z.; Liao, W.; Ao, Q.L. Expression of CD200 and INSM1 in gastrointestinal and pancreatic neuroendocrine neoplasms and its diagnostic values. Zhonghua Bing Li Xue Za Zhi 2021, 50, 1134–1138. [Google Scholar] [PubMed]
- Seok, J.Y.; Kang, M.; De Peralta-Venturina, M.; Fan, X. Diagnostic Utility of INSM1 in Medullary Thyroid Carcinoma. Int. J. Surg. Pathol. 2021, 29, 615–626. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kaira, K. Insulinoma-associated protein 1 (INSM1) expression in breast carcinomas with neuroendocrine mor-phologies: Application and future prospective. Virchows Arch. 2021, 479, 191–194. [Google Scholar] [CrossRef]
- Sakakibara, R.; Kobayashi, M.; Takahashi, N.; Inamura, K.; Ninomiya, H.; Wakejima, R.; Kitazono, S.; Yanagitani, N.; Horiike, A.; Ichinose, J.; et al. Insulinoma-associated Protein 1 (INSM1) Is a Better Marker for the Diagnosis and Prognosis Estimation of Small Cell Lung Carcinoma Than Neuroendocrine Phenotype Markers Such as Chromogranin A, Synaptophysin, and CD56. Am. J. Surg. Pathol. 2020, 44, 757–764. [Google Scholar] [CrossRef]
- Wang, M.; Abi-Raad, R.; Baldassarri, R.; Adeniran, A.J.; Cai, G. Expression of insulinoma-associated protein 1 in non–small cell lung cancers: A diagnostic pitfall for neuroendocrine tumors. Hum. Pathol. 2021, 115, 104–111. [Google Scholar] [CrossRef]
- Tsai, H.K.; Hornick, J.L.; Vivero, M. INSM1 expression in a subset of thoracic malignancies and small round cell tumors: Rare potential pitfalls for small cell carcinoma. Mod. Pathol. 2020, 33, 1571–1580. [Google Scholar] [CrossRef]
- Warmke, L.M.; Tinkham, E.G.; Ingram, D.R.; Lazar, A.J.; Panse, G.; Wang, W.-L. INSM1 Expression in Angiosarcoma. Am. J. Clin. Pathol. 2020, 155, 575–580. [Google Scholar] [CrossRef]
- Yoshida, A.; Makise, N.; Wakai, S.; Kawai, A.; Hiraoka, N. INSM1 expression and its diagnostic significance in extraskeletal myxoid chondrosarcoma. Mod. Pathol. 2018, 31, 744–752. [Google Scholar] [CrossRef] [Green Version]
- Juhlin, C.C. Second-Generation Neuroendocrine Immunohistochemical Markers: Reflections from Clinical Implementation. Biology 2021, 10, 874. [Google Scholar] [CrossRef] [PubMed]
- Juhlin, C.C.; Zedenius, J.; Höög, A. Clinical Routine Application of the Second-generation Neuroendocrine Markers ISL1, INSM1, and Secretagogin in Neuroendocrine Neoplasia: Staining Outcomes and Potential Clues for Determining Tumor Origin. Endocr. Pathol. 2020, 31, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Selberherr, A.; Koperek, O.; Riss, P.; Scheuba, C.; Kaderli, R.; Perren, A.; Niederle, B. Neuroendocrine Liver Metastasis-a Spe-cific Set of Markers to Detect Primary Tumor Sites. Endocr. Pathol. 2019, 30, 31–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, J.E.; Thompson, K.; Kilgore, M.R.; Westerhoff, M.; Murphy, C.E.; Papanicolau-Sengos, A.; McCormick, K.A.; Shankaran, V.; Vandeven, N.; Miller, F.; et al. CD200 Expression in Neuroendocrine Neoplasms. Am. J. Clin. Pathol. 2017, 148, 236–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soga, J. The life of S. Oberndorfer: The proposer of the term “carcinoid”—The outcome of a seed in the past 100 years. Nihon Rinsho. Jpn. J. Clin. Med. 2009, 67, 2201–2206. [Google Scholar]
- Williams, E.D.; Sandler, M. The classification of carcinoid tumours. Lancet 1963, 1, 238–239. [Google Scholar] [CrossRef]
- Arrigoni, M.G.; Woolner, L.B.; Bernatz, P.E. Atypical carcinoid tumors of the lung. J. Thorac. Cardiovasc. Surg. 1972, 64, 413–421. [Google Scholar] [CrossRef]
- Creutzfeldt, W. Carcinoid Tumors: Development of Our Knowledge. World J. Surg. 1996, 20, 126–131. [Google Scholar] [CrossRef]
- Klöppel, G. Classification and pathology of gastroenteropancreatic neuroendocrine neoplasms. Endocr.-Relat. Cancer 2011, 18, S1–S16. [Google Scholar] [CrossRef] [PubMed]
- Marchiò, C.; Gatti, G.; Massa, F.; Bertero, L.; Filosso, P.; Pelosi, G.; Cassoni, P.; Volante, M.; Papotti, M. Distinctive pathological and clinical features of lung carcinoids with high proliferation index. Virchows Arch. 2017, 471, 713–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasajima, A.; Konukiewitz, B.; Oka, N.; Suzuki, H.; Sakurada, A.; Okada, Y.; Kameya, T.; Ishikawa, Y.; Sasano, H.; Weichert, W.; et al. Clinicopathological Profiling of Lung Carcinoids with a Ki67 Index > 20%. Neuroendocrinology 2018, 108, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Mete, O.; Asa, S.L.; Gill, A.J.; Kimuta, N.; de Krijger, R.; Tischler, A. Overview of the 2022 WHO Classification of Paragan-gliomas and Pheochromocytomas. Endocr. Pathol. 2022, 33, 90–114. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.H.; Untch, B.R.; Reidy, D.L.; O’Reilly, E.; Dhall, D.; Jih, L.; Basturk, O.; Allen, P.J.; Klimstra, D.S. Well-Differentiated Neuroendocrine Tumors with a Morphologically Apparent High-Grade Component: A Pathway Distinct from Poorly Dif-ferentiated Neuroendocrine Carcinomas. Clin. Cancer Res. 2016, 22, 1011–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofland, J.; Kaltsas, G.; de Herder, W.W. Advances in the Diagnosis and Management of Well-Differentiated Neuroendocrine Neoplasms. Endocr. Rev. 2020, 41, 371–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uccella, S.; La Rosa, S.; Metovic, J.; Marchiori, D.; Scoazec, J.Y.; Volante, M.; Mete, O.; Papotti, M. Genomics of High-Grade Neuroendocrine Neoplasms: Well-Differentiated Neuroendocrine Tumor with High-Grade Features (G3 NET) and Neuro-endocrine Carcinomas (NEC) of Various Anatomic Sites. Endocr. Pathol. 2021, 32, 192–210. [Google Scholar] [CrossRef]
- La Rosa, S. Challenges in High-grade Neuroendocrine Neoplasms and Mixed Neuroendocrine/Non-neuroendocrine Neo-plasms. Endocr. Pathol. 2021, 32, 245–257. [Google Scholar] [CrossRef]
- Heetfeld, M.; Chougnet, C.N.; Olsen, I.H.; Rinke, A.; Borbath, I.; Crespo, G.; Barriuso, J.; Pavel, M.; O’Toole, D.; Walter, T.; et al. Characteristics and treatment of patients with G3 gastroenteropancreatic neuroendo-crine neoplasms. Endocr. Relat. Cancer 2015, 22, 657–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vélayoudom-Céphise, F.L.; Duvillard, P.; Foucan, L.; Hadoux, J.; Chougnet, C.N.; Leboulleux, S.; Malka, D.; Guigay, J.; Goere, D.; Debaere, T.; et al. Are G3 ENETS neuroendocrine neoplasms heterogeneous? Endocr. Relat. Cancer 2013, 20, 649–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milione, M.; Maisonneuve, P.; Spada, F.; Pellegrinelli, A.; Spaggiari, P.; Albarello, L.; Pisa, E.; Barberis, M.; Vanoli, A.; Buzzoni, R.; et al. The Clinicopathologic Heterogeneity of Grade 3 Gastroenteropancreatic Neuroendocrine Neoplasms: Morphological Differentiation and Proliferation Identify Dif-ferent Prognostic Categories. Neuroendocrinology 2017, 104, 85–93. [Google Scholar] [CrossRef]
- Pellat, A.; Coriat, R. Well Differentiated Grade 3 Neuroendocrine Tumors of the Digestive Tract: A Narrative Review. J. Clin. Med. 2020, 9, 1677. [Google Scholar] [CrossRef] [PubMed]
- Dromain, C.; Prior, J.O.; Schaefer, N. Functional and radiological imaging of neuroendocrine neoplasms. In The Spectrum of Neuroendocrine Neoplasia; Asa, S.L., La Rosa, S., Mete, O., Eds.; Spinder Nature: Cham, Switzerland, 2021; pp. 29–54. [Google Scholar]
- Asa, S.L.; La Rosa, S.; Basturk, O.; Adsay, V.; Minnetti, M.; Grossman, A.B. Molecular Pathology of Well-Differentiated Gas-tro-entero-pancreatic Neuroendocrine Tumors. Endocr. Pathol. 2021, 32, 169–191. [Google Scholar] [CrossRef] [PubMed]
- Duan, K.; Mete, O. Algorithmic approach to neuroendocrine tumors in targeted biopsies: Practical applications of immuno-histochemical markers. Cancer Cytopathol. 2016, 124, 871–884. [Google Scholar] [CrossRef] [Green Version]
- De Mestier, L.; Cros, J.; Neuzillet, C.; Hentic, O.; Egal, A.; Muller, N.; Bouché, O.; Cadiot, G.; Ruszniewski, P.; Couvelard, A.; et al. Digestive System Mixed Neuroendocrine-Non-Neuroendocrine Neoplasms. Neuroendocrinology 2017, 105, 412–425. [Google Scholar] [CrossRef]
- Milione, M.; Maisonneuve, P.; Pellegrinelli, A.; Grillo, F.; Albarello, L.; Spaggiari, P.; Vanoli, A.; Tagliabue, G.; Pisa, E.; Messerini, L.; et al. Ki67 proliferative index of the neuroendocrine component drives MANEC prognosis. Endocr.-Relat. Cancer 2018, 25, 583–593. [Google Scholar] [CrossRef] [Green Version]
- Milione, M.; Maisonneuve, P.; Grillo, F.; Mangogna, A.; Centonze, G.; Prinzi, N.; Pusceddu, S.; Garzone, G.; Cattaneo, L.; Busico, A.; et al. Ki-67 Index of 55% Distinguishes Two Groups of Bronchopulmonary Pure and Composite Large Cell Neuroendocrine Carcinomas with Distinct Prognosis. Neuroendocrinology 2020, 111, 475–489. [Google Scholar] [CrossRef]
- WHO Classification of Tumours Editorial Board. Thoracic Tumours, 5th ed.; WHO Classification of Tumours Series; International Agency for Research on Cancer: Lyon, France, 2021; Volume 5.
- Travis, W.D.B.E.; Burke, A.P.; Marx, A.; Nicholson, A.G. WHO Classification of Tumours of the Lung, Pleura, Thymus and Heart, 4th ed.; Bosman, F.T.J.E., Lakhani, S.R., Ohgaki, H., Eds.; International Agency for Research on Cancer: Lyon, France, 2015; p. 412.
- Moonen, L.; Derks, J.L.; Hermans, B.C.; Bunnik, I.M.; Hillen, L.M.; van Suylen, R.J.; Bakker, M.A.D.; von der Thüsen, J.H.; Damhuis, R.A.; Broek, E.C.V.D.; et al. Preoperative Biopsy Diagnosis in Pulmonary Carcinoids, a Shot in the Dark. J. Thorac. Oncol. 2020, 16, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Marchevsky, A.M.; Hendifar, A.; Walts, A.E. The use of Ki-67 labeling index to grade pulmonary well-differentiated neuro-endocrine neoplasms: Current best evidence. Mod. Pathol. 2018, 31, 1523–1531. [Google Scholar] [CrossRef]
- Dermawan, J.K.; Farver, C.F. The Role of Histologic Grading and Ki-67 Index in Predicting Outcomes in Pulmonary Carcinoid Tumors. Am. J. Surg. Pathol. 2019, 44, 224–231. [Google Scholar] [CrossRef]
- Swarts, D.R.A.; Rudelius, M.; Claessen, S.M.H.; Cleutjens, J.P.; Seidl, S.; Volante, M.; Ramaekers, F.C.S.; Speel, E.J.M. Limited additive value of the Ki-67 proliferative index on patient survival in World Health Organization-classified pulmonary carcinoids. Histopathology 2016, 70, 412–422. [Google Scholar] [CrossRef] [PubMed]
- Hermans, B.; Derks, J.; Moonen, L.; Habraken, C.; von der Thüsen, J.; Hillen, L.; Speel, E.; Dingemans, A.-M. Pulmonary neuroendocrine neoplasms with well differentiated morphology and high proliferative activity: Illustrated by a case series and review of the literature. Lung Cancer 2020, 150, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Quinn, A.M.; Chaturvedi, A.; Nonaka, D. High-grade Neuroendocrine Carcinoma of the Lung with Carcinoid Morphology: A Study of 12 Cases. Am. J. Surg. Pathol. 2017, 41, 263–270. [Google Scholar] [CrossRef]
- Rekhtman, N.; Desmeules, P.; Litvak, A.M.; Pietanza, M.C.; Santos-Zabala, M.L.; Ni, A.; Montecalvo, J.; Chang, J.C.; Beras, A.; Preeshagul, I.R.; et al. Stage IV lung carcinoids: Spectrum and evolution of proliferation rate, focusing on variants with elevated proliferation indices. Mod. Pathol. 2019, 32, 1106–1122. [Google Scholar] [CrossRef] [PubMed]
- Clay, V.; Papaxoinis, G.; Sanderson, B.; Valle, J.W.; Howell, M.; Lamarca, A.; Krysiak, P.; Bishop, P.; Nonaka, D.; Mansoor, W. Evaluation of diagnostic and prognostic significance of Ki-67 index in pulmonary carcinoid tumours. Clin. Transl. Oncol. 2016, 19, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Thunnissen, E.; Borczuk, A.C.; Flieder, D.B.; Witte, B.; Beasley, M.B.; Chung, J.H.; Dacic, S.; Lantuejoul, S.; Russell, P.A.; den Bakker, M.; et al. The Use of Immunohistochemistry Improves the Diagnosis of Small Cell Lung Cancer and Its Differential Diag-nosis. An International Reproducibility Study in a Demanding Set of Cases. J. Thorac. Oncol. 2017, 12, 334–346. [Google Scholar] [CrossRef] [Green Version]
- Rooper, L.; Sharma, R.; Li, Q.K.; Illei, P.B.; Westra, W.H. INSM1 Demonstrates Superior Performance to the Individual and Combined Use of Synaptophysin, Chromogranin and CD56 for Diagnosing Neuroendocrine Tumors of the Thoracic Cavity. Am. J. Surg. Pathol. 2017, 41, 1561–1569. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Dermawan, J.K.; Lanigan, C.P.; Farver, C.F. Insulinoma-associated protein 1 (INSM1) is a sensitive and highly specific marker of neuroendocrine differentiation in primary lung neoplasms: An immunohistochemical study of 345 cases, including 292 whole-tissue sections. Mod. Pathol. 2018, 32, 100–109. [Google Scholar] [CrossRef]
- Baine, M.K.; Hsieh, M.-S.; Lai, W.V.; Egger, J.V.; Jungbluth, A.A.; Daneshbod, Y.; Beras, A.; Spencer, R.; Lopardo, J.; Bodd, F.; et al. SCLC Subtypes Defined by ASCL1, NEUROD1, POU2F3, and YAP1: A Comprehensive Immunohistochemical and Histopathologic Characterization. J. Thorac. Oncol. 2020, 15, 1823–1835. [Google Scholar] [CrossRef]
- Huang, Y.-H.; Klingbeil, O.; He, X.; Wu, X.S.; Arun, G.; Lu, B.; Somerville, T.D.; Milazzo, J.P.; Wilkinson, J.E.; Demerdash, O.E.; et al. POU2F3 is a master regulator of a tuft cell-like variant of small cell lung cancer. Genes Dev. 2018, 32, 915–928. [Google Scholar] [CrossRef] [Green Version]
- Nagtegaal, I.D.; Odze, R.D.; Klimstra, D.; Paradis, V.; Rugge, M.; Schirmacher, P.; Washington, K.M.; Carneiro, F.; Cree, I.A. WHO Classification of Tumours Editorial Board The 2019 WHO Classification of Tumours of the Digestive System. Histopathology 2020, 76, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; An, S.; Lee, K.; Ahn, S.; Park, D.Y.; Kim, J.H.; Kang, D.W.; Kim, M.J.; Chang, M.S.; Jung, E.S.; et al. Gastrointestinal Pathology Study Group of the Korean Society of Pathologists. Pancreatic High-Grade Neuroendocrine Neoplasms in the Korean Population: A Multi-center Study. Cancer Res. Treat. 2020, 52, 263–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raj, N.; Valentino, E.; Capanu, M.; Tang, L.H.; Basturk, O.; Untch, B.R.; Allen, P.J.; Klimstra, D.S.; Reidy-Lagunes, D. Treat-ment Response and Outcomes of Grade 3 Pancreatic Neuroendocrine Neoplasms Based on Morphology: Well Differentiated Versus Poorly Differentiated. Pancreas 2017, 46, 296–301. [Google Scholar] [CrossRef]
- Inzani, F.; Rindi, G. Introduction to neuroendocrine neoplasms of the digestive system: Definition and classification. Pathologica 2021, 113, 1–4. [Google Scholar] [CrossRef]
- Fang, J.M.; Li, J.; Shi, J. An update on the diagnosis of gastroenteropancreatic neuroendocrine neoplasms. World J. Gastroenterol. 2022, 28, 1009–1023. [Google Scholar] [CrossRef] [PubMed]
- Rindi, G.; Luinetti, O.; Cornaggia, M.; Capella, C.; Solcia, E. Three subtypes of gastric argyrophil carcinoid and the gastric neuroendocrine carcinoma: A clinicopathologic study. Gastroenterology 1993, 104, 994–1006. [Google Scholar] [CrossRef]
- Köseoğlu, H.; Duzenli, T.; Sezikli, M. Gastric neuroendocrine neoplasms: A review. World J. Clin. Cases 2021, 9, 7973–7985. [Google Scholar] [CrossRef]
- Tsolakis, A.V.; Ragkousi, A.; Vujasinovic, M.; Kaltsas, G.; Daskalakis, K. Gastric neuroendocrine neoplasms type 1: A systematic review and meta-analysis. World J. Gastroenterol. 2019, 25, 5376–5387. [Google Scholar] [CrossRef]
- Delle Fave, G.; O’Toole, D.; Sundin, A.; Taal, B.; Ferolla, P.; Ramage, J.; Ferone, D.; Ito, T.; Weber, W.; Zheng-Pei, Z.; et al. ENETS Consensus Guidelines Update for Gastroduodenal Neuroendocrine Neoplasms. Neuroendocrinology 2016, 103, 119–124. [Google Scholar] [CrossRef] [Green Version]
- Panzuto, F.; Campana, D.; Massironi, S.; Faggiano, A.; Rinzivillo, M.; Lamberti, G.; Sciola, V.; Lahner, E.; Manuzzi, L.; Colao, A.; et al. Tumour type and size are prognostic factors in gastric neuroendocrine neoplasia: A multicentre retrospective study. Dig. Liver Dis. 2019, 51, 1456–1460. [Google Scholar] [CrossRef]
- Ahmed, M. Gastrointestinal neuroendocrine tumors in 2020. World J. Gastrointest. Oncol. 2020, 12, 791–807. [Google Scholar] [CrossRef] [PubMed]
- Milione, M.; Parente, P.; Grillo, F.; Zamboni, G.; Mastracci, L.; Capella, C.; Fassan, M.; Vanoli, A. Neuroendocrine neoplasms of the duodenum, ampullary region, jejunum and ileum. Pathologica 2021, 113, 12–18. [Google Scholar] [CrossRef]
- Vanoli, A.; La Rosa, S.; Klersy, C.; Grillo, F.; Albarello, L.; Inzani, F.; Maragliano, R.; Manca, R.; Luinetti, O.; Milione, M.; et al. Four Neuroendocrine Tumor Types and Neuroendocrine Carcinoma of the Duodenum: Analysis of 203 Cases. Neuroendocrinology 2016, 104, 112–125. [Google Scholar] [CrossRef]
- Assarzadegan, N.; Montgomery, E. What is New in the 2019 World Health Organization (WHO) Classification of Tumors of the Digestive System: Review of Selected Updates on Neuroendocrine Neoplasms, Appendiceal Tumors, and Molecular Testing. Arch. Pathol. Lab. Med. 2020, 145, 664–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hrabe, J. Neuroendocrine Tumors of the Appendix, Colon, and Rectum. Surg. Oncol. Clin. N. Am. 2020, 29, 267–279. [Google Scholar] [CrossRef]
- Gut, P.; Waligórska-Stachura, J.; Czarnywojtek, A.; Sawicka-Gutaj, N.; Bączyk, M.; Ziemnicka, K.; Woliński, K.; Zybek, A.; Fischbach, J.; Ruchała, M. Hindgut neuroendocrine neoplasms—Characteristics and prognosis. Arch. Med. Sci. 2017, 13, 1427–1432. [Google Scholar] [CrossRef] [Green Version]
- Pasternak, S.; Carter, M.D.; Ly, T.Y.; Doucette, S.; Walsh, N.M. Immunohistochemical profiles of different subsets of Merkel cell carcinoma. Hum. Pathol. 2018, 82, 232–238. [Google Scholar] [CrossRef] [PubMed]
- WHO Classification of Tumours Editorial Board. Head and Neck Tumours, 5th ed.; WHO Classification of Tumours Series; International Agency for Research on Cancer: Lyon, France, 2022; Volume 9.
- Asa, S.L.; Mete, O. Endocrine pathology: Past, present and future. Pathology 2017, 50, 111–118. [Google Scholar] [CrossRef]
- WHO Classification of Tumours Editorial Board. Endocrine and Neuroendocrine Tumours, 5th ed.; WHO Classification of Tumours Series; International Agency for Research on Cancer: Lyon, France, 2022; Volume 10.
- WHO Classification of Tumours Editorial Board. Breast Tumours, 5th ed.; WHO Classification of Tumours Series; International Agency for Research on Cancer: Lyon, France, 2019; Volume 2.
- WHO Classification of Tumours Editorial Board. Female Genital Tumours, 5th ed.; WHO Classification of Tumours Series; International Agency for Research on Cancer: Lyon, France, 2020; Volume 4.
- WHO Classification of Tumours Editorial Board. Urinary and Male Genital Tumours, 5th ed.; WHO Classification of Tumours Series; International Agency for Research on Cancer: Lyon, France, 2022; Volume 8.
- Shehabeldin, A.N.; Ro, J.Y. Neuroendocrine tumors of genitourinary tract: Recent advances. Ann. Diagn. Pathol. 2019, 42, 48–58. [Google Scholar] [CrossRef]
- Zaffuto, E.; Pompe, R.S.; Zanaty, M.; Bondarenko, H.D.; Leyh-Bannurah, S.-R.; Moschini, M.; Dell’Oglio, P.; Gandaglia, G.; Fossati, N.; Stabile, A.; et al. Contemporary Incidence and Cancer Control Outcomes of Primary Neuroendocrine Prostate Cancer: A SEER Database Analysis. Clin. Genitourin. Cancer 2017, 15, e793–e800. [Google Scholar] [CrossRef]
- Akamatsu, S.; Inoue, T.; Ogawa, O.; Gleave, M.E. Clinical and molecular features of treatment-related neuroendocrine prostate cancer. Int. J. Urol. 2018, 25, 345–351. [Google Scholar] [CrossRef] [PubMed]
- WHO Classification of Tumours Editorial Board. Skin Tumours, 4th ed.; WHO Classification of Tumours Series; International Agency for Research on Cancer: Lyon, France, 2018.
- Goto, K.; Anan, T.; Nakatsuka, T.; Kaku, Y.; Sakurai, T.; Fukumoto, T.; Kimura, T.; Shibata, A. Low-Grade Neuroendocrine Carcinoma of the Skin (Primary Cutaneous Carcinoid Tumor) as a Distinctive Entity of Cutaneous Neuroendocrine Tumors: A Clinicopathologic Study of 3 Cases With Literature Review. Am. J. Dermatopathol. 2017, 39, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Asa, S.L.; Mete, O.; Ezzat, S. Genomics and Epigenomics of Pituitary Tumors: What Do Pathologists Need to Know? Endocr. Pathol. 2021, 32, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Mete, O.; Asa, S.L. Structure, Function, and Morphology in the Classification of Pituitary Neuroendocrine Tumors: The Im-portance of Routine Analysis of Pituitary Transcription Factors. Endocr. Pathol. 2020, 31, 330–336. [Google Scholar] [CrossRef]
- Mete, O.; Alshaikh, O.M.; Cintosun, A.; Ezzat, S.; Asa, S.L. Synchronous Multiple Pituitary Neuroendocrine Tumors of Dif-ferent Cell Lineages. Endocr. Pathol. 2018, 29, 332–338. [Google Scholar] [CrossRef]
- Asa, S.L.; Mete, O. Cytokeratin profiles in pituitary neuroendocrine tumors. Hum. Pathol. 2020, 107, 87–95. [Google Scholar] [CrossRef]
- Grimm, F.; Maurus, R.; Beschorner, R.; Naros, G.; Stanojevic, M.; Gugel, I.; Giese, S.; Bier, G.; Bender, B.; Honegger, J. Ki-67 labeling index and expression of p53 are non-predictive for invasiveness and tumor size in functional and nonfunctional pi-tuitary adenomas. Acta Neurochir. 2019, 161, 1149–1156. [Google Scholar] [CrossRef]
- Asioli, S.; Righi, A.; Iommi, M.; Baldovini, C.; Ambrosi, F.; Guaraldi, F.; Zoli, M.; Mazzatenta, D.; Faustini-Fustini, M.; Rucci, P.; et al. Validation of a clinicopathological score for the prediction of post-surgical evolution of pituitary adenoma: Retrospective analysis on 566 patients from a tertiary care centre. Eur. J. Endocrinol. 2019, 180, 127–134. [Google Scholar] [CrossRef] [Green Version]
- Lenders, N.F.; Wilkinson, A.C.; Wong, S.J.; Shein, T.T.; Harvey, R.J.; Inder, W.J.; Earls, P.E.; McCormack, A.I. Transcription factor immunohistochemistry in the diagnosis of pituitary tumours. Eur. J. Endocrinol. 2021, 184, 891–901. [Google Scholar] [CrossRef]
- Cortez, G.M.; Monteiro, A.; Agnoletto, G.; Bit-Ivan, E.N.; Sauvageau, E.; Hanel, R.A. Aggressive Pituitary Tumor with Crooke’s Cells and Invasion of the Posterior Fossa. World Neurosurg. 2020, 138, 530–534.e1. [Google Scholar] [CrossRef]
- Dogansen, S.C.; Bilgic, B.; Yalin, G.Y.; Tanrikulu, S.; Yarman, S. Clinical significance of granulation pattern in corticotroph pituitary adenomas. Turk. J. Pathol. 2018, 35, 9–14. [Google Scholar] [CrossRef]
- Rak, B.; Maksymowicz, M.; Pękul, M.; Zieliński, G. Clinical, Biological, Radiological Pathological and Immediate Post-Operative Remission of Sparsely and Densely Granulated Corticotroph Pituitary Tumors: A Retrospective Study of a Cohort of 277 Pa-tients with Cushing’s Disease. Front. Endocrinol. 2021, 12, 672178. [Google Scholar] [CrossRef]
- Almeida, J.P.; Stephens, C.C.; Eschbacher, J.M.; Felicella, M.M.; Yuen, K.C.J.; White, W.L.; Mooney, M.A.; Bernat, A.L.; Mete, O.; Zadeh, G.; et al. Clinical, pathologic, and imaging characteristics of pituitary null cell adenomas as defined according to the 2017 World Health Organization criteria: A case series from two pituitary centers. Pituitary 2019, 22, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Haddad, A.F.; Young, J.S.; Oh, T.; Pereira, M.P.; Joshi, R.S.; Pereira, K.M.; Osorio, R.C.; Donohue, K.C.; Peeran, Z.; Sudhir, S.; et al. Clinical characteristics and outcomes of null-cell versus silent gonadotroph adenomas in a series of 1166 pituitary adenomas from a single institution. Neurosurg. Focus 2020, 48, E13. [Google Scholar] [CrossRef]
- Pierre, C.; Agopiantz, M.; Brunaud, L.; Battaglia-Hsu, S.-F.; Max, A.; Pouget, C.; Nomine, C.; Lomazzi, S.; Vignaud, J.-M.; Weryha, G.; et al. COPPS, a composite score integrating pathological features, PS100 and SDHB losses, predicts the risk of metastasis and progression-free survival in pheochromocytomas/paragangliomas. Virchows Arch. 2019, 474, 721–734. [Google Scholar] [CrossRef]
- Thompson, L.D.R.; Gill, A.J.; Asa, S.L.; Clifton-Bligh, R.J.; de Krijger, R.R.; Kimura, N.; Komminoth, P.; Lack, E.E.; Lenders, J.W.M.; Lloyd, R.V.; et al. Data set for the reporting of pheochromocytoma and paraganglioma: Explanations and recommendations of the guidelines from the International Collaboration on Cancer Re-porting. Hum. Pathol. 2021, 110, 83–97. [Google Scholar] [CrossRef]
- Crona, J.; Lamarca, A.; Ghosal, S.; Welin, S.; Skogseid, B.; Pacak, K. Genotype–phenotype correlations in pheochromocytoma and paraganglioma: A systematic review and individual patient meta-analysis. Endocr.-Relat. Cancer 2019, 26, 539–550. [Google Scholar] [CrossRef]
- Hempenstall, L.E.; Siriwardana, A.R.; Desai, D.J. Investigation of a renal mass: Diagnosing renal paraganglioma. Urol. Case Rep. 2018, 21, 8–9. [Google Scholar] [CrossRef]
- Dandpat, S.K.; Rai, S.K.R.; Shah, A.; Goel, N.; Goel, A.H. Silent stellate ganglion paraganglioma masquerading as schwan-noma: A surgical nightmare. J. Craniovertebr. Junction Spine 2020, 11, 240–242. [Google Scholar] [PubMed]
- Ruzevick, J.; Koh, E.K.; Gonzalez-Cuyar, L.F.; Cimino, P.J.; Moe, K.; Wright, L.A.; Failor, R.; Ferreira, M. Clival paragangliomas: A report of two cases involving the midline skull base and review of the literature. J. Neuro-Oncol. 2017, 132, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.D. Paragangliomas of the Head and Neck: An Overview from Diagnosis to Genetics. Head Neck Pathol. 2017, 11, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Dermawan, J.K.; Mukhopadhyay, S.; Shah, A.A. Frequency and extent of cytokeratin expression in paraganglioma: An im-munohistochemical study of 60 cases from 5 anatomic sites and review of the literature. Hum. Pathol. 2019, 93, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.Y.; Coffey, M.; Mansur, D.; Wasman, J.; Asa, S.L.; Couce, M. Images in Endocrine Pathology: Progressive Loss of Sustentacular Cells in a Case of Recurrent Jugulotympanic Paraganglioma over a Span of 5 years. Endocr. Pathol. 2020, 31, 310–314. [Google Scholar] [CrossRef]
- Chatterjee, U.; Mukherjee, S.; Mondal, A.; Sengupta, M.; Sarkar, D.; Mukhopadhyay, S. Composite phaeochromocytoma with malignant peripheral nerve sheath tumour: A case report with summary of prior published cases. Indian J. Pathol. Microbiol. 2021, 64, 571. [Google Scholar] [CrossRef]
- Dhanasekar, K.; Visakan, V.; Tahir, F.; Balasubramanian, S.P. Composite phaeochromocytomas—A systematic review of published literature. Langenbeck’s Arch. Surg. 2021, 407, 517–527. [Google Scholar] [CrossRef]
- Chen, J.; Wu, Y.; Wang, P.; Wu, H.; Tong, A.; Chang, X. Composite pheochromocytoma/paraganglioma-ganglioneuroma: Analysis of SDH and ATRX status, and identification of frequent HRAS and BRAF mutations. Endocr. Connect. 2021, 10, 926–934. [Google Scholar] [CrossRef]


{kind=link}
| Histopathological Features | NETs | NECs |
|---|---|---|
| Architecture | “Organoid” proliferation with sporadic nests, trabecular patterns, and glandular or rosette development | Solid proliferation of less monomorphic cells with small-cells or large-cells |
| Other features | Usually, scant stroma with prominent blood vessels Occasionally, cell palisading at the periphery of the nests, amyloid stroma, calcifications with psammoma bodies | Areas of necrosis, apoptotic bodies |
| NENs, especially NECs, can exhibit a non-neuroendocrine component with various differentiation, such as glandular or squamous | ||
| Cell features | Usually epithelioid and closed-packed, rarely spindle and dis-cohesive cells Medium-sized cells with abundant cytoplasm and uniform round to oval nuclei with fine to coarsely granular chromatin and inconspicuous nucleoli (classic “salt and pepper” aspect) | Small-cell: scant cytoplasm, irregular, hyperchromatic nuclei with “salt and pepper” chromatin and severe nuclear molding Large-cell: abundant cytoplasm, vesicular nuclei with conspicuous nucleolus, possibly large and intensely eosinophilic |
| Mitotic figures | Absent or rare | Frequent |
| Neuroendocrine Neoplasm | Classification | Defining Criteria |
|---|---|---|
| Lung and Thymus | ||
| Well-differentiated neuroendocrine tumor (NET) | Typical carcinoid/NET G1 | <2 mitoses/2 mm2 and no necrosis |
| Atypical carcinoid/NET G2 | 2–10 mitoses/2 mm2 and/or necrosis (usually punctate) | |
| Carcinoid/NET with elevated mitotic counts and/or elevated Ki67 proliferation Index | Atypical carcinoid morphology and elevated miotic counts (>10 mitoses/2 mm2) and/or Ki67 > 30% | |
| Poorly differentiated neuroendocrine carcinoma (NEC) | Small-cell lung carcinoma | Small-cell morphology and >10 mitoses/2 mm2 |
| Large-cell neuroendocrine lung carcinoma | Large-cell morphology, necrosis always present and >10 mitoses/2 mm2 | |
| Gastrointestinal and pancreatobiliary tract | ||
| Well-differentiated neuroendocrine tumor (NET) | NET, grade 1 | <2 mitoses/2 mm2 and/or Ki67 < 3% |
| NET, grade 2 | 2–20 mitoses/2 mm2 and/or Ki67 3–20% | |
| NET, grade 3 | >20 mitoses/2 mm2 and/or Ki67 > 20% | |
| Poorly differentiated neuroendocrine carcinoma (NEC) | Small-cell neuroendocrine carcinoma | Small-cell morphology and >20 mitoses/2 mm2 and/or Ki67 > 20% (often > 70%) |
| Large-cell neuroendocrine carcinoma | Large-cell morphology and >20 mitoses/2 mm2 and/or Ki67 > 20% (often > 70%) | |
| Upper aerodigestive tract and salivary glands | ||
| Well-differentiated neuroendocrine tumor (NET) | NET, grade 1 | <2 mitoses/2 mm2, Ki67 < 20% and no necrosis |
| NET, grade 2 | 2–10 mitoses/2 mm2, Ki67 < 20% and/or necrosis | |
| NET, grade 3 | >10 mitoses/2 mm2 and/or Ki67 > 20% | |
| Poorly differentiated neuroendocrine carcinoma (NEC) | Small-cell neuroendocrine carcinoma | Small-cell morphology and >10 mitoses/2 mm2 and/or Ki67 > 20% (often > 70%) |
| Large-cell neuroendocrine carcinoma | Large-cell morphology and >20 mitoses/2 mm2 and/or Ki67 > 20% (often > 55%) |
| Chromogranin A, Synaptophysin | Ki67 | Other Neuroendocrine Markers | Other Markers for Differential Diagnosis | |
|---|---|---|---|---|
| Lung NENs | Mandatory for LCNECs May be negative in 15–20% of SCLCs | Desirable to be reported, >30% in carcinomas | CD56, INSM1, and/or POU2F3 | CD45 for lymphoma or p40 for the basaloid version of squamous cell carcinoma |
| Gastrointesntinal&
pancreaticobiliary NENs | Chromogranin A: diffusely and intensely positive in NETs, but focally and weakly positive in NECs | Mandatory for grading NETs | SSTRs: diffusely positive in NETs but mostly absent in NECs | CK20: distinctive dot-like pattern for anal MCCs BCL10 and trypsin: for differentiating pancreatic NECs or G3 NETs from pancreatic acinar cell carcinoma |
| Head&Neck NENs | May be weakly positive in NECs | No clear cut-off value for NENs of the nasal cavity, paranasal sinuses, skull base, larynx, hypopharynx, trachea, and parapharyngeal space; Mandatory for grading MTC | INSM1: superior sensitivity for NECs; Calcitonin expression: a negative prognostic factor in MTC | For NECs: extensive analysis to exclude undifferentiated sinonasal carcinoma, NUT carcinoma, mucosal melanoma, embryonal rhabdomyosarcoma, neuroblastoma |
| Breast NENs | Positive but can also be expressed in other type of breast carcinomas | No clear cut-off value | The distinction between breast NENs and other entities should primarily be based on histological criteria | |
| Female Reproductive System NENs | Positive expression of at least one: mandatory for diagnosis | No clear cut-off value | CD56: favors the diagnosis of NENs only in the presence of clear histopathological criteria | |
| Urinary and Male Genital System NENs | Usually expressed, but not mandatory for diagnosing SCNECs | No clear cut-off value | CD56: low specificity INSM1: superior sensitivity | |
| Skin NENs | Positive but can also be expressed in other skin carcinomas | No clear cut-off value | CK20 and/or NPF: characteristic dot-like pattern in Merkel Cell Carcinoma |
| Tumor Type | Transcription Factor(s) | Hormone(s) | Keratin (CAM 5.2 or CK18) |
|---|---|---|---|
| Somatotroph tumors | PIT1 | GH, α-subunit | Perinuclear |
| GH | Fibrous bodies (>70%) | ||
| Lactotroph tumors | PIT1, ERα | PRL (paranuclear) | Weak or negative |
| PRL (diffuse cytoplasmic) | Weak or negative | ||
| Mammosomatotroph tumor | PIT1, ERα | GH (often predominant) PRL, α-subunit | Perinuclear |
| Thyrotroph tumor | PIT1, GATA2/3 | α-subunit, βTSH | Weak or negative |
| Mature plurihormonal PIT1-lineage tumor | PIT1, ERα, GATA2/3 | GH (often predominant), PRL, α-subunit, βTSH | Perinuclear |
| Acidophil stem cell tumor | PIT1, ERα | PRL (predominant), GH (focal/variable) | Scattered fibrous bodies |
| Immature PIT1-lineage tumor | PIT1, ERα GATA2/3 | GH, PRL, α-subunit, βTSH | Focal/ Variable |
| Corticotroph tumors | TPIT (TBX19), NeuroD1/β2 | ACTH and other POMC derivatives | Strong |
| Variable | |||
| Intense ring-like perinuclear | |||
| Gonadotroph tumor | SF1, ERα GATA2/3 | α-subunit, βFSH, βLH | Variable |
| Unclassified plurihormonal tumors | Multiple combinations | Multiple combinations | Variable |
| Null cell tumor | None | None | Variable |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gheorghișan-Gălățeanu, A.-A.; Ilieșiu, A.; Lambrescu, I.M.; Țăpoi, D.A. The Complex Histopathological and Immunohistochemical Spectrum of Neuroendocrine Tumors—An Overview of the Latest Classifications. Int. J. Mol. Sci. 2023, 24, 1418. https://doi.org/10.3390/ijms24021418
Gheorghișan-Gălățeanu A-A, Ilieșiu A, Lambrescu IM, Țăpoi DA. The Complex Histopathological and Immunohistochemical Spectrum of Neuroendocrine Tumors—An Overview of the Latest Classifications. International Journal of Molecular Sciences. 2023; 24(2):1418. https://doi.org/10.3390/ijms24021418
Chicago/Turabian StyleGheorghișan-Gălățeanu, Ancuța-Augustina, Andreea Ilieșiu, Ioana Maria Lambrescu, and Dana Antonia Țăpoi. 2023. "The Complex Histopathological and Immunohistochemical Spectrum of Neuroendocrine Tumors—An Overview of the Latest Classifications" International Journal of Molecular Sciences 24, no. 2: 1418. https://doi.org/10.3390/ijms24021418
APA StyleGheorghișan-Gălățeanu, A. -A., Ilieșiu, A., Lambrescu, I. M., & Țăpoi, D. A. (2023). The Complex Histopathological and Immunohistochemical Spectrum of Neuroendocrine Tumors—An Overview of the Latest Classifications. International Journal of Molecular Sciences, 24(2), 1418. https://doi.org/10.3390/ijms24021418