

Recent Advances in the Knowledge of the Mechanisms of Leptin Physiology and Actions in Neurological and Metabolic Pathologies

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Regulation of Leptin Efficiency
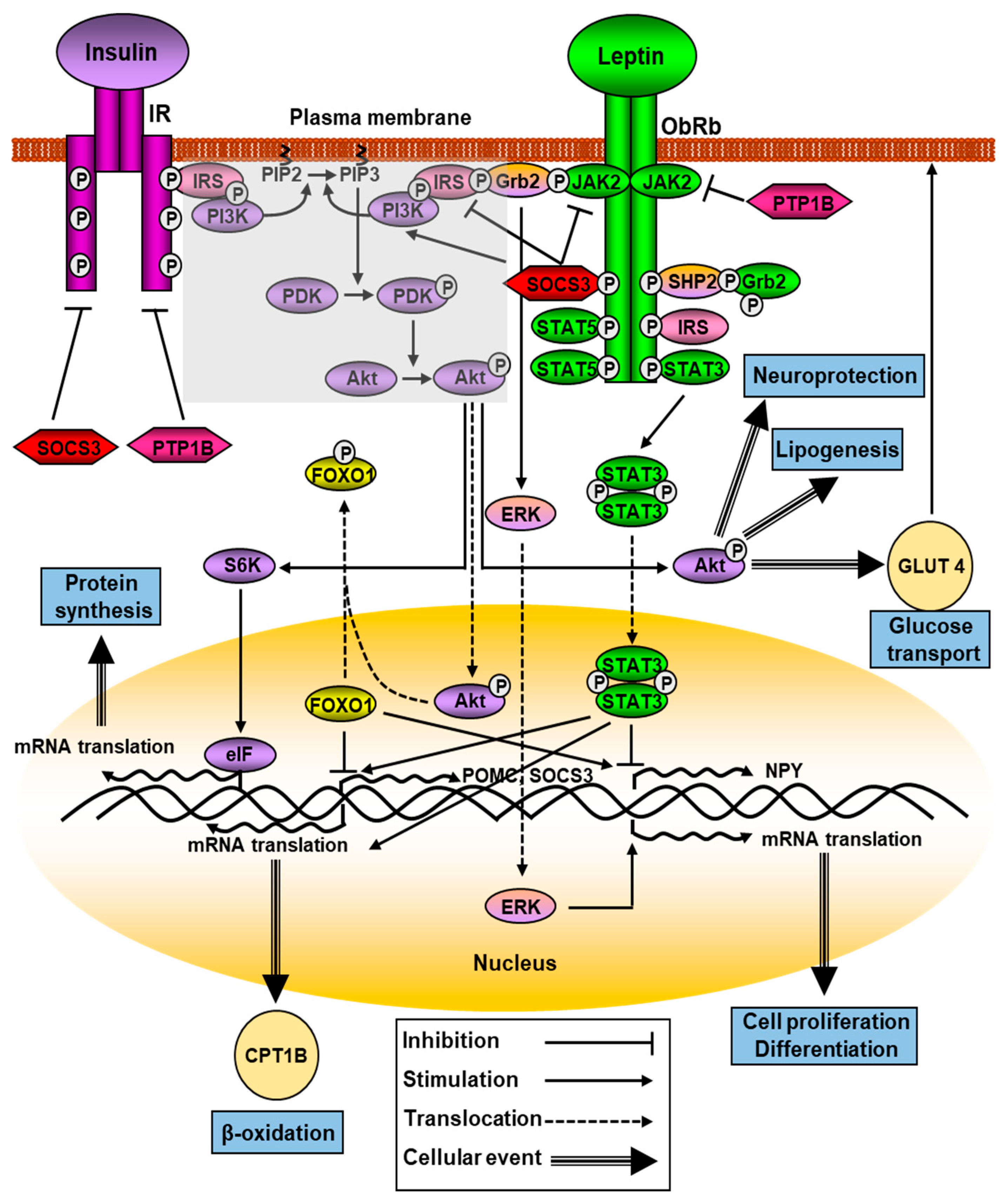
2.1. Leptin Signaling
2.2. Leptin Expression and Secretion
3. Functionality of Leptin and Its Involvement in Pathology
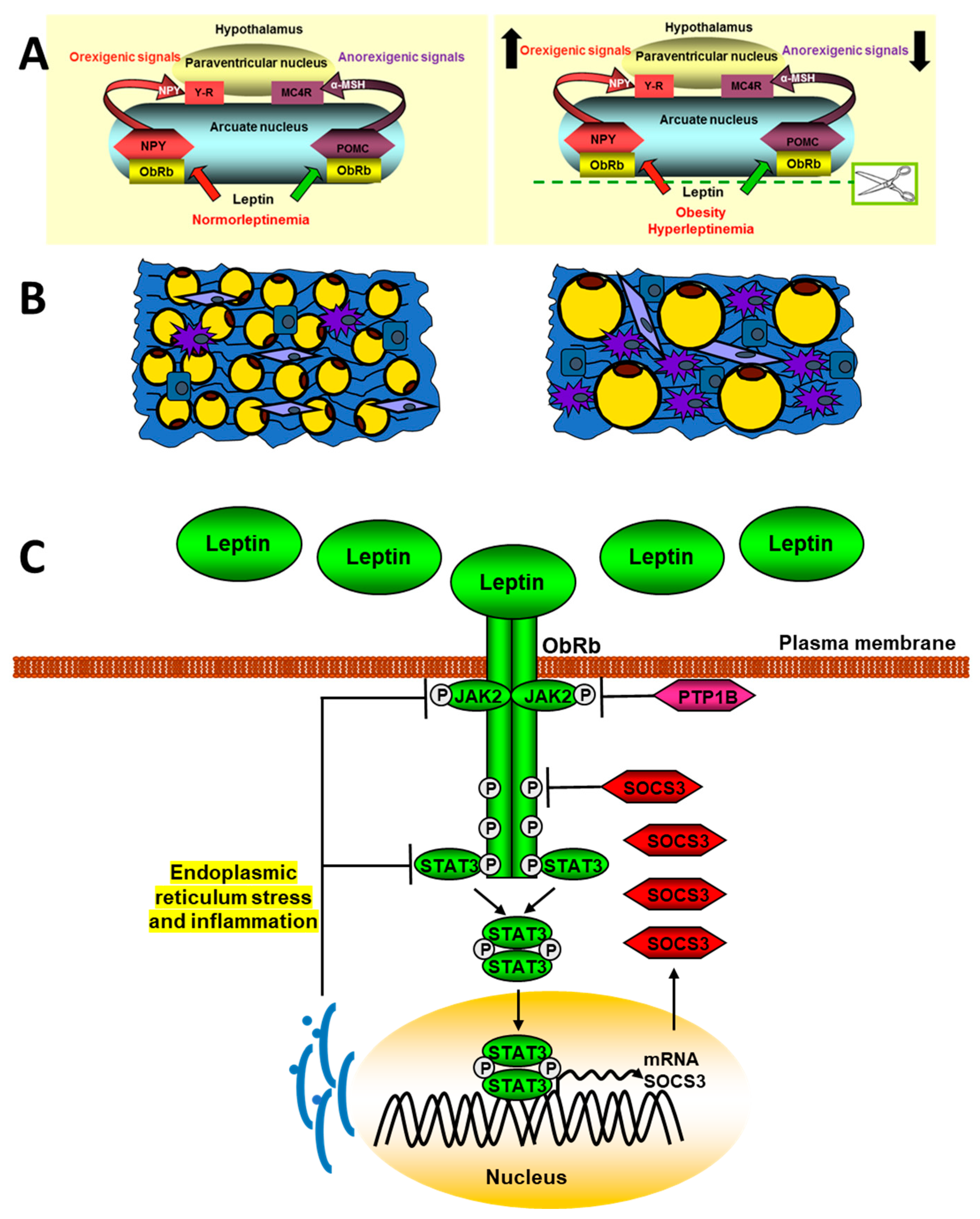
3.1. Leptin-Associated Central Dysfunctions
3.1.1. Central Leptin Resistance and Dysregulation of Energy Homeostasis
3.1.2. Leptin, Neurogenesis, and Neuroprotection
3.1.3. Neurological Diseases
3.2. Leptin Status and Its Implication in Metabolic Pathologies
3.2.1. Obesity and Lipodystrophy
3.2.2. Diabetes
3.2.3. Cancer
3.2.4. Non-Alcoholic Fatty Liver and Steatohepatitis Diseases
3.3. Other Diseases
4. Closing Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Halaas, J.L.; Gajiwala, K.S.; Maffei, M.; Cohen, S.L.; Chait, B.T.; Rabinowitz, D.; Lallone, R.L.; Burley, S.K.; Friedman, J.M. Weight-reducing effects of the plasma protein encoded by the obese gene. Science 1995, 269, 543–546. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.W.; Seeley, R.J.; Campfield, L.A.; Burn, P.; Baskin, D.G. Identification of targets of leptin action in rat hypothalamus. J. Clin. Investig. 1996, 98, 1101–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalra, S.P.; Dube, M.G.; Pu, S.; Xu, B.; Horvath, T.L.; Kalra, P.S. Interacting appetite-regulating pathways in the hypothalamic regulation of body weight. Endocr. Rev. 1999, 20, 68–100. [Google Scholar] [CrossRef] [PubMed]
- King, P.J.; Widdowson, P.S.; Doods, H.; Williams, G. Regulation of neuropeptide Y release from hypothalamic slices by melanocortin-4 agonists and leptin. Peptides 2000, 21, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Mountjoy, K.G.; Mortrud, M.T.; Low, M.J.; Simerly, R.B.; Cone, R.D. Localization of the melanocortin-4 receptor (MC4-R) in neuroendocrine and autonomic control circuits in the brain. Mol. Endocrinol. 1994, 8, 1298–1308. [Google Scholar] [CrossRef] [Green Version]
- Rajcsanyi, L.S.; Zheng, Y.; Fischer-Posovszky, P.; Wabitsch, M.; Hebebrand, J.; Hinney, A. Prevalence estimates of putatively pathogenic leptin variants in the gnomAD database. PLoS ONE 2022, 17, e0266642. [Google Scholar] [CrossRef]
- de Candia, P.; Prattichizzo, F.; Garavelli, S.; Alviggi, C.; La Cava, A.; Matarese, G. The pleiotropic roles of leptin in metabolism, immunity, and cancer. J. Exp. Med. 2021, 218, e20191593. [Google Scholar] [CrossRef]
- Cui, Q.; Zhang, Y.; Tian, N.; Yang, J.; Ya, D.; Xiang, W.; Zhou, Z.; Jiang, Y.; Deng, J.; Yang, B.; et al. Leptin promotes angiogenesis via pericyte STAT3 pathway upon intracerebral hemorrhage. Cells 2022, 11, 2755. [Google Scholar] [CrossRef]
- Cairat, M.; Rinaldi, S.; Navionis, A.S.; Romieu, I.; Biessy, C.; Viallon, V.; Olsen, A.; Tjønneland, A.; Fournier, A.; Severi, G.; et al. Circulating inflammatory biomarkers, adipokines and breast cancer risk-a case-control study nested within the EPIC cohort. BMC Med. 2022, 20, 118. [Google Scholar] [CrossRef]
- Kinoshita, Y.; Arita, S.; Ogawa, T.; Takenouchi, A.; Inagaki-Ohara, K. Augmented leptin-induced trefoil factor 3 expression and epidermal growth factor receptor transactivation differentially influences neoplasia progression in the stomach and colorectum of dietary fat-induced obese mice. Arch. Biochem. Biophys. 2022, 729, 109379. [Google Scholar] [CrossRef]
- Leifheit-Nestler, M.; Wagner, N.M.; Gogiraju, R.; Didié, M.; Konstantinides, S.; Hasenfuss, G.; Schäfer, K. Importance of leptin signaling and signal transducer and activator of transcription-3 activation in mediating the cardiac hypertrophy associated with obesity. J. Transl. Med. 2013, 11, 170. [Google Scholar] [CrossRef] [Green Version]
- Özkan, E.A.; Sadigov, A.; Öztürk, O. Evaluation of serum omentin-1, vaspin, leptin, adiponectin levels in obese/overweight children and their relationship with non-alcoholic fatty liver disease. Clin. Nutr. Res. 2022, 11, 194–203. [Google Scholar] [CrossRef]
- Bukosza, E.N.; Kaucsár, T.; Godó, M.; Lajtár, E.; Tod, P.; Koncsos, G.; Varga, Z.V.; Baranyai, T.; Nguyen, M.T.; Schachner, H.; et al. Glomerular collagen deposition and lipocalin-2 expression are early signs of renal injury in prediabetic obese rats. Int. J. Mol. Sci. 2019, 20, 4266. [Google Scholar] [CrossRef] [Green Version]
- Ranea-Robles, P.; Lund, J.; Clemmensen, C. The physiology of experimental overfeeding in animals. Mol. Metab. 2022, 64, 101573. [Google Scholar] [CrossRef]
- Huang, X.; He, Q.; Zhu, H.; Fang, Z.; Che, L.; Lin, Y.; Xu, S.; Zhuo, Y.; Hua, L.; Wang, J.; et al. Hepatic leptin signaling improves hyperglycemia by stimulating mapk phosphatase-3 protein degradation via STAT3. Cell. Mol. Gastroenterol. Hepatol. 2022, 14, 983–1001. [Google Scholar] [CrossRef]
- Kim, G.H.; Szabo, A.; King, E.M.; Ayala, J.; Ayala, J.E.; Altarejos, J.Y. Leptin recruits Creb-regulated transcriptional coactivator 1 to improve hyperglycemia in insulin-deficient diabetes. Mol. Metab. 2014, 4, 227–236. [Google Scholar] [CrossRef]
- Barrios, V.; Guerra-Cantera, S.; Martín-Rivada, Á.; Canelles, S.; Campillo-Calatayud, A.; Arilla-Ferreiro, E.; Frago, L.M.; Chowen, J.A.; Argente, J. Chronic central leptin infusion promotes an anti-inflammatory cytokine profile related to the activation of insulin signaling in the gastrocnemius of male rats. Biomedicines 2022, 10, 1465. [Google Scholar] [CrossRef]
- Lee, M.J.; Wang, Y.; Ricci, M.R.; Sullivan, S.; Russell, C.D.; Fried, S.K. Acute and chronic regulation of leptin synthesis, storage, and secretion by insulin and dexamethasone in human adipose tissue. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E858–E864. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Chen, Y.; Heiman, M.; Dimarchi, R. Leptin: Structure, function and biology. Vitam. Horm. 2005, 71, 345–372. [Google Scholar] [CrossRef]
- Greco, M.; De Santo, M.; Comandè, A.; Belsito, E.L.; Andò, S.; Liguori, A.; Leggio, A. Leptin-activity modulators and their potential pharmaceutical applications. Biomolecules 2021, 11, 1045. [Google Scholar] [CrossRef]
- Peelman, F.; Iserentant, H.; De Smet, A.S.; Vandekerckhove, J.; Zabeau, L.; Tavernier, J. Mapping of binding site III in the leptin receptor and modeling of a hexameric leptin.leptin receptor complex. J. Biol. Chem. 2006, 281, 15496–15504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wauman, J.; Zabeau, L.; Tavernier, J. The leptin receptor complex: Heavier than expected? Front. Endocrinol. 2017, 8, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, C.; Heyne, H.O.; Heiland, T.; Dommel, S.; Höfling, C.; Guiu-Jurado, E.; Lorenz, J.; Roßner, S.; Dannemann, M.; Kelso, J.; et al. A novel compound heterozygous leptin receptor mutation causes more severe obesity than in Leprdb/db mice. J. Lipid Res. 2021, 62, 100105. [Google Scholar] [CrossRef] [PubMed]
- Clément, K.; Vaisse, C.; Lahlou, N.; Cabrol, S.; Pelloux, V.; Cassuto, D.; Gourmelen, M.; Dina, C.; Chambaz, J.; Lacorte, J.M.; et al. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature 1998, 392, 398–401. [Google Scholar] [CrossRef] [PubMed]
- Lou, P.H.; Yang, G.; Huang, L.; Cui, Y.; Pourbahrami, T.; Radda, G.K.; Li, C.; Han, W. Reduced body weight and increased energy expenditure in transgenic mice over-expressing soluble leptin receptor. PLoS ONE 2010, 5, e11669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glassman, C.R.; Tsutsumi, N.; Saxton, R.A.; Lupardus, P.J.; Jude, K.M.; Garcia, K.C. Structure of a Janus kinase cytokine receptor complex reveals the basis for dimeric activation. Science 2022, 376, 163–169. [Google Scholar] [CrossRef]
- Bjorbak, C.; Lavery, H.J.; Bates, S.H.; Olson, R.K.; Davis, S.M.; Flier, J.S.; Myers, M.G., Jr. SOCS3 mediates feedback inhibition of the leptin receptor via Tyr985. J. Biol. Chem. 2000, 275, 40649–40657. [Google Scholar] [CrossRef] [Green Version]
- Shi, S.Y.; Martin, R.G.; Duncan, R.E.; Choi, D.; Lu, S.Y.; Schroer, S.A.; Cai, E.P.; Luk, C.T.; Hopperton, K.E.; Domenichiello, A.F.; et al. Hepatocyte-specific deletion of Janus kinase 2 (JAK2) protects against diet-induced steatohepatitis and glucose intolerance. J. Biol. Chem. 2012, 287, 10277–10288. [Google Scholar] [CrossRef] [Green Version]
- Rabie, H.; Othman, W.; Elsabaawy, D.M.; Elshemy, E.E.; Abdelmageed, N.; Khalaf, F.A.; Bedair, H.M. Janus kinase-2 mutation associated portal vein thrombosis complicating liver cirrhosis and hepatocellular carcinoma. Asian Pac. J. Cancer Prev. 2021, 22, 267–275. [Google Scholar] [CrossRef]
- Rolles, B.; Mullally, A. Molecular pathogenesis of myeloproliferative neoplasms. Curr. Hematol. Malig. Rep. 2022, 17, 319–329. [Google Scholar] [CrossRef]
- Calabretto, G.; Teramo, A.; Barilà, G.; Vicenzetto, C.; Gasparini, V.R.; Semenzato, G.; Zambello, R. Neutropenia and large granular lymphocyte leukemia: From pathogenesis to therapeutic options. Cells 2021, 10, 2800. [Google Scholar] [CrossRef]
- El-Tanani, M.; Al Khatib, A.O.; Aladwan, S.M.; Abuelhana, A.; McCarron, P.A.; Tambuwala, M.M. Importance of STAT3 signalling in cancer, metastasis and therapeutic interventions. Cell. Signal. 2022, 92, 110275. [Google Scholar] [CrossRef]
- Tsilifis, C.; Freeman, A.F.; Gennery, A.R. STAT3 hyper-IgE syndrome-an update and unanswered questions. J. Clin. Immunol. 2021, 41, 864–880. [Google Scholar] [CrossRef]
- Pedroso, J.A.B.; Silva, I.B.D.; Zampieri, T.T.; Totola, L.T.; Moreira, T.S.; Taniguti, A.P.T.; Diniz, G.P.; Barreto-Chaves, M.L.M.; Donato, J., Jr. SOCS3 Ablation in leptin receptor-expressing cells causes autonomic and cardiac dysfunctions in middle-aged mice despite improving energy and glucose metabolism. Int. J. Mol. Sci. 2022, 23, 6484. [Google Scholar] [CrossRef]
- Ornellas, F.; Souza-Mello, V.; Mandarim-de-Lacerda, C.A.; Aguila, M.B. Combined parental obesity augments single-parent obesity effects on hypothalamus inflammation, leptin signaling (JAK/STAT), hyperphagia, and obesity in the adult mice offspring. Physiol. Behav. 2016, 153, 47–55. [Google Scholar] [CrossRef]
- Al Saqri, A.; Malgundkar, S.H.; Al Kindi, F.; Gupta, I.; Al Moundhri, M.; Tamimi, Y. SOCS3 gene silencing does not occur through methylation and mutations in gastric cancer. Hum. Cell. 2022, 35, 1114–1125. [Google Scholar] [CrossRef]
- Zheng, S.; Li, Z. Identification of a cullin5-RING E3 ligase transcriptome signature in glioblastoma multiforme. Aging 2020, 12, 17380–17392. [Google Scholar] [CrossRef]
- Altinkiliç, E.M.; Bayrakdar, S.; Seymen Karabulut, G.; Haliloğlu, B.; Attar, R. The role of circulating miRNAs in leptin resistance in obese children. J. Pediatr. Endocrinol. Metab. 2022, 35, 761–766. [Google Scholar] [CrossRef]
- Kwon, O.; Kim, K.W.; Kim, M.S. Leptin signalling pathways in hypothalamic neurons. Cell. Mol. Life Sci. 2016, 73, 1457–1477. [Google Scholar] [CrossRef]
- do Carmo, J.M.; da Silva, A.A.; Ebaady, S.E.; Sessums, P.O.; Abraham, R.S.; Elmquist, J.K.; Lowell, B.B.; Hall, J.E. Shp2 signaling in POMC neurons is important for leptin’s actions on blood pressure, energy balance, and glucose regulation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 307, R1438–R1447. [Google Scholar] [CrossRef]
- Kanumuri, R.; Kumar Pasupuleti, S.; Burns, S.S.; Ramdas, B.; Kapur, R. Targeting SHP2 phosphatase in hematological malignancies. Expert. Opin. Ther. Targets 2022, 26, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Roshanzadeh, A.; Yadav, A.K.; Pydi, S.P.; Kimura, T.; Jang, B.C. Expression and role of β3-adrenergic receptor during the differentiation of 3T3-L1 preadipocytes into adipocytes. Biology 2022, 11, 772. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Allison, M.B.; Sabatini, P.; Rupp, A.; Adams, J.; Patterson, C.; Jones, J.C.; Olson, D.P.; Myers, M.G., Jr. Transcriptional and physiological roles for STAT proteins in leptin action. Mol. Metab. 2019, 22, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Beghini, M.; Wagner, T.; Luca, A.C.; Metz, M.; Kaltenecker, D.; Spirk, K.; Hackl, M.T.; Haybaeck, J.; Moriggl, R.; Kautzky-Willer, A.; et al. Adipocyte STAT5 deficiency does not affect blood glucose homeostasis in obese mice. PLoS ONE 2021, 16, e0260501. [Google Scholar] [CrossRef] [PubMed]
- D’souza, A.M.; Asadi, A.; Johnson, J.D.; Covey, S.D.; Kieffer, T.J. Leptin deficiency in rats results in hyperinsulinemia and impaired glucose homeostasis. Endocrinology 2014, 155, 1268–1279. [Google Scholar] [CrossRef]
- Jiang, L.; Su, H.; Wu, X.; Shen, H.; Kim, M.H.; Li, Y.; Myers, M.G., Jr.; Owyang, C.; Rui, L. Leptin receptor-expressing neuron Sh2b1 supports sympathetic nervous system and protects against obesity and metabolic disease. Nat. Commun. 2020, 11, 1517. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zhou, Q.; Hart, J.R.; Xu, Y.; Yang, S.; Yang, D.; Vogt, P.K.; Wang, M.W. Cryo-EM structures of cancer-specific helical and kinase domain mutations of PI3Kα. Proc. Natl. Acad. Sci. USA 2022, 119, e2215621119. [Google Scholar] [CrossRef]
- Ramasubbu, K.; Devi Rajeswari, V. Impairment of insulin signaling pathway PI3K/Akt/mTOR and insulin resistance induced AGEs on diabetes mellitus and neurodegenerative diseases: A perspective review. Mol. Cell. Biochem. 2022. [Google Scholar] [CrossRef]
- Ma, W.; Fuentes, G.; Shi, X.; Verma, C.; Radda, G.K.; Han, W. FoxO1 negatively regulates leptin-induced POMC transcription through its direct interaction with STAT3. Biochem. J. 2015, 466, 291–298. [Google Scholar] [CrossRef]
- Stefani, C.; Miricescu, D.; Stanescu-Spinu, I.I.; Nica, R.I.; Greabu, M.; Totan, A.R.; Jinga, M. Growth factors, PI3K/AKT/mTOR and MAPK signaling pathways in colorectal cancer pathogenesis: Where are we now? Int. J. Mol. Sci. 2021, 22, 10260. [Google Scholar] [CrossRef]
- Tsunekawa, T.; Banno, R.; Mizoguchi, A.; Sugiyama, M.; Tominaga, T.; Onoue, T.; Hagiwara, D.; Ito, Y.; Iwama, S.; Goto, M.; et al. Deficiency of PTP1B attenuates hypothalamic inflammation via activation of the JAK2-STAT3 pathway in microglia. EBioMedicine 2017, 16, 172–183. [Google Scholar] [CrossRef] [Green Version]
- Ono, H.; Pocai, A.; Wang, Y.; Sakoda, H.; Asano, T.; Backer, J.M.; Schwartz, G.J.; Rossetti, L. Activation of hypothalamic S6 kinase mediates diet-induced hepatic insulin resistance in rats. J. Clin. Investig. 2008, 118, 2959–2968. [Google Scholar] [CrossRef]
- Sahu, M.; Anamthathmakula, P.; Sahu, A. Hypothalamic PDE3B deficiency alters body weight and glucose homeostasis in mouse. J. Endocrinol. 2018, 239, 93–105. [Google Scholar] [CrossRef]
- Heidema, A.G.; Wang, P.; van Rossum, C.T.; Feskens, E.J.; Boer, J.M.; Bouwman, F.G.; Van’t Veer, P.; Mariman, E.C. Sex-specific effects of CNTF, IL6 and UCP2 polymorphisms on weight gain. Physiol. Behav. 2010, 99, 1–7. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, B.T.; Jiang, Q.L.; Zhao, H.Q.; Xu, Q.; Zeng, Y.; Xu, J.Y.; Jiang, J. Leptin receptor antagonist attenuates experimental autoimmune thyroiditis in mice by regulating Treg/Th17 cell differentiation. Front. Endocrinol. 2022, 13, 1042511. [Google Scholar] [CrossRef]
- Zhang, Y.; Jin, W.; Zhang, D.; Lin, C.; He, H.; Xie, F.; Gan, L.; Fu, W.; Wu, L.; Wu, Y. TNF-α antagonizes the effect of leptin on insulin secretion through FOXO1-dependent transcriptional suppression of LepRb in INS-1 cells. Oxid. Med. Cell. Longev. 2022, 2022, 9142798. [Google Scholar] [CrossRef]
- Le Foll, C.; Johnson, M.D.; Dunn-Meynell, A.A.; Boyle, C.N.; Lutz, T.A.; Levin, B.E. Amylin-induced central IL-6 production enhances ventromedial hypothalamic leptin signaling. Diabetes 2015, 64, 1621–1631. [Google Scholar] [CrossRef] [Green Version]
- Caron, A.; Lee, S.; Elmquist, J.K.; Gautron, L. Leptin and brain-adipose crosstalks. Nat. Rev. Neurosci. 2018, 19, 153–165. [Google Scholar] [CrossRef]
- Andreoli, M.F.; Donato, J.; Cakir, I.; Perello, M. Leptin resensitisation: A reversion of leptin-resistant states. J. Endocrinol. 2019, 241, R81–R96. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Guan, D.; Auen, T.; Choi, J.W.; Salazar Hernández, M.A.; Lee, J.; Chun, H.; Faruk, F.; Kaplun, E.; Herbert, Z.; et al. IL1R1 is required for celastrol’s leptin-sensitization and antiobesity effects. Nat. Med. 2019, 25, 575–582. [Google Scholar] [CrossRef]
- Mason, M.M.; He, Y.; Chen, H.; Quon, M.J.; Reitman, M. Regulation of leptin promoter function by Sp1, C/EBP, and a novel factor. Endocrinology 1998, 139, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Wrann, C.D.; Rosen, E.D. New insights into adipocyte-specific leptin gene expression. Adipocyte 2012, 1, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Dallner, O.S.; Marinis, J.M.; Lu, Y.H.; Birsoy, K.; Werner, E.; Fayzikhodjaeva, G.; Dill, B.D.; Molina, H.; Moscati, A.; Kutalik, Z.; et al. Dysregulation of a long noncoding RNA reduces leptin leading to a leptin-responsive form of obesity. Nat. Med. 2019, 25, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Kilpeläinen, T.O.; Carli, J.F.; Skowronski, A.A.; Sun, Q.; Kriebel, J.; Feitosa, M.F.; Hedman, Å.K.; Drong, A.W.; Hayes, J.E.; Zhao, J.; et al. Genome-wide meta-analysis uncovers novel loci influencing circulating leptin levels. Nat. Commun. 2016, 7, 10494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csanova, A.; Hlavacova, N.; Hasiec, M.; Pokusa, M.; Prokopova, B.; Jezova, D. β3-Adrenergic receptors, adipokines and neuroendocrine activation during stress induced by repeated immune challenge in male and female rats. Stress 2017, 20, 294–302. [Google Scholar] [CrossRef]
- Marques-Oliveira, G.H.; Silva, T.M.; Lima, W.G.; Valadares, H.M.S.; Chaves, V.E. Insulin as a hormone regulator of the synthesis and release of leptin by white adipose tissue. Peptides 2018, 106, 49–58. [Google Scholar] [CrossRef]
- Meriin, A.B.; Zaarur, N.; Roy, D.; Kandror, K.V. Egr1 plays a major role in the transcriptional response of white adipocytes to insulin and environmental cues. Front. Cell. Dev. Biol. 2022, 10, 1003030. [Google Scholar] [CrossRef]
- Bakshi, A.; Singh, R.; Rai, U. Trajectory of leptin and leptin receptor in vertebrates: Structure, function and their regulation. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2022, 257, 110652. [Google Scholar] [CrossRef]
- Lee, M.J.; Fried, S.K. Integration of hormonal and nutrient signals that regulate leptin synthesis and secretion. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E1230–E1238. [Google Scholar] [CrossRef]
- Wróblewski, A.; Strycharz, J.; Świderska, E.; Drewniak, K.; Drzewoski, J.; Szemraj, J.; Kasznicki, J.; Śliwińska, A. Molecular insight into the interaction between epigenetics and leptin in metabolic disorders. Nutrients 2019, 11, 1872. [Google Scholar] [CrossRef]
- Melzner, I.; Scott, V.; Dorsch, K.; Fischer, P.; Wabitsch, M.; Brüderlein, S.; Hasel, C.; Möller, P. Leptin gene expression in human preadipocytes is switched on by maturation-induced demethylation of distinct CpGs in its proximal promoter. J. Biol. Chem. 2002, 277, 45420–45427. [Google Scholar] [CrossRef] [Green Version]
- Kuroda, M.; Tominaga, A.; Nakagawa, K.; Nishiguchi, M.; Sebe, M.; Miyatake, Y.; Kitamura, T.; Tsutsumi, R.; Harada, N.; Nakaya, Y.; et al. DNA methylation suppresses leptin gene in 3T3-L1 adipocytes. PLoS ONE 2016, 11, e0160532. [Google Scholar] [CrossRef] [Green Version]
- Sadashiv; Modi, A.; Khokhar, M.; Sharma, P.; Joshi, R.; Mishra, S.S.; Bharshankar, R.N.; Tiwari, S.; Singh, P.K.; Bhosale, V.V.; et al. Leptin DNA methylation and its association with metabolic risk factors in a northwest Indian obese population. J. Obes. Metab. Syndr. 2021, 30, 304–311. [Google Scholar] [CrossRef]
- Çakır, I.; Hadley, C.K.; Pan, P.L.; Bagchi, R.A.; Ghamari-Langroudi, M.; Porter, D.T.; Wang, Q.; Litt, M.J.; Jana, S.; Hagen, S.; et al. Histone deacetylase 6 inhibition restores leptin sensitivity and reduces obesity. Nat. Metab. 2022, 4, 44–59. [Google Scholar] [CrossRef]
- Lee, M.J.; Yang, R.; Gong, D.W.; Fried, S.K. Feeding and insulin increase leptin translation. Importance of the leptin mRNA untranslated regions. J. Biol. Chem. 2007, 282, 72–80. [Google Scholar] [CrossRef]
- Fernández-Fígares, I.; Lachica, M.; Martínez-Pérez, M.; Ramsay, T.G. Conjugated linoleic acid and betaine affect lipolysis in pig adipose tissue explants. Animal 2019, 13, 2840–2846. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, S.; Murphy, E.A.; Szwed, S.K.; Kanke, M.; Marchildon, F.; Sethupathy, P.; Darnell, R.B.; Cohen, P. AGO HITS-CLIP reveals distinct miRNA regulation of white and brown adipose tissue identity. Genes Dev. 2021, 35, 771–781. [Google Scholar] [CrossRef]
- Jasinski-Bergner, S.; Kielstein, H. Adipokine regulated the expression of tumor relevant microRNA. Obes. Fact. 2019, 12, 211–225. [Google Scholar] [CrossRef]
- Dattilo, A.; Ceccarini, G.; Scabia, G.; Magno, S.; Quintino, L.; Pelosini, C.; Salvetti, G.; Cusano, R.; Massidda, M.; Montanelli, L.; et al. Circulating levels of miRNAs from 320 family in subjects with lipodystrophy: Disclosing novel signatures of the disease. Front. Endocrinol. 2022, 13, 866679. [Google Scholar] [CrossRef]
- Mak, K.W.Y.; Mustafa, A.F.; Belsham, D.D. Neuroendocrine microRNAs linked to energy homeostasis: Future therapeutic potential. Pharmacol. Rep. 2022, 74, 774–789. [Google Scholar] [CrossRef]
- Kasiappan, R.; Rajarajan, D. Role of microRNA regulation in obesity-associated breast cancer: Nutritional perspectives. Adv. Nutr. 2017, 8, 868–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakrabarti, P.; Anno, T.; Manning, B.D.; Luo, Z.; Kandror, K.V. The mammalian target of rapamycin complex 1 regulates leptin biosynthesis in adipocytes at the level of translation: The role of the 5′-untranslated region in the expression of leptin messenger ribonucleic acid. Mol. Endocrinol. 2008, 22, 2260–2267. [Google Scholar] [CrossRef] [PubMed]
- Robertson, L.T.; Treviño-Villarreal, J.H.; Mejia, P.; Grondin, Y.; Harputlugil, E.; Hine, C.; Vargas, D.; Zheng, H.; Ozaki, C.K.; Kristal, B.S.; et al. Protein and calorie restriction contribute additively to protection from renal ischemia reperfusion injury partly via leptin reduction in male mice. J. Nutr. 2015, 145, 1717–1727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bond, S.T.; Calkin, A.C.; Drew, B.G. Adipose-derived extracellular vesicles: Systemic messengers and metabolic regulators in health and disease. Front. Physiol. 2022, 13, 837001. [Google Scholar] [CrossRef] [PubMed]
- Baltaci, A.K.; Mogulkoc, R. Leptin, NPY, melatonin and zinc levels in experimental hypothyroidism and hyperthyroidism: The relation to zinc. Biochem. Genet. 2017, 55, 223–233. [Google Scholar] [CrossRef]
- Cammisotto, P.G.; Gélinas, Y.; Deshaies, Y.; Bukowiecki, L.J. Regulation of leptin secretion from white adipocytes by insulin, glycolytic substrates, and amino acids. Am. J. Physiol. Endocrinol. Metab. 2005, 289, E166–E171. [Google Scholar] [CrossRef] [Green Version]
- Assinder, S.J.; Boumelhem, B.B. Oxytocin stimulates lipolysis, prostaglandin E2 synthesis, and leptin secretion in 3T3-L1 adipocytes. Mol. Cell. Endocrinol. 2021, 534, 111381. [Google Scholar] [CrossRef]
- Coppola, A.; Capuani, B.; Pacifici, F.; Pastore, D.; Arriga, R.; Bellia, A.; Andreadi, A.; Di Daniele, N.; Lauro, R.; Della-Morte, D.; et al. Activation of peripheral blood mononuclear cells and leptin secretion: New potential role of interleukin-2 and high mobility group box (HMGB)1. Int. J. Mol. Sci. 2021, 22, 7988. [Google Scholar] [CrossRef]
- Mankiewicz, J.L.; Deck, C.A.; Taylor, J.D.; Douros, J.D.; Borski, R.J. Epinephrine and glucose regulation of leptin synthesis and secretion in a teleost fish, the tilapia (Oreochromis mossambicus). Gen. Comp. Endocrinol. 2021, 302, 113669. [Google Scholar] [CrossRef]
- Rösch, G.; Muschter, D.; Taheri, S.; El Bagdadi, K.; Dorn, C.; Meurer, A.; Zaucke, F.; Schilling, A.F.; Grässel, S.; Straub, R.H.; et al. β2-adrenoceptor deficiency results in increased calcified cartilage thickness and subchondral bone remodeling in murine experimental osteoarthritis. Front. Immunol. 2022, 12, 801505. [Google Scholar] [CrossRef]
- Cowley, M.A.; Smart, J.L.; Rubinstein, M.; Cerdán, M.G.; Diano, S.; Horvath, T.L.; Cone, R.D.; Low, M.J. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature 2001, 411, 480–484. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Cheng, D.; Jie, C.; Liu, T.; Huang, S.; Hu, S. Leptin alleviates endoplasmic reticulum stress induced by cerebral ischemia/reperfusion injury via the PI3K/Akt signaling pathway. Biosci. Rep. 2022, 42, BSR20221443. [Google Scholar] [CrossRef]
- Bjørbaek, C. Central leptin receptor action and resistance in obesity. J. Investig. Med. 2009, 57, 789–794. [Google Scholar] [CrossRef]
- Liu, H.; Du, T.; Li, C.; Yang, G. STAT3 phosphorylation in central leptin resistance. Nutr. Metab. 2021, 18, 39. [Google Scholar] [CrossRef]
- Ren, D.; Zhou, Y.; Morris, D.; Li, M.; Li, Z.; Rui, L. Neuronal SH2B1 is essential for controlling energy and glucose homeostasis. J. Clin. Investig. 2007, 117, 397–406. [Google Scholar] [CrossRef]
- Saeed, S.; Janjua, Q.M.; Haseeb, A.; Khanam, R.; Durand, E.; Vaillant, E.; Ning, L.; Badreddine, A.; Berberian, L.; Boissel, M.; et al. Rare variant analysis of obesity-associated genes in young adults with severe obesity from a consanguineous population of Pakistan. Diabetes 2022, 71, 694–705. [Google Scholar] [CrossRef]
- Kaushik, S.; Rodriguez-Navarro, J.A.; Arias, E.; Kiffin, R.; Sahu, S.; Schwartz, G.J.; Cuervo, A.M.; Singh, R. Autophagy in hypothalamic AgRP neurons regulates food intake and energy balance. Cell Metab. 2011, 14, 173–183. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Aintablian, A.; Coupe, B.; Bouret, S.G. The endoplasmic reticulum stress-autophagy pathway controls hypothalamic development and energy balance regulation in leptin-deficient neonates. Nat. Commun. 2020, 11, 1914. [Google Scholar] [CrossRef] [Green Version]
- Boucsein, A.; Kamstra, K.; Tups, A. Central signalling cross-talk between insulin and leptin in glucose and energy homeostasis. J. Neuroendocrinol. 2021, 33, e12944. [Google Scholar] [CrossRef]
- Ye, L.; Jia, G.; Li, Y.; Wang, Y.; Chen, H.; Yu, L.; Wu, D. C1q/TNF-related protein 4 restores leptin sensitivity by downregulating NF-κB signaling and microglial activation. J. Neuroinflamm. 2021, 18, 159. [Google Scholar] [CrossRef]
- Crujeiras, A.B.; Carreira, M.C.; Cabia, B.; Andrade, S.; Amil, M.; Casanueva, F.F. Leptin resistance in obesity: An epigenetic landscape. Life Sci. 2015, 140, 57–63. [Google Scholar] [CrossRef]
- Hsuchou, H.; Kastin, A.J.; Tu, H.; Joan Abbott, N.; Couraud, P.O.; Pan, W. Role of astrocytic leptin receptor subtypes on leptin permeation across hCMEC/D3 human brain endothelial cells. J. Neurochem. 2010, 115, 1288–1298. [Google Scholar] [CrossRef] [Green Version]
- Banks, W.A.; Coon, A.B.; Robinson, S.M.; Moinuddin, A.; Shultz, J.M.; Nakaoke, R.; Morley, J.E. Triglycerides induce leptin resistance at the blood-brain barrier. Diabetes 2004, 53, 1253–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, S.; Cha, D.; Kim, D.W.; Hoang, T.V.; Blackshaw, S. Tanycyte-independent control of hypothalamic leptin signaling. Front. Neurosci. 2019, 13, 240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Kim, H.; Hamann, C.A.; Rhea, E.M.; Brunger, J.M.; Lippmann, E.S. Nuclear receptor ligand screening in an iPSC-derived in vitro blood-brain barrier model identifies new contributors to leptin transport. Fluids Barriers CNS 2022, 19, 77. [Google Scholar] [CrossRef] [PubMed]
- Nabil, M.; El Demellawy, M.A.; Mahmoud, M.F.; Mahmoud, A.A.A. Prolonged overnutrition with fructose or fat induces metabolic derangements in rats by disrupting the crosstalk between the hypothalamus and periphery: Possible amelioration with fenofibrate. Eur. J. Pharmacol. 2020, 879, 173136. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Liu, Y.; Hong, W.; Pan, X.; Gong, P.; Liu, Q.; Zhou, G.; Qin, S. Conditional knockout of leptin receptor in neural stem cells leads to obesity in mice and affects neuronal differentiation in the hypothalamus early after birth. Mol. Brain. 2020, 13, 109. [Google Scholar] [CrossRef]
- Dhar, M.; Zhu, M.; Impey, S.; Lambert, T.J.; Bland, T.; Karatsoreos, I.N.; Nakazawa, T.; Appleyard, S.M.; Wayman, G.A. Leptin induces hippocampal synaptogenesis via CREB-regulated microRNA-132 suppression of p250GAP. Mol. Endocrinol. 2014, 28, 1073–1087. [Google Scholar] [CrossRef] [Green Version]
- Sahin, G.S.; Dhar, M.; Dillon, C.; Zhu, M.; Shiina, H.; Winters, B.D.; Lambert, T.J.; Impey, S.; Appleyard, S.M.; Wayman, G.A. Leptin stimulates synaptogenesis in hippocampal neurons via KLF4 and SOCS3 inhibition of STAT3 signaling. Mol. Cell. Neurosci. 2020, 106, 103500. [Google Scholar] [CrossRef]
- Bland, T.; Sahin, G.S.; Zhu, M.; Dillon, C.; Impey, S.; Appleyard, S.M.; Wayman, G.A. USP8 deubiquitinates the leptin receptor and is necessary for leptin-mediated synapse formation. Endocrinology 2019, 160, 1982–1998. [Google Scholar] [CrossRef]
- Bland, T.; Zhu, M.; Dillon, C.; Sahin, G.S.; Rodriguez-Llamas, J.L.; Appleyard, S.M.; Wayman, G.A. Leptin controls glutamatergic synaptogenesis and NMDA-receptor trafficking via fyn kinase regulation of NR2B. Endocrinology 2020, 161, bqz030. [Google Scholar] [CrossRef]
- McGregor, G.; Harvey, J. Leptin regulation of synaptic function at hippocampal TA-CA1 and SC-CA1 synapses: Implications for health and disease. Neurochem. Res. 2019, 44, 650–660. [Google Scholar] [CrossRef] [Green Version]
- Luo, X.; McGregor, G.; Irving, A.J.; Harvey, J. Leptin induces a novel form of NMDA receptor-dependent LTP at hippocampal temporoammonic-CA1 synapses. eNeuro 2015, 2, ENEURO.0007-15.2015. [Google Scholar] [CrossRef] [Green Version]
- Ho, P.W.; Ho, J.W.; Liu, H.F.; So, D.H.; Tse, Z.H.; Chan, K.H.; Ramsden, D.B.; Ho, S.L. Mitochondrial neuronal uncoupling proteins: A target for potential disease-modification in Parkinson’s disease. Transl. Neurodegener. 2012, 1, 3. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Zou, W.; Hu, M.; Tian, Q.; Xiao, F.; Li, M.; Zhang, P.; Chen, Y.J.; Jiang, J.M. Hydrogen sulphide attenuates neuronal apoptosis of substantia nigra by re-establishing autophagic flux via promoting leptin signalling in a 6-hydroxydopamine rat model of Parkinson’s disease. Clin. Exp. Pharmacol. Physiol. 2022, 49, 122–133. [Google Scholar] [CrossRef]
- Cheng, Y.; Buchan, M.; Vitanova, K.; Aitken, L.; Gunn-Moore, F.J.; Ramsay, R.R.; Doherty, G. Neuroprotective actions of leptin facilitated through balancing mitochondrial morphology and improving mitochondrial function. J. Neurochem. 2020, 155, 191–206. [Google Scholar] [CrossRef] [Green Version]
- Greco, S.J.; Bryan, K.J.; Sakar, S.; Zhu, X.; Smith, M.A.; Ashford, J.W.; Johnston, J.M.; Tezapsidis, N.; Casadesus, G. Leptin reduces pathology and improves memory in a transgenic mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2010, 19, 1155–1167. [Google Scholar] [CrossRef] [Green Version]
- Perianes-Cachero, A.; Burgos-Ramos, E.; Puebla-Jiménez, L.; Canelles, S.; Frago, L.M.; Hervás-Aguilar, A.; de Frutos, S.; Toledo-Lobo, M.V.; Mela, V.; Viveros, M.P.; et al. Acute up-regulation of the rat brain somatostatin receptor-effector system by leptin is related to activation of insulin signaling and may counteract central leptin actions. Neuroscience 2013, 252, 289–301. [Google Scholar] [CrossRef]
- Aguado-Llera, D.; Canelles, S.; Fernández-Mendívil, C.; Frago, L.M.; Argente, J.; Arilla-Ferreiro, E.; López, M.G.; Barrios, V. Improvement in inflammation is associated with the protective effect of Gly-Pro-Glu and cycloprolylglycine against Aβ-induced depletion of the hippocampal somatostatinergic system. Neuropharmacology 2019, 151, 112–126. [Google Scholar] [CrossRef]
- Gonzalez-Rodriguez, M.; Astillero-Lopez, V.; Villanueva-Anguita, P.; Paya-Rodriguez, M.E.; Flores-Cuadrado, A.; Villar-Conde, S.; Ubeda-Banon, I.; Martinez-Marcos, A.; Saiz-Sanchez, D. Somatostatin and astroglial involvement in the human limbic system in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 8434. [Google Scholar] [CrossRef]
- Tong, J.Q.; Zhang, J.; Hao, M.; Yang, J.; Han, Y.F.; Liu, X.J.; Shi, H.; Wu, M.N.; Liu, Q.S.; Qi, J.S. Leptin attenuates the detrimental effects of β-amyloid on spatial memory and hippocampal later-phase long term potentiation in rats. Horm. Behav. 2015, 73, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Ji, M.; Gong, W.; Wang, S.; Zhang, D.; Chen, B.; Li, X.; Wu, X.; Cui, L.; Feng, Y.; Verkhratsky, A.; et al. Leptin attenuates fear memory by inhibiting astrocytic NLRP3 inflammasome in post-traumatic stress disorder model. Neurochem. Res. 2022. [Google Scholar] [CrossRef] [PubMed]
- Erichsen, J.M.; Fadel, J.R.; Reagan, L.P. Peripheral versus central insulin and leptin resistance: Role in metabolic disorders, cognition, and neuropsychiatric diseases. Neuropharmacology 2022, 203, 108877. [Google Scholar] [CrossRef] [PubMed]
- Weng, Z.; Signore, A.P.; Gao, Y.; Wang, S.; Zhang, F.; Hastings, T.; Yin, X.M.; Chen, J. Leptin protects against 6-hydroxydopamine-induced dopaminergic cell death via mitogen-activated protein kinase signaling. J. Biol. Chem. 2007, 282, 34479–34491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, V.; Singh, T.G.; Kaur, A.; Mannan, A.; Dhiman, S. Brain-derived neurotrophic factor: A novel dynamically regulated therapeutic modulator in neurological disorders. Neurochem. Res. 2022. [Google Scholar] [CrossRef]
- Yin, X.; Wang, M.; Wang, W.; Chen, T.; Song, G.; Niu, Y.; Jiang, Z.; Gao, Z.; Wang, Z. Identification of potential miRNA-mRNA regulatory network contributing to Parkinson’s Disease. Parkinsons Dis. 2022, 2022, 2877728. [Google Scholar] [CrossRef]
- Ho, P.W.; Liu, H.F.; Ho, J.W.; Zhang, W.Y.; Chu, A.C.; Kwok, K.H.; Ge, X.; Chan, K.H.; Ramsden, D.B.; Ho, S.L. Mitochondrial uncoupling protein-2 (UCP2) mediates leptin protection against MPP+ toxicity in neuronal cells. Neurotox. Res. 2010, 17, 332–343. [Google Scholar] [CrossRef] [Green Version]
- Rahnemayan, S.; Mirghafourvand, M.; Fathalizadeh, A.; Faramarzi, E.; Reyhanifard, A.; Mahmoodpoor, A.; Sanaie, S. Leptin levels in patients with Parkinson’s disease: A systematic review and meta-analysis. Clin. Nutr. ESPEN 2021, 41, 104–109. [Google Scholar] [CrossRef]
- Zou, X.; Zhong, L.; Zhu, C.; Zhao, H.; Zhao, F.; Cui, R.; Gao, S.; Li, B. Role of leptin in mood disorder and neurodegenerative disease. Front. Neurosci. 2019, 13, 378. [Google Scholar] [CrossRef]
- Guo, M.; Huang, T.Y.; Garza, J.C.; Chua, S.C.; Lu, X.Y. Selective deletion of leptin receptors in adult hippocampus induces depression-related behaviours. Int. J. Neuropsychopharmacol. 2013, 16, 857–867. [Google Scholar] [CrossRef]
- Milaneschi, Y.; Lamers, F.; Bot, M.; Drent, M.L.; Penninx, B.W. Leptin dysregulation is specifically associated with major depression with atypical features: Evidence for a mechanism connecting obesity and depression. Biol. Psychiatry 2017, 81, 807–814. [Google Scholar] [CrossRef]
- Choi, W.; Kim, J.W.; Kang, H.J.; Kim, H.K.; Kang, H.C.; Lee, J.Y.; Kim, S.W.; Stewart, R.; Kim, J.M. Interactive effects of serum leptin levels and physical comorbidity on the pharmacotherapeutic response of depressive disorders. Clin. Psychopharmacol. Neurosci. 2022, 20, 662–674. [Google Scholar] [CrossRef]
- Tian, J.; Wang, T.; Jia, K.; Guo, L.; Swerdlow, R.H.; Du, H. Nonobese male patients with Alzheimer’s Disease are vulnerable to decrease in plasma leptin. J. Alzheimers Dis. 2022, 88, 1017–1027. [Google Scholar] [CrossRef]
- Bonda, D.J.; Stone, J.G.; Torres, S.L.; Siedlak, S.L.; Perry, G.; Kryscio, R.; Jicha, G.; Casadesus, G.; Smith, M.A.; Zhu, X.; et al. Dysregulation of leptin signaling in Alzheimer disease: Evidence for neuronal leptin resistance. J. Neurochem. 2014, 128, 162–172. [Google Scholar] [CrossRef]
- Perianes-Cachero, A.; Canelles, S.; Aguado-Llera, D.; Frago, L.M.; Toledo-Lobo, M.V.; Carrera, I.; Cacabelos, R.; Chowen, J.A.; Argente, J.; Arilla-Ferreiro, E.; et al. Reduction in Aβ-induced cell death in the hippocampus of 17β-estradiol-treated female rats is associated with an increase in IGF-I signaling and somatostatinergic tone. J. Neurochem. 2015, 135, 1257–1271. [Google Scholar] [CrossRef] [Green Version]
- Christensen, A.; Liu, J.; Pike, C.J. Aging reduces estradiol protection against neural but not metabolic effects of obesity in female 3xTg-AD mice. Front. Aging Neurosci. 2020, 12, 113. [Google Scholar] [CrossRef]
- Conley, A.C.; Albert, K.M.; McDonald, B.C.; Saykin, A.J.; Dumas, J.A.; Newhouse, P.A. Estradiol treatment in young postmenopausal women with self-reported cognitive complaints: Effects on cholinergic-mediated cognitive performance. Hum. Psychopharmacol. 2022, 37, e2838. [Google Scholar] [CrossRef]
- Cecon, E.; Lhomme, T.; Maurice, T.; Luka, M.; Chen, M.; Silva, A.; Wauman, J.; Zabeau, L.; Tavernier, J.; Prévot, V.; et al. Amyloid beta peptide is an endogenous negative allosteric modulator of leptin receptor. Neuroendocrinology 2021, 111, 370–387. [Google Scholar] [CrossRef]
- Calió, M.L.; Mosini, A.C.; Marinho, D.S.; Salles, G.N.; Massinhani, F.H.; Ko, G.M.; Porcionatto, M.A. Leptin enhances adult neurogenesis and reduces pathological features in a transgenic mouse model of Alzheimer’s disease. Neurobiol. Dis. 2021, 148, 105219. [Google Scholar] [CrossRef]
- Lai, K.S.P.; Liu, C.S.; Rau, A.; Lanctôt, K.L.; Köhler, C.A.; Pakosh, M.; Carvalho, A.F.; Herrmann, N. Peripheral inflammatory markers in Alzheimer’s disease: A systematic review and meta-analysis of 175 studies. J. Neurol. Neurosurg. Psychiatry 2017, 88, 876–882. [Google Scholar] [CrossRef]
- Mejido, D.C.P.; Peny, J.A.; Vieira, M.N.N.; Ferreira, S.T.; De Felice, F.G. Insulin and leptin as potential cognitive enhancers in metabolic disorders and Alzheimer’s disease. Neuropharmacology 2020, 171, 108115. [Google Scholar] [CrossRef] [PubMed]
- Corrigan, R.R.; Piontkivska, H.; Casadesus, G. Amylin pharmacology in Alzheimer’s disease pathogenesis and treatment. Curr. Neuropharmacol. 2022, 20, 1894–1907. [Google Scholar] [CrossRef] [PubMed]
- Menendez, A.; Wanczyk, H.; Walker, J.; Zhou, B.; Santos, M.; Finck, C. Obesity and adipose tissue dysfunction: From pediatrics to adults. Genes 2022, 13, 1866. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, R.; Kimonis, V.; Butler, M.G. Genetics of obesity in humans: A clinical review. Int. J. Mol. Sci. 2022, 23, 11005. [Google Scholar] [CrossRef] [PubMed]
- Baxter, J.; Armijo, P.R.; Flores, L.; Krause, C.; Samreen, S.; Tanner, T. Updates on monogenic obesity in a multifactorial disease. Obes. Surg. 2019, 29, 4077–4083. [Google Scholar] [CrossRef]
- Funcke, J.B.; von Schnurbein, J.; Lennerz, B.; Lahr, G.; Debatin, K.M.; Fischer-Posovszky, P.; Wabitsch, M. Monogenic forms of childhood obesity due to mutations in the leptin gene. Mol. Cell. Pediatr. 2014, 1, 3. [Google Scholar] [CrossRef] [Green Version]
- Farooqi, I.S.; Wangensteen, T.; Collins, S.; Kimber, W.; Matarese, G.; Keogh, J.M.; Lank, E.; Bottomley, B.; Lopez-Fernandez, J.; Ferraz-Amaro, I.; et al. Clinical and molecular genetic spectrum of congenital deficiency of the leptin receptor. N. Engl. J. Med. 2007, 356, 237–247. [Google Scholar] [CrossRef] [Green Version]
- Chaves, C.; Kay, T.; Anselmo, J. Early onset obesity due to a mutation in the human leptin receptor gene. Endocrinol. Diabetes Metab. Case Rep. 2022, 2022, 21-0124. [Google Scholar] [CrossRef]
- Yang, J.; Wang, M.; Tobias, D.K.; Rich-Edwards, J.W.; Darling, A.M.; Abioye, A.I.; Pembe, A.B.; Madzorera, I.; Fawzi, W.W. Gestational weight gain during the second and third trimesters and adverse pregnancy outcomes, results from a prospective pregnancy cohort in urban Tanzania. Reprod. Health 2022, 19, 140. [Google Scholar] [CrossRef]
- Lin, X.H.; Gao, L.; Tian, S.; Klausen, C.; Guo, M.X.; Gao, Q.; Liu, M.E.; Wang, H.; Wu, D.D.; Zhou, C.L.; et al. Maternal high-fat-diet exposure is associated with elevated blood pressure and sustained increased leptin levels through epigenetic memory in offspring. Sci. Rep. 2021, 11, 316. [Google Scholar] [CrossRef]
- Zwamborn, R.A.; Slieker, R.C.; Mulder, P.C.; Zoetemelk, I.; Verschuren, L.; Suchiman, H.E.; Toet, K.H.; Droog, S.; Slagboom, P.E.; Kooistra, T.; et al. Prolonged high-fat diet induces gradual and fat depot-specific DNA methylation changes in adult mice. Sci. Rep. 2017, 7, 43261. [Google Scholar] [CrossRef] [Green Version]
- Harvey, N.C.; Sheppard, A.; Godfrey, K.M.; McLean, C.; Garratt, E.; Ntani, G.; Davies, L.; Murray, R.; Inskip, H.M.; Gluckman, P.D.; et al. Childhood bone mineral content is associated with methylation status of the RXRA promoter at birth. J. Bone Miner. Res. 2014, 29, 600–607. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.C.; Melton, P.E.; Burton, M.A.; Beilin, L.J.; Clarke-Harris, R.; Cook, E.; Godfrey, K.M.; Burdge, G.C.; Mori, T.A.; Anderson, D.; et al. Adiposity associated DNA methylation signatures in adolescents are related to leptin and perinatal factors. Epigenetics 2022, 17, 819–836. [Google Scholar] [CrossRef]
- Mansell, T.; Ponsonby, A.L.; Collier, F.; Burgner, D.; Pezic, A.; Vuillermin, P.; Ryan, J.; Saffery, R. Methylation of the LEP gene promoter in blood at 12 months and BMI at 4 years of age-a population-based cohort study. Int. J. Obes. 2020, 44, 842–847. [Google Scholar] [CrossRef]
- Mazzucco, M.B.; Higa, R.; Capobianco, E.; Kurtz, M.; Jawerbaum, A.; White, V. Saturated fat-rich diet increases fetal lipids and modulates LPL and leptin receptor expression in rat placentas. J. Endocrinol. 2013, 217, 303–315. [Google Scholar] [CrossRef] [Green Version]
- Nogues, P.; Dos Santos, E.; Jammes, H.; Berveiller, P.; Arnould, L.; Vialard, F.; Dieudonné, M.N. Maternal obesity influences expression and DNA methylation of the adiponectin and leptin systems in human third-trimester placenta. Clin. Epigenetics 2019, 11, 20. [Google Scholar] [CrossRef] [Green Version]
- Eyckerman, S.; Broekaert, D.; Verhee, A.; Vandekerckhove, J.; Tavernier, J. Identification of the Y985 and Y1077 motifs as SOCS3 recruitment sites in the murine leptin receptor. FEBS Lett. 2000, 486, 33–37. [Google Scholar] [CrossRef] [Green Version]
- McEwen, H.J.; Inglis, M.A.; Quennell, J.H.; Grattan, D.R.; Anderson, G.M. Deletion of suppressor of cytokine signaling 3 from forebrain neurons delays infertility and onset of hypothalamic leptin resistance in response to a high caloric diet. J. Neurosci. 2016, 36, 7142–7153. [Google Scholar] [CrossRef] [Green Version]
- Ancel, C.M.; Evans, M.C.; Kerbus, R.I.; Wallace, E.G.; Anderson, G.M. Deletion of PTP1B from brain neurons partly protects mice from diet-induced obesity and minimally improves fertility. Endocrinology 2022, 163, bqab266. [Google Scholar] [CrossRef]
- Hsu, Y.H.; Wu, C.H.; Chiu, C.J.; Chen, W.T.; Chang, Y.C.; Wabitsch, M.; Chang, M.S. IL-20 is involved in obesity by modulation of adipogenesis and macrophage dysregulation. Immunology 2021, 164, 817–833. [Google Scholar] [CrossRef]
- Cakir, I.; Nillni, E.A. Endoplasmic reticulum stress, the hypothalamus, and energy balance. Trends Endocrinol. Metab. 2019, 30, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Barrios, V.; Campillo-Calatayud, A.; Guerra-Cantera, S.; Canelles, S.; Martín-Rivada, Á.; Frago, L.M.; Chowen, J.A.; Argente, J. Opposite effects of chronic central leptin infusion on activation of insulin signaling pathways in adipose tissue and liver are related to changes in the inflammatory environment. Biomolecules 2021, 11, 1734. [Google Scholar] [CrossRef] [PubMed]
- Ahi, E.P.; Brunel, M.; Tsakoumis, E.; Chen, J.; Schmitz, M. Appetite regulating genes in zebrafish gut; a gene expression study. PLoS ONE 2022, 17, e0255201. [Google Scholar] [CrossRef] [PubMed]
- Roujeau, C.; Jockers, R.; Dam, J. Endospanin 1 determines the balance of leptin-regulated hypothalamic functions. Neuroendocrinology 2019, 108, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Vauthier, V.; Swartz, T.D.; Chen, P.; Roujeau, C.; Pagnon, M.; Mallet, J.; Sarkis, C.; Jockers, R.; Dam, J. Endospanin 1 silencing in the hypothalamic arcuate nucleus contributes to sustained weight loss of high fat diet obese mice. Gene Ther. 2014, 21, 638–644. [Google Scholar] [CrossRef]
- Li, Z.; Ceccarini, G.; Eisenstein, M.; Tan, K.; Friedman, J.M. Phenotypic effects of an induced mutation of the ObRa isoform of the leptin receptor. Mol. Metab. 2013, 2, 364–375. [Google Scholar] [CrossRef]
- Guillebaud, F.; Duquenne, M.; Djelloul, M.; Pierre, C.; Poirot, K.; Roussel, G.; Riad, S.; Lanfray, D.; Morin, F.; Jean, A.; et al. Glial endozepines reverse high-fat diet-induced obesity by enhancing hypothalamic response to peripheral leptin. Mol. Neurobiol. 2020, 57, 3307–3333. [Google Scholar] [CrossRef]
- Stürzebecher, P.E.; Kralisch, S.; Schubert, M.R.; Filipova, V.; Hoffmann, A.; Oliveira, F.; Sheikh, B.N.; Blüher, M.; Kogel, A.; Scholz, M.; et al. Leptin treatment has vasculo-protective effects in lipodystrophic mice. Proc. Natl. Acad. Sci. USA 2022, 119, e2110374119. [Google Scholar] [CrossRef]
- Mainieri, F.; Tagi, V.M.; Chiarelli, F. Treatment options for lipodystrophy in children. Front. Endocrinol. 2022, 13, 879979. [Google Scholar] [CrossRef]
- Moon, H.S.; Matarese, G.; Brennan, A.M.; Chamberland, J.P.; Liu, X.; Fiorenza, C.G.; Mylvaganam, G.H.; Abanni, L.; Carbone, F.; Williams, C.J.; et al. Efficacy of metreleptin in obese patients with type 2 diabetes: Cellular and molecular pathways underlying leptin tolerance. Diabetes 2011, 60, 1647–1656. [Google Scholar] [CrossRef]
- Crépin, D.; Benomar, Y.; Riffault, L.; Amine, H.; Gertler, A.; Taouis, M. The over-expression of miR-200a in the hypothalamus of ob/ob mice is linked to leptin and insulin signaling impairment. Mol. Cell. Endocrinol. 2014, 384, 1–11. [Google Scholar] [CrossRef]
- Zhang, N.; Zhang, N.; Song, L.; Xie, H.; Zhao, C.; Li, S.; Zhao, W.; Zhao, Y.; Gao, C.; Xu, G. Adipokines and free fatty acids regulate insulin sensitivity by increasing microRNA-21 expression in human mature adipocytes. Mol. Med. Rep. 2017, 16, 2254–2258. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.J.; Hwang, S.J.; Bae, Y.C.; Jung, J.S. MiR-21 regulates adipogenic differentiation through the modulation of TGF-beta signaling in mesenchymal stem cells derived from human adipose tissue. Stem Cells 2009, 27, 3093–3102. [Google Scholar] [CrossRef]
- Kim, Y.J.; Hwang, S.H.; Cho, H.H.; Shin, K.K.; Bae, Y.C.; Jung, J.S. MicroRNA 21 regulates the proliferation of human adipose tissue derived mesenchymal stem cells and high-fat diet-induced obesity alters microRNA 21 expression in white adipose tissues. J. Cell. Physiol. 2012, 227, 183–193. [Google Scholar] [CrossRef]
- Dong, T. Anticancer activities of PPARγ in breast cancer are context dependent. Am. J. Pathol. 2013, 182, 1–4. [Google Scholar] [CrossRef]
- Yagai, T.; Yan, T.; Luo, Y.; Takahashi, S.; Aibara, D.; Kim, D.; Brocker, C.N.; Levi, M.; Motohashi, H.; Gonzalez, F.J. Feedback repression of PPARα signaling by Let-7 microRNA. Cell. Rep. 2021, 36, 109506. [Google Scholar] [CrossRef]
- Kollari, E.; Zografou, I.; Sampanis, C.; Athyros, V.G.; Didangelos, T.; Mantzoros, C.S.; Karagiannis, A. Serum adipokine levels in patients with type 1 diabetes are associated with degree of obesity but only resistin is independently associated with atherosclerosis markers. Hormones 2022, 21, 91–101. [Google Scholar] [CrossRef]
- Szalecki, M.; Pańkowska, E.; Wysocka-Mincewicz, M.; Klupa, T.; Janas, R. Leptin and soluble leptin receptor in children with type 1 diabetes mellitus. Pediatr. Endocrinol. Diabetes Metab. 2010, 16, 262–269. [Google Scholar]
- Gamarra, J.R.; Haeusler, R.A. Hepatocentric leptin signaling modulates gluconeogenesis via MKP-3. Cell. Mol. Gastroenterol. Hepatol. 2022, 14, 1166–1167. [Google Scholar] [CrossRef]
- Denroche, H.C.; Levi, J.; Wideman, R.D.; Sequeira, R.M.; Huynh, F.K.; Covey, S.D.; Kieffer, T.J. Leptin therapy reverses hyperglycemia in mice with streptozotocin-induced diabetes, independent of hepatic leptin signaling. Diabetes 2011, 60, 1414–1423. [Google Scholar] [CrossRef] [Green Version]
- Ebihara, K.; Nakao, K. Translational research of leptin in lipodystrophy and its related diseases. In Innovative Medicine: Basic Research and Development [Internet]; Nakao, K., Minato, N., Uemoto, S., Eds.; Springer: Tokyo, Japan, 2015. [Google Scholar] [CrossRef]
- Ito, Y.; Sun, R.; Yagimuma, H.; Taki, K.; Mizoguchi, A.; Kobayashi, T.; Sugiyama, M.; Onoue, T.; Tsunekawa, T.; Takagi, H.; et al. Protein tyrosine phosphatase 1B deficiency improves glucose homeostasis in type 1 diabetes treated with leptin. Diabetes 2022, 71, 1902–1914. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Chang, J.T.; Myers, M.G., Jr.; Xu, Y.; Tong, Q. Euglycemia restoration by central leptin in type 1 diabetes requires STAT3 signaling but not fast-acting neurotransmitter release. Diabetes 2016, 65, 1040–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, S.; Xu, Y.; Lu, Y.; Jiang, Z.; Li, H.; Morrill, J.C.; Cai, J.; Wu, Q.; Xu, Y.; Xue, M.; et al. A neural basis for brain leptin action on reducing type 1 diabetic hyperglycemia. Nat. Commun. 2021, 12, 2662. [Google Scholar] [CrossRef] [PubMed]
- Bonnefond, A.; Semple, R.K. Achievements, prospects and challenges in precision care for monogenic insulin-deficient and insulin-resistant diabetes. Diabetologia 2022, 65, 1782–1795. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.J.; Petersen, K.F.; Shulman, G.I. Pleotropic effects of leptin to reverse insulin resistance and diabetic ketoacidosis. Diabetologia 2016, 59, 933–937. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, Y.; Huang, E.S. Changes in racial/ethnic disparities in the prevalence of Type 2 diabetes by obesity level among US adults. Ethn. Health 2009, 14, 439–457. [Google Scholar] [CrossRef] [Green Version]
- Jais, A.; Brüning, J.C. Arcuate nucleus-dependent regulation of metabolism-pathways to obesity and diabetes mellitus. Endocr. Rev. 2022, 43, 314–328. [Google Scholar] [CrossRef]
- Agrawal, R.; Reno, C.M.; Sharma, S.; Christensen, C.; Huang, Y.; Fisher, S.J. Insulin action in the brain regulates both central and peripheral functions. Am. J. Physiol. Endocrinol. Metab. 2021, 321, E156–E163. [Google Scholar] [CrossRef]
- Hayden, M.R.; Banks, W.A. Deficient leptin cellular signaling plays a key role in brain ultrastructural remodeling in obesity and type 2 diabetes mellitus. Int. J. Mol. Sci. 2021, 22, 5427. [Google Scholar] [CrossRef]
- Wang, A.N.; Carlos, J.; Fraser, G.M.; McGuire, J.J. Zucker Diabetic-Sprague Dawley (ZDSD) rat: Type 2 diabetes translational research model. Exp. Physiol. 2022, 107, 265–282. [Google Scholar] [CrossRef]
- Suckow, M.A.; Gobbett, T.A.; Peterson, R.G. Wound healing delay in the ZDSD rat. In Vivo 2017, 31, 55–60. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Lin, C.; Chen, R.; Luo, L.; Huang, J.; Liu, H.; Chen, W.; Xu, J.; Yu, H.; Ding, Y. Association analysis of SOCS3, JAK2 and STAT3 gene polymorphisms and genetic susceptibility to type 2 diabetes mellitus in Chinese population. Diabetol. Metab. Syndr. 2022, 14, 4. [Google Scholar] [CrossRef]
- van Doorn, M.; Kemme, M.; Ouwens, M.; van Hoogdalem, E.J.; Jones, R.; Romijn, H.; de Kam, M.; Schoemaker, R.; Burggraaf, K.; Cohen, A. Evaluation of proinflammatory cytokines and inflammation markers as biomarkers for the action of thiazolidinediones in Type 2 diabetes mellitus patients and healthy volunteers. Br. J. Clin. Pharmacol. 2006, 62, 391–402. [Google Scholar] [CrossRef]
- Li, Y.; Chen, S.; Zhao, T.; Li, M. Serum IL-36 cytokines levels in type 2 diabetes mellitus patients and their association with obesity, insulin resistance, and inflammation. J. Clin. Lab. Anal. 2021, 35, e23611. [Google Scholar] [CrossRef]
- Yang, M.; Tian, M.; Zhang, X.; Xu, J.; Yang, B.; Yu, J.; Li, F.; Li, Y.; Li, S.; Li, X. Role of the JAK2/STAT3 signaling pathway in the pathogenesis of type 2 diabetes mellitus with macrovascular complications. Oncotarget 2017, 8, 96958–96969. [Google Scholar] [CrossRef] [Green Version]
- Maskarinec, G.; Fontaine, A.; Torfadottir, J.E.; Lipscombe, L.L.; Lega, I.C.; Figueroa, J.; Wild, S. The relation of type 2 diabetes and breast cancer incidence in Asian, Hispanic and African American populations—A review. Can. J. Diabetes 2018, 42, 100–105. [Google Scholar] [CrossRef] [Green Version]
- Karra, P.; Winn, M.; Pauleck, S.; Bulsiewicz-Jacobsen, A.; Peterson, L.; Coletta, A.; Doherty, J.; Ulrich, C.M.; Summers, S.A.; Gunter, M.; et al. Metabolic dysfunction and obesity-related cancer: Beyond obesity and metabolic syndrome. Obesity 2022, 30, 1323–1334. [Google Scholar] [CrossRef]
- Rossi, M.; Turati, F.; Lagiou, P.; Trichopoulos, D.; Augustin, L.S.; La Vecchia, C.; Trichopoulou, A. Mediterranean diet and glycaemic load in relation to incidence of type 2 diabetes: Results from the Greek cohort of the population-based European Prospective Investigation into Cancer and Nutrition (EPIC). Diabetologia 2013, 56, 2405–2413. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Li, Y.; Yin, L.; Qi, Y.; Sun, H.; Sun, P.; Xu, M.; Tang, Z.; Peng, J. miR-125a-5p ameliorates hepatic glycolipid metabolism disorder in type 2 diabetes mellitus through targeting of STAT3. Theranostics 2018, 8, 5593–5609. [Google Scholar] [CrossRef]
- Arner, P.; Kulyte, A. MicroRNA regulatory networks in human adipose tissue and obesity. Nat. Rev. Endocrinol. 2015, 11, 276–288. [Google Scholar] [CrossRef]
- Zhu, H.; Shyh-Chang, N.; Segrè, A.V.; Shinoda, G.; Shah, S.P.; Einhorn, W.S.; Takeuchi, A.; Engreitz, J.M.; Hagan, J.P.; Kharas, M.G.; et al. The Lin28/let-7 axis regulates glucose metabolism. Cell 2011, 147, 81–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, G.; Ji, C.; Shi, C.; Fu, H.; Zhu, L.; Xu, L.; Xu, L.; Chen, L.; Feng, Y.; Zhao, Y.; et al. Modulation of hsa-mir-26b levels following adipokine stimulation. Mol. Biol. Rep. 2013, 40, 3577–3582. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Ji, C.; Song, G.; Zhao, C.; Shi, C.; Song, L.; Chen, L.; Yang, L.; Huang, F.; Pang, L.; et al. MiR-26b modulates insulin sensitivity in adipocytes by interrupting the PTEN/PI3K/AKT pathway. Int. J. Obes. 2015, 39, 1523–1530. [Google Scholar] [CrossRef] [PubMed]
- Sletner, L.; Moen, A.E.F.; Yajnik, C.S.; Lekanova, N.; Sommer, C.; Birkeland, K.I.; Jenum, A.K.; Böttcher, Y. Maternal glucose and LDL-cholesterol levels are related to placental leptin gene methylation, and, together with nutritional factors, largely explain a higher methylation level among ethnic South Asians. Front. Endocrinol. 2021, 12, 809916. [Google Scholar] [CrossRef] [PubMed]
- Fanelli, G.; Mota, N.R.; Salas-Salvadó, J.; Bulló, M.; Fernandez-Aranda, F.; Camacho-Barcia, L.; Testa, G.; Jiménez-Murcia, S.; Bertaina-Anglade, V.; Franke, B.; et al. The link between cognition and somatic conditions related to insulin resistance in the UK Biobank study cohort: A systematic review. Neurosci. Biobehav. Rev. 2022, 143, 104927. [Google Scholar] [CrossRef]
- Lin, L.; Wang, Y.; Xu, W.; Huang, C.; Hu, J.; Chen, X.; Lv, X.; Qin, Y.; Zhao, X.; Li, H. Aerobic exercise improves type 2 diabetes mellitus-related cognitive impairment by inhibiting JAK2/STAT3 and enhancing AMPK/SIRT1 pathways in mice. Dis. Markers 2022, 2022, 6010504. [Google Scholar] [CrossRef]
- Behl, T.; Gupta, A.; Sehgal, A.; Albarrati, A.; Albratty, M.; Meraya, A.M.; Najmi, A.; Bhatia, S.; Bungau, S. Exploring protein tyrosine phosphatases (PTP) and PTP-1B inhibitors in management of diabetes mellitus. Biomed. Pharmacother. 2022, 153, 113405. [Google Scholar] [CrossRef]
- Salazar, J.; Chávez-Castillo, M.; Rojas, J.; Ortega, A.; Nava, M.; Pérez, J.; Rojas, M.; Espinoza, C.; Chacin, M.; Herazo, Y.; et al. Is “leptin resistance” another key resistance to manage type 2 diabetes? Curr. Diabetes Rev. 2020, 16, 733–749. [Google Scholar] [CrossRef]
- O’Brien, S.N.; Welter, B.H.; Price, T.M. Presence of leptin in breast cell lines and breast tumors. Biochem. Biophys. Res. Commun. 1999, 259, 695–698. [Google Scholar] [CrossRef]
- Yuan, S.S.; Tsai, K.B.; Chung, Y.F.; Chan, T.F.; Yeh, Y.T.; Tsai, L.Y.; Su, J.H. Aberrant expression and possible involvement of the leptin receptor in endometrial cancer. Gynecol. Oncol. 2004, 92, 769–775. [Google Scholar] [CrossRef]
- Sharma, D.; Saxena, N.K.; Vertino, P.M.; Anania, F.A. Leptin promotes the proliferative response and invasiveness in human endometrial cancer cells by activating multiple signal-transduction pathways. Endocr. Relat. Cancer 2006, 13, 629–640. [Google Scholar] [CrossRef] [Green Version]
- Yeung, C.Y.; Tso, A.W.; Xu, A.; Wang, Y.; Woo, Y.C.; Lam, T.H.; Lo, S.V.; Fong, C.H.; Wat, N.M.; Woo, J.; et al. Pro-inflammatory adipokines as predictors of incident cancers in a Chinese cohort of low obesity prevalence in Hong Kong. PLoS ONE 2013, 8, e78594. [Google Scholar] [CrossRef] [Green Version]
- Acedo, S.C.; Gambero, S.; Pereira Cunha, F.G.; Lorand-Metze, I.; Gambero, A. Participation of leptin in the determination of the macrophage phenotype: An additional role in adipocyte and macrophage crosstalk. In Vitro Cell. Dev. Biol. Anim. 2013, 49, 473–478. [Google Scholar] [CrossRef]
- Tiwari, P.; Blank, A.; Cui, C.; Schoenfelt, K.Q.; Zhou, G.; Xu, Y.; Khramtsova, G.; Olopade, F.; Shah, A.M.; Khan, S.A.; et al. Metabolically activated adipose tissue macrophages link obesity to triple-negative breast cancer. J. Exp. Med. 2019, 216, 1345–1358. [Google Scholar] [CrossRef]
- Pérez-Pérez, A.; Sánchez-Jiménez, F.; Vilariño-García, T.; Sánchez-Margalet, V. Role of leptin in inflammation and vice versa. Int. J. Mol. Sci. 2020, 21, 5887. [Google Scholar] [CrossRef]
- García-Miranda, A.; Garcia-Hernandez, A.; Castañeda-Saucedo, E.; Navarro-Tito, N.; Maycotte, P. Adipokines as regulators of autophagy in obesity-linked cancer. Cells 2022, 11, 3230. [Google Scholar] [CrossRef]
- Lipsey, C.C.; Harbuzariu, A.; Daley-Brown, D.; Gonzalez-Perez, R.R. Oncogenic role of leptin and Notch interleukin-1 leptin crosstalk outcome in cancer. World, J. Methodol. 2016, 6, 43–55. [Google Scholar] [CrossRef]
- Atoum, M.F.; Alzoughool, F.; Al-Hourani, H. Linkage between obesity leptin and breast cancer. Breast Cancer 2020, 14, 178223419898458. [Google Scholar] [CrossRef]
- Huang, Y.; Jin, Q.; Su, M.; Ji, F.; Wang, N.; Zhong, C.; Jiang, Y.; Liu, Y.; Zhang, Z.; Yang, J.; et al. Leptin promotes the migration and invasion of breast cancer cells by upregulating ACAT2. Cell Oncol. 2017, 40, 537–547. [Google Scholar] [CrossRef]
- Flores-López, L.A.; Martínez-Hernández, M.G.; Viedma-Rodríguez, R.; Díaz-Flores, M.; Baiza-Gutman, L.A. High glucose and insulin enhance uPA expression, ROS formation and invasiveness in breast cancer-derived cells. Cell Oncol. 2016, 39, 365–378. [Google Scholar] [CrossRef]
- Nepal, S.; Kim, M.J.; Hong, J.T.; Kim, S.H.; Sohn, D.H.; Lee, S.H.; Song, K.; Choi, D.Y.; Lee, E.S.; Park, P.H. Autophagy induction by leptin contributes to suppression of apoptosis in cancer cells and xenograft model: Involvement of p53/FoxO3A axis. Oncotarget 2015, 6, 7166–7181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrera-Vargas, A.K.; García-Rodríguez, E.; Olea-Flores, M.; Mendoza-Catalán, M.A.; Flores-Alfaro, E.; Navarro-Tito, N. Pro-angiogenic activity and vasculogenic mimicry in the tumor microenvironment by leptin in cancer. Cytokine Growth Factor Rev. 2021, 62, 23–41. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Guo, S.; Gonzalez-Perez, R.R. Leptin pro-angiogenic signature in breast cancer is linked to IL-1 signalling. Br. J. Cancer 2011, 104, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Bieńkiewicz, J.; Romanowicz, H.; Wilczyński, M.; Jabłoński, G.; Stepowicz, A.; Obłękowska, A.; Malinowski, A.; Smolarz, B. Association of single nucleotide polymorphism LEP-R c.668A>G (p.Gln223Arg, rs1137101) of leptin receptor gene with endometrial cancer. BMC Cancer 2021, 21, 925. [Google Scholar] [CrossRef] [PubMed]
- Tayel, S.I.; Alhanafy, A.M.; Ahmed, S.M.; Eltorgoman, A.A.; Elsayed, I.E. Biochemical study on modifying role of variants of leptin gene and its receptor on serum leptin levels in breast cancer. Mol. Biol. Rep. 2020, 47, 3807–3820. [Google Scholar] [CrossRef] [PubMed]
- Paes, J.; Silva, G.A.V.; Tarragô, A.M.; Mourão, L.P.S. The contribution of JAK2 46/1 haplotype in the predisposition to myeloproliferative neoplasms. Int. J. Mol. Sci. 2022, 23, 12582. [Google Scholar] [CrossRef]
- Jadid, F.Z.; Chihab, H.; Alj, H.S.; Elfihry, R.; Zaidane, I.; Tazi, S.; Badre, W.; Marchio, A.; El Filali, K.M.; Tahiri, M.; et al. Control of progression towards liver fibrosis and hepatocellular carcinoma by SOCS3 polymorphisms in chronic HCV-infected patients. Infect. Genet. Evol. 2018, 66, 1–8. [Google Scholar] [CrossRef]
- Zheng, H.; Yan, Y.; Cheng, J.; Yu, S.; Wang, Y. Association between SOCS3 hypermethylation and HBV-related hepatocellular carcinoma and effect of sex and age: A meta-analysis. Medicine 2021, 100, e27604. [Google Scholar] [CrossRef]
- Jiménez-Cortegana, C.; López-Saavedra, A.; Sánchez-Jiménez, F.; Pérez-Pérez, A.; Castiñeiras, J.; Virizuela-Echaburu, J.A.; de la Cruz-Merino, L.; Sánchez-Margalet, V. Leptin, both bad and good actor in cancer. Biomolecules 2021, 11, 913. [Google Scholar] [CrossRef]
- Thompson, K.J.; Lau, K.N.; Johnson, S.; Martinie, J.B.; Iannitti, D.A.; McKillop, I.H.; Sindram, D. Leptin inhibits hepatocellular carcinoma proliferation via p38-MAPK-dependent signalling. HPB 2011, 13, 225–233. [Google Scholar] [CrossRef]
- van Schooneveld, E.; Wildiers, H.; Vergote, I.; Vermeulen, P.B.; Dirix, L.Y.; Van Laere, S.J. Dysregulation of microRNAs in breast cancer and their potential role as prognostic and predictive biomarkers in patient management. Breast Cancer Res. 2015, 17, 21. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.F.; Zhang, Y.; Li, X.Y.; Li, C.; Tian, W.; Liu, L. Expression of miR-31, miR-125b-5p, and miR-326 in the adipogenic differentiation process of adipose-derived stem cells. OMICS 2009, 13, 331–336. [Google Scholar] [CrossRef]
- Yu, F.; Yao, H.; Zhu, P.; Zhang, X.; Pan, Q.; Gong, C.; Huang, Y.; Hu, X.; Su, F.; Lieberman, J.; et al. let-7 regulates self-renewal and tumorigenicity of breast cancer cells. Cell 2007, 131, 1109–1123. [Google Scholar] [CrossRef] [Green Version]
- Al-Rawaf, H. Circulating microRNAs and adipokines as markers of metabolic syndrome in adolescents with obesity. Clin. Nutr. 2019, 38, 2231–2238. [Google Scholar] [CrossRef]
- Zhang, R.; Wang, D.; Xia, Z.; Chen, C.; Cheng, P.; Xie, H.; Luo, X. The role of microRNAs in adipocyte differentiation. Front. Med. 2013, 7, 223–230. [Google Scholar] [CrossRef]
- Xia, C.; Yang, Y.; Kong, F.; Kong, Q.; Shan, C. MiR-143-3p inhibits the proliferation, cell migration and invasion of human breast cancer cells by modulating the expression of MAPK7. Biochimie 2014, 147, 98–104. [Google Scholar] [CrossRef]
- Bartholomeusz, C.; Gonzalez-Angulo, A.M.; Liu, P.; Hayashi, N.; Lluch, A.; Ferrer-Lozano, J.; Hortobágyi, G.N. High ERK protein expression levels correlate with shorter survival in triple-negative breast cancer patients. Oncologist 2012, 17, 766–774. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; Li, H.; Shi, J. miR-27 inhibits the NF-κB signaling pathway by targeting leptin in osteoarthritic chondrocytes. Int. J. Mol. Med. 2017, 40, 523–530. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.H.; Chang, A.C.; Wang, S.W.; Wang, S.J.; Chang, Y.S.; Chang, T.M.; Hsu, S.K.; Fong, Y.C.; Tang, C.H. Leptin promotes VEGF-C production and induces lymphangiogenesis by suppressing miR-27b in human chondrosarcoma cells. Sci. Rep. 2016, 6, 28647. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Si, W.; Shen, J.; Du, C.; Lou, W.; Bao, C.; Zheng, H.; Pan, J.; Zhong, G.; Xu, L.; et al. miR-27b-3p inhibits proliferation and potentially reverses multi-chemoresistance by targeting CBLB/GRB2 in breast cancer cells. Cell Death Dis. 2018, 9, 188. [Google Scholar] [CrossRef]
- Chen, J.; Xu, T.; Chen, C. The critical roles of miR-21 in anti-cancer effects of curcumin. Ann. Transl. Med. 2015, 3, 330. [Google Scholar] [CrossRef] [PubMed]
- Khaidakov, M.; Mehta, J.L. Oxidized LDL triggers pro-oncogenic signaling in human breast mammary epithelial cells partly via stimulation of MiR-21. PLoS ONE 2012, 7, e46973. [Google Scholar] [CrossRef] [PubMed]
- Katsarou, A.; Moustakas, I.I.; Pyrina, I.; Lembessis, P.; Koutsilieris, M.; Chatzigeorgiou, A. Metabolic inflammation as an instigator of fibrosis during non-alcoholic fatty liver disease. World J. Gastroenterol. 2020, 26, 1993–2011. [Google Scholar] [CrossRef] [PubMed]
- Khanmohammadi, S.; Kuchay, M.S. Toll-like receptors and metabolic (dysfunction)-associated fatty liver disease. Pharmacol. Res. 2022, 185, 106507. [Google Scholar] [CrossRef] [PubMed]
- Arias-Loste, M.T.; Iruzubieta, P.; Puente, Á.; Ramos, D.; Santa Cruz, C.; Estébanez, Á.; Llerena, S.; Alonso-Martín, C.; San Segundo, D.; Álvarez, L.; et al. Increased expression profile and functionality of TLR6 in peripheral blood mononuclear cells and hepatocytes of morbidly obese patients with non-alcoholic fatty liver disease. Int. J. Mol. Sci. 2016, 17, 1878. [Google Scholar] [CrossRef] [Green Version]
- Wong, V.W.; Wong, G.L.; Choi, P.C.; Chan, A.W.; Li, M.K.; Chan, H.Y.; Chim, A.M.; Yu, J.; Sung, J.J.; Chan, H.L. Disease progression of non-alcoholic fatty liver disease: A prospective study with paired liver biopsies at 3 years. Gut 2010, 59, 969–974. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Aronis, K.N.; Kountouras, J.; Raptis, D.D.; Vasiloglou, M.F.; Mantzoros, C.S. Circulating leptin in non-alcoholic fatty liver disease: A systematic review and meta-analysis. Diabetologia 2016, 59, 30–43. [Google Scholar] [CrossRef]
- Martínez-Uña, M.; López-Mancheño, Y.; Diéguez, C.; Fernández-Rojo, M.A.; Novelle, M.G. Unraveling the role of leptin in liver function and its relationship with liver diseases. Int. J. Mol. Sci. 2020, 21, 9368. [Google Scholar] [CrossRef]
- Petrescu, A.D.; Grant, S.; Williams, E.; An, S.Y.; Seth, N.; Shell, M.; Amundsen, T.; Tan, C.; Nadeem, Y.; Tjahja, M.; et al. Leptin enhances hepatic fibrosis and inflammation in a mouse model of cholestasis. Am. J. Pathol. 2022, 192, 484–502. [Google Scholar] [CrossRef]
- Akinci, G.; Celik, M.; Akinci, B. Complications of lipodystrophy syndromes. Presse Med. 2021, 50, 104085. [Google Scholar] [CrossRef]
- Kořínková, L.; Pražienková, V.; Černá, L.; Karnošová, A.; Železná, B.; Kuneš, J.; Maletínská, L. Pathophysiology of NAFLD and NASH in experimental models: The role of food intake regulating peptides. Front. Endocrinol. 2020, 11, 597583. [Google Scholar] [CrossRef]
- Guan, L.J.; Xu, K.X.; Xu, S.Y.; Li, N.N.; Wang, X.R.; Xia, Y.K.; Wu, D. Profiles of metabolic gene expression in the white adipose tissue, liver and hypothalamus in leptin knockout (LepΔI14/ΔI14) rats. J. Biomed. Res. 2017, 31, 528–540. [Google Scholar] [CrossRef]
- Banini, B.A.; Kumar, D.P.; Cazanave, S.; Seneshaw, M.; Mirshahi, F.; Santhekadur, P.K.; Wang, L.; Guan, H.P.; Oseini, A.M.; Alonso, C.; et al. Identification of a metabolic, transcriptomic, and molecular signature of patatin-like phospholipase domain containing 3-mediated acceleration of steatohepatitis. Hepatology 2021, 73, 1290–1306. [Google Scholar] [CrossRef]
- Pontes-da-Silva, R.M.; de Souza Marinho, T.; de Macedo Cardoso, L.E.; Mandarim-de-Lacerda, C.A.; Aguila, M.B. Obese mice weight loss role on nonalcoholic fatty liver disease and endoplasmic reticulum stress treated by a GLP-1 receptor agonist. Int. J. Obes. 2022, 46, 21–29. [Google Scholar] [CrossRef]
- Bi, J.; Sun, K.; Wu, H.; Chen, X.; Tang, H.; Mao, J. PPARγ alleviated hepatocyte steatosis through reducing SOCS3 by inhibiting JAK2/STAT3 pathway. Biochem. Biophys. Res. Commun. 2018, 498, 1037–1044. [Google Scholar] [CrossRef]
- Pan, X.; Zheng, M.; Zou, T.; Liu, W.; Gu, X.; Zhang, X.; Cheng, X. The LEPR K109R and Q223R might contribute to the risk of NAFLD: A meta-analysis. Curr. Mol. Med. 2018, 18, 91–99. [Google Scholar] [CrossRef]
- Lopez-Yus, M.; Lorente-Cebrian, S.; Del Moral-Bergos, R.; Hörndler, C.; Garcia-Sobreviela, M.P.; Casamayor, C.; Sanz-Paris, A.; Bernal-Monterde, V.; Arbones-Mainar, J.M. Identification of novel targets in adipose tissue involved in non-alcoholic fatty liver disease progression. FASEB J. 2022, 36, e22429. [Google Scholar] [CrossRef]
- Cao, C.; Duan, P.; Li, W.; Guo, Y.; Zhang, J.; Gui, Y.; Yuan, S. Lack of miR-379/miR-544 cluster resists high-fat diet-induced obesity and prevents hepatic triglyceride accumulation in mice. Front. Cell. Dev. Biol. 2021, 9, 720900. [Google Scholar] [CrossRef]
- Ge, Y.; Gu, P.; Wang, W.; Cao, L.; Zhang, L.; Li, J.; Mu, W.; Wang, H. Benzo[a]pyrene stimulates miR-650 expression to promote the pathogenesis of fatty liver disease and hepatocellular carcinoma via SOCS3/JAK/STAT3 cascades. J. Mol. Cell. Biol. 2021, 13, 556–564. [Google Scholar] [CrossRef]
- Pogoda, P.; Egermann, M.; Schnell, J.C.; Priemel, M.; Schilling, A.F.; Alini, M.; Schinke, T.; Rueger, J.M.; Schneider, E.; Clarke, I.; et al. Leptin inhibits bone formation not only in rodents, but also in sheep. J. Bone Miner. Res. 2006, 21, 1591–1599. [Google Scholar] [CrossRef]
- Karsenty, G.; Khosla, S. The crosstalk between bone remodeling and energy metabolism: A translational perspective. Cell Metab. 2022, 34, 805–817. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Rohatgi, N.; Brestoff, J.R.; Zhang, Y.; Scheller, E.L.; Craft, C.S.; Brodt, M.D.; Migotsky, N.; Silva, M.J.; Harris, C.A.; et al. Congenital lipodystrophy induces severe osteosclerosis. PLoS Genet. 2019, 15, e1008244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordeladze, J.O.; Reseland, J.E. A unified model for the action of leptin on bone turnover. J. Cell. Biochem. 2003, 88, 706–712. [Google Scholar] [CrossRef] [PubMed]
- Cirmanova, V.; Zofkova, I.; Kasalicky, P.; Lanska, V.; Bayer, M.; Starka, L.; Kanceva, R. Hormonal and bone parameters in pubertal girls. Physiol. Res. 2017, 66, S419–S424. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, M.T.; de la Piedra, C.; Barrios, V.; Garrido, G.; Argente, J. Changes in bone density and bone markers in rhythmic gymnasts and ballet dancers: Implications for puberty and leptin levels. Eur. J. Endocrinol. 2004, 151, 491–496. [Google Scholar] [CrossRef] [Green Version]
- Bartell, S.M.; Rayalam, S.; Ambati, S.; Gaddam, D.R.; Hartzell, D.L.; Hamrick, M.; She, J.X.; Della-Fera, M.A.; Baile, C.A. Central (ICV) leptin injection increases bone formation, bone mineral density, muscle mass, serum IGF-1, and the expression of osteogenic genes in leptin-deficient ob/ob mice. J. Bone Miner. Res. 2011, 26, 1710–1720. [Google Scholar] [CrossRef]
- Zhou, H.; Newnum, A.B.; Martin, J.R.; Li, P.; Nelson, M.T.; Moh, A.; Fu, X.Y.; Yokota, H.; Li, J. Osteoblast/osteocyte-specific inactivation of Stat3 decreases load-driven bone formation and accumulates reactive oxygen species. Bone 2011, 49, 404–411. [Google Scholar] [CrossRef]
- Mitchell, A.L.; Urban, A.K.; Freeman, A.F.; Hammoud, D.A. An unusual pattern of premature cervical spine degeneration in STAT3-LOF. J. Clin. Immunol. 2021, 41, 576–584. [Google Scholar] [CrossRef]
- McGaffin, K.R.; Witham, W.G.; Yester, K.A.; Romano, L.C.; O’Doherty, R.M.; McTiernan, C.F.; O’Donnell, C.P. Cardiac-specific leptin receptor deletion exacerbates ischaemic heart failure in mice. Cardiovasc. Res. 2011, 89, 60–71. [Google Scholar] [CrossRef] [Green Version]
- Kamareddine, L.; Ghantous, C.M.; Allouch, S.; Al-Ashmar, S.A.; Anlar, G.; Kannan, S.; Djouhri, L.; Korashy, H.M.; Agouni, A.; Zeidan, A. Between inflammation and autophagy: The role of leptin-adiponectin axis in cardiac remodeling. J. Inflamm. Res. 2021, 14, 5349–5365. [Google Scholar] [CrossRef]
- Momken, I.; Chabowski, A.; Dirkx, E.; Nabben, M.; Jain, S.S.; McFarlan, J.T.; Glatz, J.F.; Luiken, J.J.; Bonen, A. A new leptin-mediated mechanism for stimulating fatty acid oxidation: A pivotal role for sarcolemmal FAT/CD36. Biochem. J. 2017, 474, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Burgos-Ramos, E.; Canelles, S.; Rodríguez, A.; Frago, L.M.; Gómez-Ambrosi, J.; Chowen, J.A.; Frühbeck, G.; Argente, J.; Barrios, V. The increase in fiber size in male rat gastrocnemius after chronic central leptin infusion is related to activation of insulin signaling. Mol. Cell. Endocrinol. 2018, 470, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Schönke, M.; Björnholm, M.; Chibalin, A.V.; Zierath, J.R.; Deshmukh, A.S. Proteomics analysis of skeletal muscle from leptin-deficient ob/ob mice reveals adaptive remodeling of metabolic characteristics and fiber type composition. Proteomics 2018, 18, e1700375. [Google Scholar] [CrossRef] [PubMed]
- Balatsky, V.; Bankovska, I.; Pena, R.N.; Saienko, A.; Buslyk, T.; Korinnyi, S.; Doran, O. Polymorphisms of the porcine cathepsins, growth hormone-releasing hormone and leptin receptor genes and their association with meat quality traits in Ukrainian large white breed. Mol. Biol. Rep. 2016, 43, 517–526. [Google Scholar] [CrossRef] [Green Version]
- Kripa, E.; Rizzo, V.; Galati, F.; Moffa, G.; Cicciarelli, F.; Catalano, C.; Pediconi, F. Do body composition parameters correlate with response to targeted therapy in ER+/HER2- metastatic breast cancer patients? Role of sarcopenia and obesity. Front. Oncol. 2022, 12, 987012. [Google Scholar] [CrossRef]
- Ng, T.P.; Lu, Y.; Choo, R.W.M.; Tan, C.T.Y.; Nyunt, M.S.Z.; Gao, Q.; Mok, E.W.H.; Larbi, A. Dysregulated homeostatic pathways in sarcopenia among frail older adults. Aging Cell 2018, 17, e12842. [Google Scholar] [CrossRef] [Green Version]
- Hamrick, M.W.; Herberg, S.; Arounleut, P.; He, H.Z.; Shiver, A.; Qi, R.Q.; Zhou, L.; Isales, C.M.; Mi, Q.S. The adipokine leptin increases skeletal muscle mass and significantly alters skeletal muscle miRNA expression profile in aged mice. Biochem. Biophys. Res. Commun. 2010, 400, 379–383. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Casado, M.E.; Collado-Pérez, R.; Frago, L.M.; Barrios, V. Recent Advances in the Knowledge of the Mechanisms of Leptin Physiology and Actions in Neurological and Metabolic Pathologies. Int. J. Mol. Sci. 2023, 24, 1422. https://doi.org/10.3390/ijms24021422
Casado ME, Collado-Pérez R, Frago LM, Barrios V. Recent Advances in the Knowledge of the Mechanisms of Leptin Physiology and Actions in Neurological and Metabolic Pathologies. International Journal of Molecular Sciences. 2023; 24(2):1422. https://doi.org/10.3390/ijms24021422
Chicago/Turabian StyleCasado, María E., Roberto Collado-Pérez, Laura M. Frago, and Vicente Barrios. 2023. "Recent Advances in the Knowledge of the Mechanisms of Leptin Physiology and Actions in Neurological and Metabolic Pathologies" International Journal of Molecular Sciences 24, no. 2: 1422. https://doi.org/10.3390/ijms24021422
APA StyleCasado, M. E., Collado-Pérez, R., Frago, L. M., & Barrios, V. (2023). Recent Advances in the Knowledge of the Mechanisms of Leptin Physiology and Actions in Neurological and Metabolic Pathologies. International Journal of Molecular Sciences, 24(2), 1422. https://doi.org/10.3390/ijms24021422