
Treg Therapy for the Induction of Immune Tolerance in Transplantation—Not Lost in Translation?


Abstract
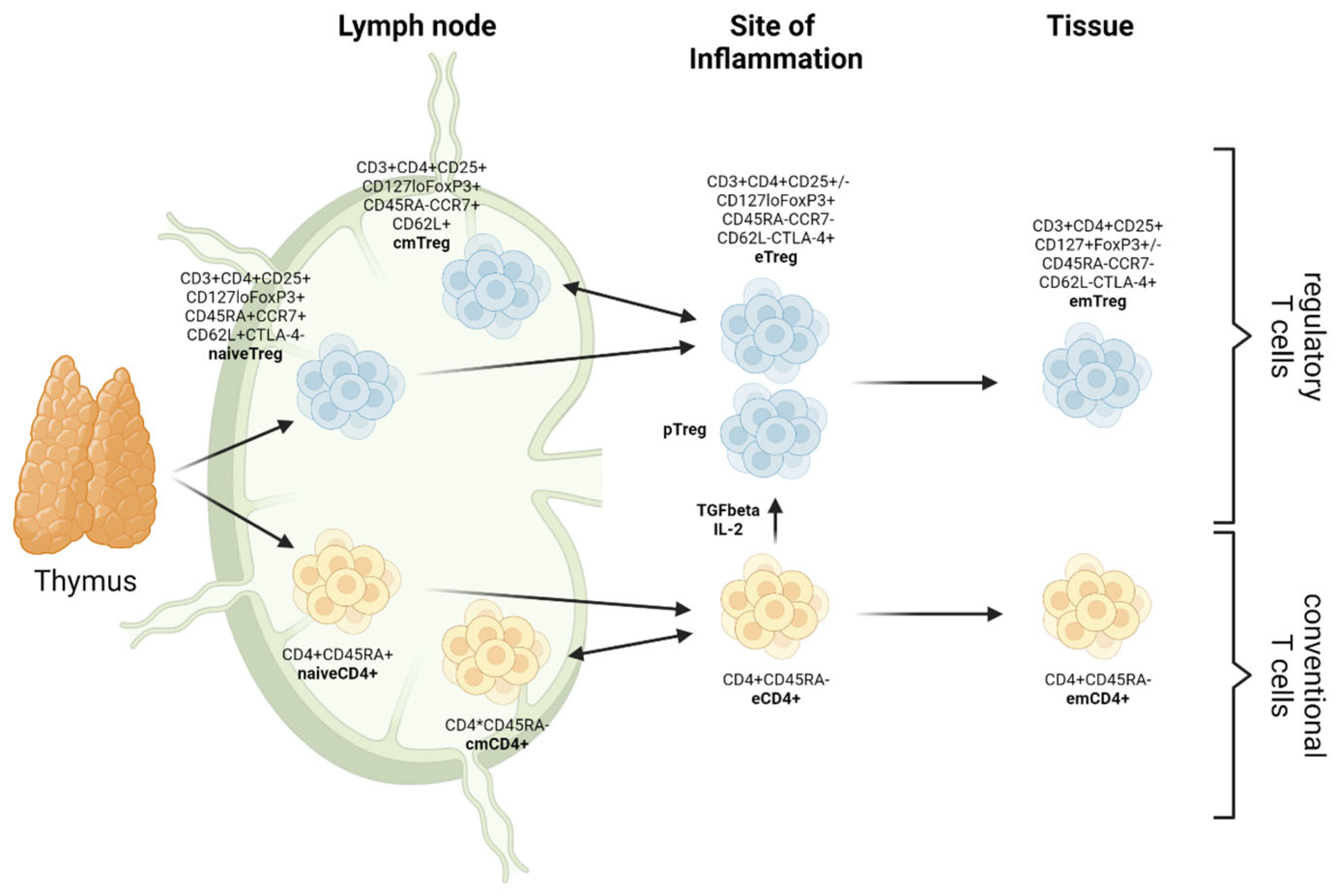
:1. Introduction
2. Current Status and Ongoing Studies

{kind=link}
{kind=link}
{kind=link}
| Study ID | Age | Title | Product | Dose | Status | Location | Aim/Results | |
|---|---|---|---|---|---|---|---|---|
| Renal Transplantation—endogenous Treg expansion | ||||||||
| NCT02417870 | I/II | 18–75 (adult, older adult) | Ultra-low Dose Subcutaneous IL-2 in Renal Transplantation | Low-dose recombinant IL-2 (proleukin) | - | Terminated (June 2021) | Brigham and Women’s Hospital, Boston, MA, US | Safety and efficacy of treatment with low-dose rIL-2 in renal transplant recipients. |
| Renal Transplantation—adoptive Treg therapy | ||||||||
| NCT02088931 | I | 18–50 (adult) | Treg Adoptive Therapy for Subclinical Inflammation in Kidney Transplantation (TASK) | CD4+CD127lo/-CD25+ polyclonally expanded Tregs | 3.2 × 108 | Completed (July 2022) | University of California, San Francisco, CA, US | Results: Approach is safe and feasible. One patient developed acute cellular rejection. Infused Tregs remained detectable for 1 month. [15] |
| NCT02091232 | I | >18 (adult, older adult) | Infusion of T-Regulatory Cells in Kidney Transplant Recipients (The ONE Study) | Tregs (recipient) stimulated with donor PBMCs and belatacept | 4–9 × 108 | Completed (Nov 2021) | Massachusetts General Hospital, Boston, MA, US | To examine in living donor renal transplant recipients the safety and feasibility of administering T regulatory cells derived from recipient PBMC stimulated with kidney donor PBMC in the presence of costimulatory blockade with belatacept. [21] |
| NCT04817774 | I/II | 18–70 (adult, older adult) | Safety & Tolerability Study of Chimeric Antigen Receptor T-Reg Cell Therapy in Living Donor Renal Transplant Recipients | CD4+ CD45RA+ CD25+ CD127low/- HLA-A*02-specific CAR Tregs | - | Recruiting (Dec 2021) | University Hospitals Leuven, Leuven, Belgium (and 3 other centers) | A multicenter, first-in-human, open-label, single-ascending-dose, dose-ranging study of autologous, chimeric antigen receptor T regulatory cells (CAR-Treg) in HLA-A2-mismatched living-donor kidney transplant recipients. |
| NCT03943238 | I | 18–65 (adult, older adult) | TLI, TBI, ATG & Hematopoietic Stem Cell Transplantation and Recipient T Regs Therapy in Living Donor Kidney Transplantation | Autologous polyclonally expanded Tregs | Starting at 25 × 106/kg | Recruiting (May 2022) | Stanford University, Palo Alto, Northwestern University, Chicago, IL, US | To determine if total lymphoid irradiation (TLI) in combination with anti-thymocyte globulin (ATG) and infusion of donor hematopoietic stem cells along with recipient Tregs will allow for discontinuation of immunosuppressive treatment after living-donor kidney transplantation. |
| NCT03284242 | n/a | 18–65 (adult, older adult) | A Pilot Study Using Autologous Regulatory T Cell Infusion Zortress (Everolimus) in Renal Transplant Recipients | Autologous polyclonally expanded Tregs | n/a | Recruiting (May 2022) | University of Kentucky Medical Center, Lexington, Kentucky, US | Safety and effectiveness of infusion of autologous polyclonally expanded Tregs to renal transplant recipients receiving Zortress (Everolimus) as immunosuppressive therapy. |
| NCT02711826 | I/II | >18 (adult, older adult) | Treg Therapy in Subclinical Inflammation in Kidney Transplantation | Autologous polyclonally expanded Tregs | 5.5 ± 4.5 × 108 | Recruiting (March 2022) | University of California at San Francisco, CA, US (and 5 other centers) | To determine the safety and efficacy of a single dose of autologous polyclonal Tregs in renal transplant recipients with subclinical inflammation (SCI) in the 3–7 months post-transplant allograft protocol biopsy compared to control patients treated with CNI-based immunosuppression. |
| NCT02145325 | I | 18–65 (adult, older adult) | Trial of Adoptive Immunotherapy with TRACT to Prevent Rejection in Living Donor Kidney Transplant Recipients | Autologous polyclonal expanded CD4+CD25+ nTregs | 0.5–5 × 106 | Completed (Oct 2019) | Northwestern University Comprehensive Transplant Center, Chicago, Illinois, US | Results: Approach is safe and feasible. Circulating levels of Tregs were increased for 1-year follow-up. [16] |
| NCT03867617 | I/II | >18 (adult, older adult) | Cell Therapy for Immunomodulation in Kidney Transplantation | Autologous polyclonally expanded CD4+ CD127lo/- CD25+ CD45RA Tregs | 0.3–1.5 × 107 | Recruiting (Sep 2019) | Medical University of Vienna, Vienna, Austria | Treatment combining ex vivo expanded recipient regulatory T cells with donor bone marrow and Tocilizumab for feasible, safe and efficacious induction of transient chimerism in living-donor kidney transplant recipients. |
| NCT01446484 | I/II | 1–18 (child) | Treatment of Children with Kidney Transplants by Injection of CD4+CD25+FoxP3+ T Cells to Prevent Organ Rejection | Autologous CD4+ CD25+ CD127low FoxP3+ Tregs | 2 × 108 | Unknown (Nov 2011) | Russian State Medical University, Moscow, Russian Federation | This study will evaluate the treatment of children who received a kidney transplant with Alemtuzumab or other immunosuppressing medications in combination with injection of autologous ex vivo expanded Tregs. |
| NCT02371434 | I/II | 18–65 (adult, older adult) | The ONE Study nTreg Trial (ONEnTreg13) | Autologous polyclonally expanded CD4+ CD25+ FoxP3+ nTregs | 0.5–3 × 106 | Completed (Feb 2020) | Charité University Medicine, Berlin, Germany | Results: Tapering of immunosuppressive medication to low-dose tacrolimus is safe and feasible. (n = 8 patients) [21] |
| NCT02244801 | I | 18–70 (adult, older adult) | Donor-Alloantigen-Reactive Regulatory T Cell (darTreg) Therapy in Renal Transplantation (The ONE Study) | Donor-alloantigen-reactive Tregs (darTregs) | 3 × 108; 9 × 108 | Completed (Oct 2018) | University of California San Francisco, CA, US | Results: Treg-based therapy is achievable and safe in living-donor kidney transplant recipients. This approach is associated with fewer infectious complications, but similar rejection rates in the first year. darTregs: Tregs stimulated with B cells that had been activated with K562 cells expressing hCD40L. [21] |
| NCT02129881 | I/II | >18 (adult, older adult) | The ONE Study UK Treg Trial | Autologous polyclonally expanded Tregs | 1–10 × 106/kg | Completed (Jan 2019) | Guy’s Hospital, London, UK | Results: Approach is safe and feasible. Mycophenolate Mofetil (MMF) withdrawn and on Tacrolimus monotherapy. (n = 4 patients) Less opportunistic infections and transient increase of Treg cell numbers were detected. [21] |
| ISRCTN 11038572 | II | >18 (adult, older adult) | TWO study: cell therapy trial in renal transplantation | Autologous polyclonally expanded Tregs | 5–10 × 106/kg | Recruiting (June 2022) | Oxford Transplant Centre, Churchill Hospital, Oxford, UK | This study aims to demonstrate the efficacy of polyclonal Tregs with the goal of allowing for reduction in immunosuppression to a single drug by 6 months post-transplantation. [23] |
| Liver Transplantation—endogenous Treg expansion | ||||||||
| NCT02739412 | II | 18–65 (adult, older adult) | Efficacy of Low Dose, SubQ Interleukin-2 (IL-2) to Expand Endogenous Regulatory T-Cells in Liver Transplant Recipients | Low-dose recombinant IL-2 (proleukin) | 0.30MIU/m2 body surface area; for 4 weeks | Active, not recruiting (May 2021) | Beth Israel Deaconess Medical Center, Boston, MA, US | Aim of this study is to investigate if very-low-dose IL-2, given to liver transplant patients by subcutaneous injections, over a 4-week period of time, will cause an increase in the number of Treg cells in the blood. Includes analysis regarding safety of treatment. |
| NCT02949492 | IV | 18–50 (adult) | Low-dose IL-2 for Treg Expansion and Tolerance (LITE) | Low-dose recombinant IL-2 (proleukin) | - | Terminated (Aug 2019) | King’s College Hospital London, UK | Results: Stable patients 2–6 years post-liver transplantation were treated with low-dose IL-2 to facilitate discontinuation of immunosuppression. Patients achieved a 2-fold increase in circulating Tregs; the trial was terminated after 6 patients developed rejection requiring immunosuppression reinstitution. [24] |
| Liver Transplantation—adoptive cell therapy | ||||||||
| NCT01624077 | I | 10–60 (child, adult) | Safety Study of Using Regulatory T Cells Induce Liver Transplantation Tolerance | Autologous polyclonally TGF-β induced CD4+ CD25+ CD127- Tregs | 1 × 106/kg | Unknown (Feb 2015) | Nanjing Medical University, Nanjing, Jiangsu, China | Generation of donor-alloantigen-specific CD4+CD25+ Tregs from peripheral blood of pretransplant patients, for graft-specific tolerance induction. |
| NCT03654040 | I/II | 18–70 (adult, older adult) | Liver Transplantation with Tregs at UCSF | Autologous expanded donor-alloantigen-reactive Tregs (arTregs) | 30–90 × 106 total Treg cells | Recruiting (Aug 2021) | University of California, San Francisco, San Francisco, California, US | A single-center, prospective, open-label, nonrandomized clinical trial using alloantigen-specific Tregs to facilitate immunosuppression withdrawal in liver transplant recipients. |
| NCT03577431 | I/II | 18–70 (adult, older adult) | Liver Transplantation with Tregs at MGH | Autologous expanded donor-alloantigen-reactive CD4+ CD25+ CD127- Treg cells (arTregs) | 2.5–125 × 106 | Recruiting (Nov 2021) | Massachusetts General Hospital: Transplantation, Boston, MA, United States | A single-center, prospective, open-label, nonrandomized clinical trial exploring cellular therapy to facilitate immunosuppression withdrawal in liver transplant recipients. |
| NCT02474199 | I/II | 18–70 (adult, older adult) | Donor Alloantigen Reactive Tregs (darTregs) for Calcineurin Inhibitor (CNI) Reduction (ARTEMIS) | Autologous donor-alloantigen-reactive Tregs (darTregs) | 3–5 × 108 | Completed (Feb 2021) | University of California at San Francisco, San Francisco, CA, US Northwestern University Comprehensive Transplant Ctr, Chicago, IL, US, Mayo Clinic in Rochester, Rochester, NY, US | Safety of donor-alloantigen-reactive Tregs to facilitate minimization and/or discontinuation of immunosuppression in adult liver transplant recipients. Results: Problems with Treg product manufacturing; discontinuation of immunosuppression not possible. |
| NCT02188719 | I | 21–70 (adult, older adult) | Donor-Alloantigen-Reactive Regulatory T Cell (darTregs) in Liver Transplantation (deLTa) | Autologous donor-alloantigen-reactive Tregs (darTregs) | 2.5–96 × 107 | Terminated (Sep 2020)—has results | University of California at San Francisco, San Francisco, CA, US; Northwestern University Comprehensive Transplant Ctr, Chicago, IL, US, Mayo Clinic in Rochester, Rochester, NY, US | Safety of receiving one or three different doses of donor-alloantigen-reactive Tregs (darTregs) while receiving a specific drug combination. Results: issues regarding donor-specific Treg manufacturing using donor B-cells led to termination. |
| NCT02166177 | I/II | 18–70 (adult, older adult) | Safety and Efficacy Study of Regulatory T Cell Therapy in Liver Transplant Patients (ThRIL) | Autologous polyclonally expanded Tregs | 0.5–1; 3–4.5 × 106/kg | Completed (Jan 2019) | King’s College Hospital, London, UK | Results: Safety of Treg transfer was confirmed. Transient increase of the pool of circulating Tregs and reduced anti-donor T-cell responses were detected. Low applicability of earlier Treg dose (3 months post-transplant). [25] |
| UMIN-000015789 | I/II | 18–65 (adult, older adult) | Tolerance induction by a regulatory T cell-based cell therapy in living donor liver transplantation | Donor-reactive Treg-enriched cell product | 0.23–6.37 × 106 Tregs/kg | Recruiting (until July 2012) Data published 2016 | Hokkaidou University Graduate School of Medicine, Japan | Results: 7 of 10 patients are immunosuppressant-free for >6 years. [13] |
| NCT05234190 | I/II | 18–70 (adult, older adult) | Safety and Clinical Activity of QEL-001 in A2-mismatch Liver Transplant Patients (LIBERATE) | Autologous CAR Tregs targeting HLA-A2 (HLA-A2 CAR-Treg) | - | Recruiting (Feb 2022) | Cambridge University Hospitals NHS Foundation Trust, Cambridge, UK Royal Free London NHS Foundation Trust, London, UK King’s College Hospital NHS Foundation Trust London, UK | A multicenter, first-in-human, open-label, single-arm study of an autologous CAR T regulatory (CAR-Treg) in HLA-A2-mismatched liver transplant recipients. The aim is for the CAR-Tregs to be activated on recognition of HLA-A2 antigens present on the donated liver and subsequently induce and maintain immunological tolerance to the organ. |
| Heart Transplantation—adoptive cell therapy | ||||||||
| NCT04924491 | I/II | 0–2 (child) | Cell Therapy with Treg Cells Obtained from Thymic Tissue (thyTreg) to Prevent Rejection in Heart Transplant Children (THYTECH) | Autologous thyTreg | 10–20 × 106 thyTreg /kg | Recruiting (Aug 2022) | Hospital General Universitario Gregorio Marañon Madrid, Spain | A phase I/II clinical trial testing the safety and efficacy of the adoptive transfer of autologous Treg cells from thymic tissue (thyTreg) discarded in pediatric cardiac surgeries to prevent rejection in heart transplant children. |
3. Open Questions
3.1. Specificity and Efficacy
3.2. Personalized Immunosuppression—Realistic Goals of Treg Therapy?
3.3. Clinical Tolerance: The Criteria for Withdrawing Immunosuppression
4. Future Strategies
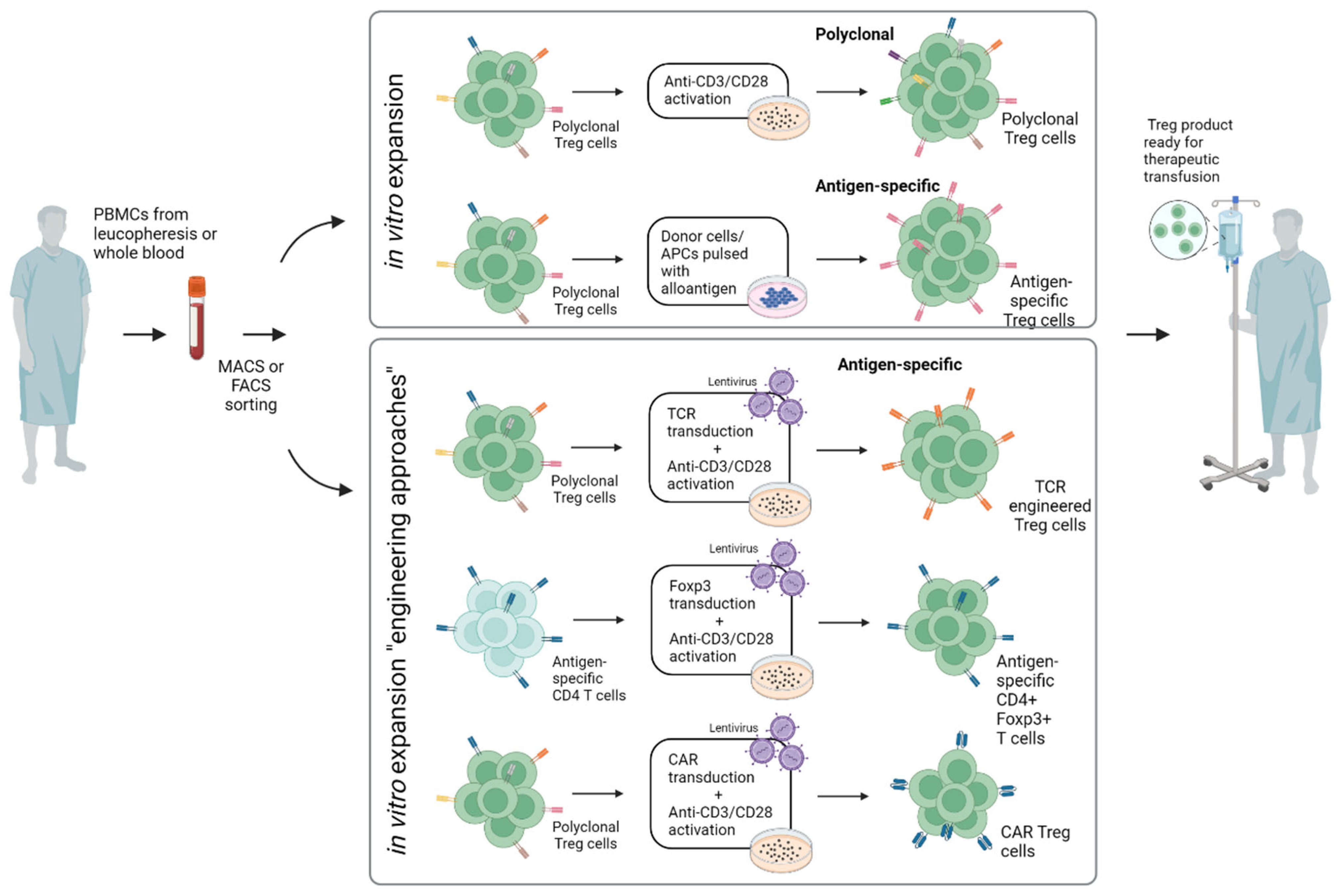
4.1. Clinical Use of CAR Tregs
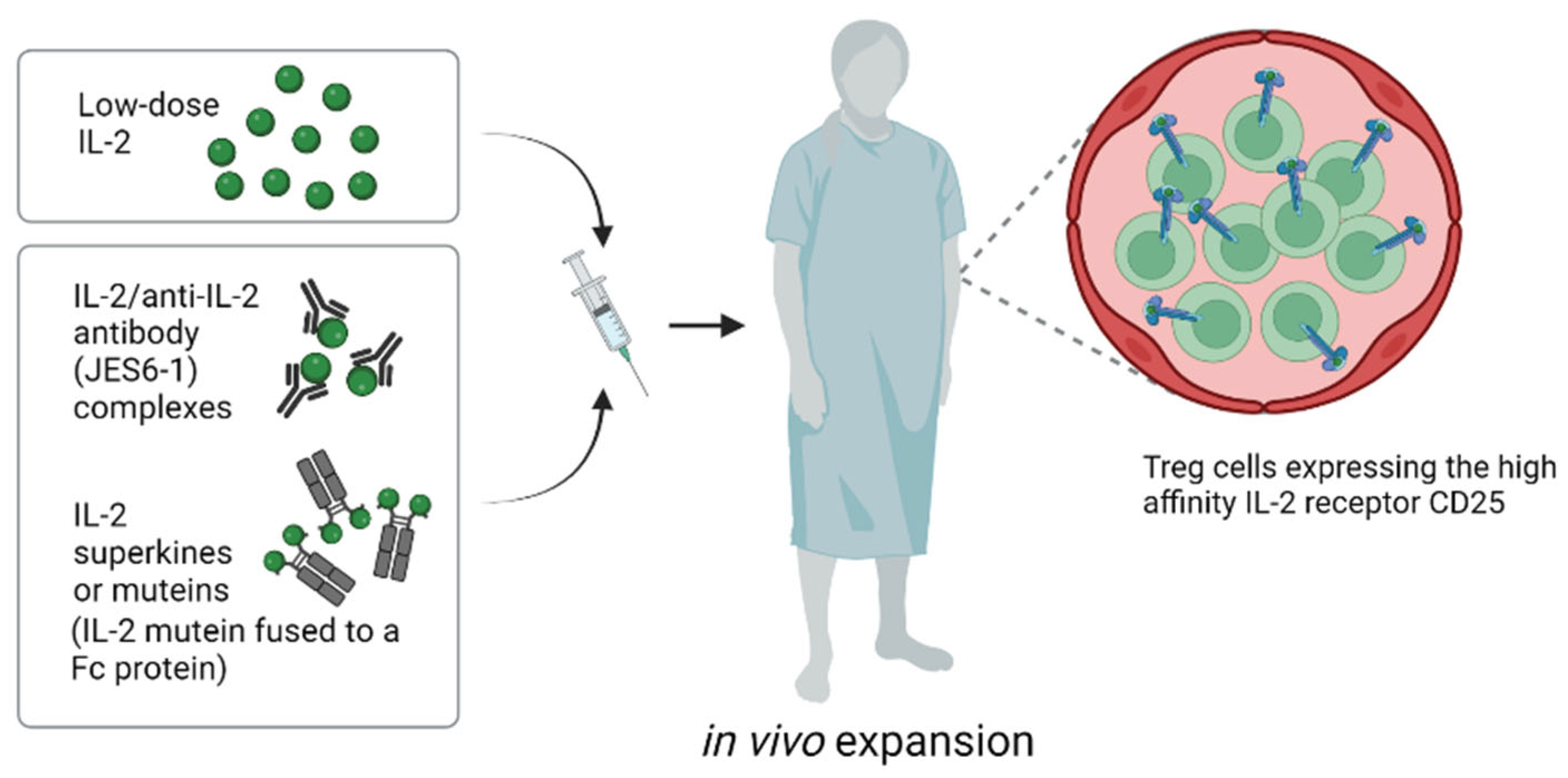
4.2. In Vivo Approaches for Expanding Tregs: IL-2 Complexes and IL-2 Engineering
4.3. Treg Cell-Derived Extracellular Vesicles/Exosomes
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| APC | antigen-presenting cell |
| CAR | chimeric antigen receptor |
| CNI | calineurin inhibitor |
| CTG | cell therapy group |
| CTLA4Ig | cytotoxic T-Lymphocyte-Associated Protein 4 immunoglobulin |
| darTreg | donor-antigen-reactive Treg |
| DSA | donor-specific antibodies |
| EV | extracellular vesicle |
| GMP | good manufacturing practice |
| GvHD | graft-versus-host disease |
| MHC | major histocompatibility complex |
| miRNa | microRNA |
| MS | multiple sclerosis |
| NHP | nonhuman primate |
| Nrp-1 | neuropilin-1 |
| nTreg | natural Treg |
| pTreg | peripheral Treg |
| RGT | reference group trial |
| SOT | solid organ transplantation |
| T1D | type 1 diabetes |
| TCR | T-cell receptor |
| TDSR | Treg-specific demethylated region |
| Tregs | regulatory T cells |
| tTreg | thymic Treg |
References
- Billingham, R.E.; Brent, L.; Medawar, P.B. Actively acquired tolerance of foreign cells. Nature 1953, 172, 603–606. [Google Scholar] [CrossRef] [PubMed]
- Slepicka, P.F.; Yazdanifar, M.; Bertaina, A. Harnessing Mechanisms of Immune Tolerance to Improve Outcomes in Solid Organ Transplantation: A Review. Front. Immunol. 2021, 12, 688460. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Sakaguchi, N.; Asano, M.; Itoh, M.; Toda, M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 1995, 155, 1151–1164. [Google Scholar] [CrossRef]
- Shevach, E.M. From vanilla to 28 flavors: Multiple varieties of T regulatory cells. Immunity 2006, 25, 195–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbas, A.K.; Trotta, E.; Simeonov, D.R.; Marson, A.; Bluestone, J.A. Revisiting IL-2: Biology and therapeutic prospects. Sci. Immunol. 2018, 3, eaat1482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thornton, A.M.; Korty, P.E.; Tran, D.Q.; Wohlfert, E.A.; Murray, P.E.; Belkaid, Y.; Shevach, E.M. Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. J. Immunol. 2010, 184, 3433–3441. [Google Scholar] [CrossRef] [Green Version]
- Yadav, M.; Louvet, C.; Davini, D.; Gardner, J.M.; Martinez-Llordella, M.; Bailey-Bucktrout, S.; Anthony, B.A.; Sverdrup, F.M.; Head, R.; Kuster, D.J.; et al. Neuropilin-1 distinguishes natural and inducible regulatory T cells among regulatory T cell subsets in vivo. J. Exp. Med. 2012, 209, 1713–1722. [Google Scholar] [CrossRef] [Green Version]
- Yadav, M.; Stephan, S.; Bluestone, J.A. Peripherally induced tregs—Role in immune homeostasis and autoimmunity. Front. Immunol. 2013, 4, 232. [Google Scholar] [CrossRef] [Green Version]
- Elkord, E. Helios Should Not Be Cited as a Marker of Human Thymus-Derived Tregs. Commentary: Helios(+) and Helios(-) Cells Coexist within the Natural FOXP3(+) T Regulatory Cell Subset in Humans. Front. Immunol. 2016, 7, 276. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, N.; Okamoto, K.; Sawa, S.; Nakashima, T.; Oh-hora, M.; Kodama, T.; Tanaka, S.; Bluestone, J.A.; Takayanagi, H. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat. Med. 2014, 20, 62–68. [Google Scholar] [CrossRef]
- Shevyrev, D.; Tereshchenko, V. Treg Heterogeneity, Function, and Homeostasis. Front. Immunol. 2019, 10, 3100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raffin, C.; Vo, L.T.; Bluestone, J.A. Treg cell-based therapies: Challenges and perspectives. Nat. Rev. Immunol. 2020, 20, 158–172. [Google Scholar] [CrossRef] [PubMed]
- Todo, S.; Yamashita, K.; Goto, R.; Zaitsu, M.; Nagatsu, A.; Oura, T.; Watanabe, M.; Aoyagi, T.; Suzuki, T.; Shimamura, T.; et al. A pilot study of operational tolerance with a regulatory T-cell-based cell therapy in living donor liver transplantation. Hepatology 2016, 64, 632–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trzonkowski, P.; Bieniaszewska, M.; Juścińska, J.; Dobyszuk, A.; Krzystyniak, A.; Marek, N.; Myśliwska, J.; Hellmann, A. First-in-man clinical results of the treatment of patients with graft versus host disease with human ex vivo expanded CD4+CD25+CD127- T regulatory cells. Clin. Immunol. 2009, 133, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Chandran, S.; Tang, Q.; Sarwal, M.; Laszik, Z.G.; Putnam, A.L.; Lee, K.; Leung, J.; Nguyen, V.; Sigdel, T.; Tavares, E.C.; et al. Polyclonal Regulatory T Cell Therapy for Control of Inflammation in Kidney Transplants. Am. J. Transplant. 2017, 17, 2945–2954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathew, J.M.; H-Voss, J.; LeFever, A.; Konieczna, I.; Stratton, C.; He, J.; Huang, X.; Gallon, L.; Skaro, A.; Ansari, M.J.; et al. A Phase I Clinical Trial with Ex Vivo Expanded Recipient Regulatory T cells in Living Donor Kidney Transplants. Sci. Rep. 2018, 8, 7428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraser, H.; Safinia, N.; Grageda, N.; Thirkell, S.; Lowe, K.; Fry, L.J.; Scotta, C.; Hope, A.; Fisher, C.; Hilton, R.; et al. A Rapamycin-Based GMP-Compatible Process for the Isolation and Expansion of Regulatory T Cells for Clinical Trials. Mol. Ther. Methods Clin. Dev. 2018, 8, 198–209. [Google Scholar] [CrossRef] [Green Version]
- Landwehr-Kenzel, S.; Zobel, A.; Hoffmann, H.; Landwehr, N.; Schmueck-Henneresse, M.; Schachtner, T.; Roemhild, A.; Reinke, P. Ex vivo expanded natural regulatory T cells from patients with end-stage renal disease or kidney transplantation are useful for autologous cell therapy. Kidney Int. 2018, 93, 1452–1464. [Google Scholar] [CrossRef]
- Putnam, A.L.; Safinia, N.; Medvec, A.; Laszkowska, M.; Wray, M.; Mintz, M.A.; Trotta, E.; Szot, G.L.; Liu, W.; Lares, A.; et al. Clinical grade manufacturing of human alloantigen-reactive regulatory T cells for use in transplantation. Am. J. Transplant. 2013, 13, 3010–3020. [Google Scholar] [CrossRef] [Green Version]
- Guinan, E.C.; Cole, G.A.; Wylie, W.H.; Kelner, R.H.; Janec, K.J.; Yuan, H.; Oppatt, J.; Brennan, L.L.; Turka, L.A.; Markmann, J. Ex Vivo Costimulatory Blockade to Generate Regulatory T Cells From Patients Awaiting Kidney Transplantation. Am. J. Transplant. 2016, 16, 2187–2195. [Google Scholar] [CrossRef]
- Sawitzki, B.; Harden, P.N.; Reinke, P.; Moreau, A.; Hutchinson, J.A.; Game, D.S.; Tang, Q.; Guinan, E.C.; Battaglia, M.; Burlingham, W.J.; et al. Regulatory cell therapy in kidney transplantation (The ONE Study): A harmonised design and analysis of seven non-randomised, single-arm, phase 1/2A trials. Lancet 2020, 395, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Ezzelarab, M.B.; Zhang, H.; Sasaki, K.; Lu, L.; Zahorchak, A.F.; van der Windt, D.J.; Dai, H.; Perez-Gutierrez, A.; Bhama, J.K.; Thomson, A.W. Ex Vivo Expanded Donor Alloreactive Regulatory T Cells Lose Immunoregulatory, Proliferation, and Antiapoptotic Markers After Infusion Into ATG-lymphodepleted, Nonhuman Primate Heart Allograft Recipients. Transplantation 2021, 105, 1965–1979. [Google Scholar] [CrossRef] [PubMed]
- Brook, M.O.; Hester, J.; Petchey, W.; Rombach, I.; Dutton, S.; Bottomley, M.J.; Black, J.; Abdul-Wahab, S.; Bushell, A.; Lombardi, G.; et al. Transplantation Without Overimmunosuppression (TWO) study protocol: A phase 2b randomised controlled single-centre trial of regulatory T cell therapy to facilitate immunosuppression reduction in living donor kidney transplant recipients. BMJ Open 2022, 12, e061864. [Google Scholar] [CrossRef] [PubMed]
- Lim, T.Y.; Perpinan, E.; Londono, M.C.; Miquel, R.; Ruiz, P.; Kurt, A.S.; Kodela, E.; Cross, A.R.; Berlin, C.; Hester, J.; et al. Low dose interleukin-2 selectively expands circulating regulatory T cells but fails to promote liver allograft tolerance in humans. J. Hepatol. 2023, 78, 153–164. [Google Scholar] [CrossRef]
- Sánchez-Fueyo, A.; Whitehouse, G.; Grageda, N.; Cramp, M.E.; Lim, T.Y.; Romano, M.; Thirkell, S.; Lowe, K.; Fry, L.; Heward, J.; et al. Applicability, safety, and biological activity of regulatory T cell therapy in liver transplantation. Am. J. Transplant. 2020, 20, 1125–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, L.M.; Zhang, H.; Lee, K.; Liang, H.; Merleev, A.; Vincenti, F.; Maverakis, E.; Thomson, A.W.; Tang, Q. A Comparison of Ex Vivo Expanded Human Regulatory T Cells Using Allogeneic Stimulated B Cells or Monocyte-Derived Dendritic Cells. Front. Immunol. 2021, 12, 679675. [Google Scholar] [CrossRef] [PubMed]
- Tsang, J.Y.; Tanriver, Y.; Jiang, S.; Xue, S.A.; Ratnasothy, K.; Chen, D.; Stauss, H.J.; Bucy, R.P.; Lombardi, G.; Lechler, R. Conferring indirect allospecificity on CD4+CD25+ Tregs by TCR gene transfer favors transplantation tolerance in mice. J. Clin. Investig. 2008, 118, 3619–3628. [Google Scholar] [CrossRef]
- Zhao, L.; Cao, Y.J. Engineered T Cell Therapy for Cancer in the Clinic. Front. Immunol. 2019, 10, 2250. [Google Scholar] [CrossRef] [Green Version]
- Jaeckel, E.; von Boehmer, H.; Manns, M.P. Antigen-specific FoxP3-transduced T-cells can control established type 1 diabetes. Diabetes 2005, 54, 306–310. [Google Scholar] [CrossRef]
- MacDonald, K.G.; Hoeppli, R.E.; Huang, Q.; Gillies, J.; Luciani, D.S.; Orban, P.C.; Broady, R.; Levings, M.K. Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor. J. Clin. Investig. 2016, 126, 1413–1424. [Google Scholar] [CrossRef]
- Noyan, F.; Zimmermann, K.; Hardtke-Wolenski, M.; Knoefel, A.; Schulde, E.; Geffers, R.; Hust, M.; Huehn, J.; Galla, M.; Morgan, M.; et al. Prevention of Allograft Rejection by Use of Regulatory T Cells With an MHC-Specific Chimeric Antigen Receptor. Am. J. Transplant. 2017, 17, 917–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sicard, A.; Lamarche, C.; Speck, M.; Wong, M.; Rosado-Sánchez, I.; Blois, M.; Glaichenhaus, N.; Mojibian, M.; Levings, M.K. Donor-specific chimeric antigen receptor Tregs limit rejection in naive but not sensitized allograft recipients. Am. J. Transplant. 2020, 20, 1562–1573. [Google Scholar] [CrossRef] [PubMed]
- Mohseni, Y.R.; Tung, S.L.; Dudreuilh, C.; Lechler, R.I.; Fruhwirth, G.O.; Lombardi, G. The Future of Regulatory T Cell Therapy: Promises and Challenges of Implementing CAR Technology. Front. Immunol. 2020, 11, 1608. [Google Scholar] [CrossRef] [PubMed]
- Joffre, O.; Santolaria, T.; Calise, D.; Al Saati, T.; Hudrisier, D.; Romagnoli, P.; van Meerwijk, J.P. Prevention of acute and chronic allograft rejection with CD4+CD25+Foxp3+ regulatory T lymphocytes. Nat. Med. 2008, 14, 88–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webster, K.E.; Walters, S.; Kohler, R.E.; Mrkvan, T.; Boyman, O.; Surh, C.D.; Grey, S.T.; Sprent, J. In vivo expansion of T reg cells with IL-2-mAb complexes: Induction of resistance to EAE and long-term acceptance of islet allografts without immunosuppression. J. Exp. Med. 2009, 206, 751–760. [Google Scholar] [CrossRef]
- Pilat, N.; Wiletel, M.; Weijler, A.M.; Steiner, R.; Mahr, B.; Warren, J.; Corpuz, T.M.; Wekerle, T.; Webster, K.E.; Sprent, J. Treg-mediated prolonged survival of skin allografts without immunosuppression. Proc. Natl. Acad. Sci. USA 2019, 116, 13508–13516. [Google Scholar] [CrossRef] [Green Version]
- Segundo, D.S.; Ruiz, J.C.; Izquierdo, M.; Fernandez-Fresnedo, G.; Gomez-Alamillo, C.; Merino, R.; Benito, M.J.; Cacho, E.; Rodrigo, E.; Palomar, R.; et al. Calcineurin inhibitors, but not rapamycin, reduce percentages of CD4+CD25+FOXP3+ regulatory T cells in renal transplant recipients. Transplantation 2006, 82, 550–557. [Google Scholar] [CrossRef]
- San Segundo, D.; Fábrega, E.; López-Hoyos, M.; Pons, F. Reduced Numbers of Blood Natural Regulatory T Cells in Stable Liver Transplant Recipients With High Levels of Calcineurin Inhibitors. Transplant. Proc. 2007, 39, 2290–2292. [Google Scholar] [CrossRef]
- Azzi, J.R.; Sayegh, M.H.; Mallat, S.G. Calcineurin Inhibitors: 40 Years Later, Can’t Live Without …. J. Immunol. 2013, 191, 5785. [Google Scholar] [CrossRef] [Green Version]
- van Kooten, C. Counteracting dysfunction of regulatory T cells in organ transplantation. Proc. Natl. Acad. Sci. USA 2017, 114, 6883–6884. [Google Scholar] [CrossRef]
- Whitehouse, G.; Gray, E.; Mastoridis, S.; Merritt, E.; Kodela, E.; Yang, J.H.M.; Danger, R.; Mairal, M.; Christakoudi, S.; Lozano, J.J.; et al. IL-2 therapy restores regulatory T-cell dysfunction induced by calcineurin inhibitors. Proc. Natl. Acad. Sci. USA 2017, 114, 7083–7088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noble, J.; Jouve, T.; Janbon, B.; Rostaing, L.; Malvezzi, P. Belatacept in kidney transplantation and its limitations. Expert. Rev. Clin. Immunol. 2019, 15, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Camirand, G.; Riella, L.V. Treg-Centric View of Immunosuppressive Drugs in Transplantation: A Balancing Act. Am. J. Transplant. 2017, 17, 601–610. [Google Scholar] [CrossRef]
- Schwarz, C.; Unger, L.; Mahr, B.; Aumayr, K.; Regele, H.; Farkas, A.M.; Hock, K.; Pilat, N.; Wekerle, T. The Immunosuppressive Effect of CTLA4 Immunoglobulin Is Dependent on Regulatory T Cells at Low But Not High Doses. Am. J. Transplant. 2016, 16, 3404–3415. [Google Scholar] [CrossRef] [PubMed]
- Riella, L.V.; Liu, T.; Yang, J.; Chock, S.; Shimizu, T.; Mfarrej, B.; Batal, I.; Xiao, X.; Sayegh, M.H.; Chandraker, A. Deleterious effect of CTLA4-Ig on a Treg-dependent transplant model. Am. J. Transplant. 2012, 12, 846–855. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, C.; Mahr, B.; Muckenhuber, M.; Weijler, A.M.; Unger, L.W.; Pilat, N.; Latus, M.; Regele, H.; Wekerle, T. In vivo Treg expansion under costimulation blockade targets early rejection and improves long-term outcome. Am. J. Transplant. 2021, 21, 3765–3774. [Google Scholar] [CrossRef]
- Alvarez Salazar, E.K.; Cortes-Hernandez, A.; Aleman-Muench, G.R.; Alberu, J.; Rodriguez-Aguilera, J.R.; Recillas-Targa, F.; Chagoya de Sanchez, V.; Cuevas, E.; Mancilla-Urrea, E.; Perez Garcia, M.; et al. Methylation of FOXP3 TSDR Underlies the Impaired Suppressive Function of Tregs from Long-term Belatacept-Treated Kidney Transplant Patients. Front. Immunol. 2017, 8, 219. [Google Scholar] [CrossRef] [Green Version]
- Cortes-Hernandez, A.; Alvarez-Salazar, E.; Arteaga-Cruz, S.; Alberu-Gomez, J.; Soldevila, G. Ex vivo expansion of regulatory T cells from long-term Belatacept-treated kidney transplant patients restores their phenotype and suppressive function but not their FOXP3 TSDR demethylation status. Cell. Immunol. 2020, 348, 104044. [Google Scholar] [CrossRef]
- Tanimine, N.; Ohira, M.; Tahara, H.; Ide, K.; Tanaka, Y.; Onoe, T.; Ohdan, H. Strategies for Deliberate Induction of Immune Tolerance in Liver Transplantation: From Preclinical Models to Clinical Application. Front. Immunol. 2020, 11, 1615. [Google Scholar] [CrossRef]
- Massart, A.; Pallier, A.; Pascual, J.; Viklicky, O.; Budde, K.; Spasovski, G.; Klinger, M.; Sever, M.S.; Sorensen, S.S.; Hadaya, K.; et al. The DESCARTES-Nantes survey of kidney transplant recipients displaying clinical operational tolerance identifies 35 new tolerant patients and 34 almost tolerant patients. Nephrol. Dial. Transplant. 2016, 31, 1002–1013. [Google Scholar] [CrossRef]
- Roodnat, J.I.; Hilbrands, L.B.; Hene, R.J.; de Sevaux, R.G.; Smak Gregoor, P.J.; Kal-van Gestel, J.A.; Konijn, C.; van Zuilen, A.; van Gelder, T.; Hoitsma, A.J.; et al. 15-year follow-up of a multicenter, randomized, calcineurin inhibitor withdrawal study in kidney transplantation. Transplantation 2014, 98, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Lerut, J.; Sanchez-Fueyo, A. An appraisal of tolerance in liver transplantation. Am. J. Transplant. 2006, 6, 1774–1780. [Google Scholar] [CrossRef] [PubMed]
- Newell, K.A. Clinical transplantation tolerance. Semin. Immunopathol. 2011, 33, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Baron, D.; Ramstein, G.; Chesneau, M.; Echasseriau, Y.; Pallier, A.; Paul, C.; Degauque, N.; Hernandez-Fuentes, M.P.; Sanchez-Fueyo, A.; Newell, K.A.; et al. A common gene signature across multiple studies relate biomarkers and functional regulation in tolerance to renal allograft. Kidney Int. 2015, 87, 984–995. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Llordella, M.; Lozano, J.J.; Puig-Pey, I.; Orlando, G.; Tisone, G.; Lerut, J.; Benitez, C.; Pons, J.A.; Parrilla, P.; Ramirez, P.; et al. Using transcriptional profiling to develop a diagnostic test of operational tolerance in liver transplant recipients. J. Clin. Investig. 2008, 118, 2845–2857. [Google Scholar] [CrossRef] [PubMed]
- Appenzeller-Herzog, C.; Hartleif, S.; Vionnet, J. Clinical parameters and biomarkers predicting spontaneous operational tolerance after liver transplantation: A scoping review. Am. J. Transplant. 2021, 21, 3312–3323. [Google Scholar] [CrossRef]
- Hu, M.; Rogers, N.M.; Li, J.; Zhang, G.Y.; Wang, Y.M.; Shaw, K.; O’Connell, P.J.; Alexander, S.I. Antigen Specific Regulatory T Cells in Kidney Transplantation and Other Tolerance Settings. Front. Immunol. 2021, 12, 717594. [Google Scholar] [CrossRef]
- Wagner, J.C.; Tang, Q. CAR-Tregs as a Strategy for Inducing Graft Tolerance. Curr. Transplant. Rep. 2020, 7, 205–214. [Google Scholar] [CrossRef]
- Ellis, G.I.; Coker, K.E.; Winn, D.W.; Deng, M.Z.; Shukla, D.; Bhoj, V.; Milone, M.C.; Wang, W.; Liu, C.; Naji, A.; et al. Trafficking and persistence of alloantigen-specific chimeric antigen receptor regulatory T cells in Cynomolgus macaque. Cell. Rep. Med. 2022, 3, 100614. [Google Scholar] [CrossRef]
- Spolski, R.; Li, P.; Leonard, W.J. Biology and regulation of IL-2: From molecular mechanisms to human therapy. Nat. Rev. Immunol. 2018, 18, 648–659. [Google Scholar] [CrossRef]
- Finkelman, F.D.; Madden, K.B.; Morris, S.C.; Holmes, J.M.; Boiani, N.; Katona, I.M.; Maliszewski, C.R. Anti-cytokine antibodies as carrier proteins. Prolongation of in vivo effects of exogenous cytokines by injection of cytokine-anti-cytokine antibody complexes. J. Immunol. 1993, 151, 1235–1244. [Google Scholar] [CrossRef] [PubMed]
- Boyman, O.; Kovar, M.; Rubinstein, M.P.; Surh, C.D.; Sprent, J. Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science 2006, 311, 1924–1927. [Google Scholar] [CrossRef] [Green Version]
- Boyman, O.; Krieg, C.; Letourneau, S.; Webster, K.; Surh, C.D.; Sprent, J. Selectively expanding subsets of T cells in mice by injection of interleukin-2/antibody complexes: Implications for transplantation tolerance. Transplant. Proc. 2012, 44, 1032–1034. [Google Scholar] [CrossRef] [PubMed]
- Trotta, E.; Bessette, P.H.; Silveria, S.L.; Ely, L.K.; Jude, K.M.; Le, D.T.; Holst, C.R.; Coyle, A.; Potempa, M.; Lanier, L.L.; et al. A human anti-IL-2 antibody that potentiates regulatory T cells by a structure-based mechanism. Nat. Med. 2018, 24, 1005–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karakus, U.; Sahin, D.; Mittl, P.R.E.; Mooij, P.; Koopman, G.; Boyman, O. Receptor-gated IL-2 delivery by an anti-human IL-2 antibody activates regulatory T cells in three different species. Sci. Transl. Med. 2020, 12, eabb9283. [Google Scholar] [CrossRef] [PubMed]
- Spangler, J.B.; Tomala, J.; Luca, V.C.; Jude, K.M.; Dong, S.; Ring, A.M.; Votavova, P.; Pepper, M.; Kovar, M.; Garcia, K.C. Antibodies to Interleukin-2 Elicit Selective T Cell Subset Potentiation through Distinct Conformational Mechanisms. Immunity 2015, 42, 815–825. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Ai, X.; Wu, C.; Wang, H.; Zeng, G.; Yang, P.; Liu, G. A novel human IL-2 mutein with minimal systemic toxicity exerts greater antitumor efficacy than wild-type IL-2. Cell Death Dis. 2018, 9, 989. [Google Scholar] [CrossRef] [Green Version]
- Carmenate, T.; Montalvo, G.; Lozada, S.L.; Rodriguez, Y.; Ortiz, Y.; Diaz, C.; Avellanet, J.; Kim, J.; Surh, C.D.; Graca, L.; et al. The antitumor effect induced by an IL-2 ‘no-alpha’ mutein depends on changes in the CD8(+) T lymphocyte/Treg cell balance. Front. Immunol. 2022, 13, 974188. [Google Scholar] [CrossRef]
- Khoryati, L.; Pham, M.N.; Sherve, M.; Kumari, S.; Cook, K.; Pearson, J.; Bogdani, M.; Campbell, D.J.; Gavin, M.A. An IL-2 mutein engineered to promote expansion of regulatory T cells arrests ongoing autoimmunity in mice. Sci. Immunol. 2020, 5, eaba5264. [Google Scholar] [CrossRef]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Prunevieille, A.; Babiker-Mohamed, M.H.; Aslami, C.; Gonzalez-Nolasco, B.; Mooney, N.; Benichou, G. T cell antigenicity and immunogenicity of allogeneic exosomes. Am. J. Transplant. 2021, 21, 2583–2589. [Google Scholar] [CrossRef] [PubMed]
- Golebiewska, J.E.; Wardowska, A.; Pietrowska, M.; Wojakowska, A.; Debska-Slizien, A. Small Extracellular Vesicles in Transplant Rejection. Cells 2021, 10, 2989. [Google Scholar] [CrossRef] [PubMed]
- Pelissier Vatter, F.A.; Cioffi, M.; Hanna, S.J.; Castarede, I.; Caielli, S.; Pascual, V.; Matei, I.; Lyden, D. Extracellular vesicle- and particle-mediated communication shapes innate and adaptive immune responses. J. Exp. Med. 2021, 218, e20202579. [Google Scholar] [CrossRef]
- Smyth, L.A.; Ratnasothy, K.; Tsang, J.Y.; Boardman, D.; Warley, A.; Lechler, R.; Lombardi, G. CD73 expression on extracellular vesicles derived from CD4+ CD25+ Foxp3+ T cells contributes to their regulatory function. Eur. J. Immunol. 2013, 43, 2430–2440. [Google Scholar] [CrossRef]
- Okoye, I.S.; Coomes, S.M.; Pelly, V.S.; Czieso, S.; Papayannopoulos, V.; Tolmachova, T.; Seabra, M.C.; Wilson, M.S. MicroRNA-containing T-regulatory-cell-derived exosomes suppress pathogenic T helper 1 cells. Immunity 2014, 41, 89–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Huang, C.; Song, B.; Xiao, Y.; Fang, M.; Feng, J.; Wang, P. CD4+CD25+ regulatory T cells-derived exosomes prolonged kidney allograft survival in a rat model. Cell Immunol. 2013, 285, 62–68. [Google Scholar] [CrossRef]
- Aiello, S.; Rocchetta, F.; Longaretti, L.; Faravelli, S.; Todeschini, M.; Cassis, L.; Pezzuto, F.; Tomasoni, S.; Azzollini, N.; Mister, M.; et al. Extracellular vesicles derived from T regulatory cells suppress T cell proliferation and prolong allograft survival. Sci. Rep. 2017, 7, 11518. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Huang, H.; Zhang, W.; Ding, F.; Fan, Z.; Zeng, Z. Exosomes Derived From T Regulatory Cells Suppress CD8+ Cytotoxic T Lymphocyte Proliferation and Prolong Liver Allograft Survival. Med. Sci. Monit. 2019, 25, 4877–4884. [Google Scholar] [CrossRef]
- Azimi, M.; Ghabaee, M.; Moghadasi, A.N.; Noorbakhsh, F.; Izad, M. Immunomodulatory function of Treg-derived exosomes is impaired in patients with relapsing-remitting multiple sclerosis. Immunol. Res. 2018, 66, 513–520. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pilat, N.; Steiner, R.; Sprent, J. Treg Therapy for the Induction of Immune Tolerance in Transplantation—Not Lost in Translation? Int. J. Mol. Sci. 2023, 24, 1752. https://doi.org/10.3390/ijms24021752
Pilat N, Steiner R, Sprent J. Treg Therapy for the Induction of Immune Tolerance in Transplantation—Not Lost in Translation? International Journal of Molecular Sciences. 2023; 24(2):1752. https://doi.org/10.3390/ijms24021752
Chicago/Turabian StylePilat, Nina, Romy Steiner, and Jonathan Sprent. 2023. "Treg Therapy for the Induction of Immune Tolerance in Transplantation—Not Lost in Translation?" International Journal of Molecular Sciences 24, no. 2: 1752. https://doi.org/10.3390/ijms24021752
APA StylePilat, N., Steiner, R., & Sprent, J. (2023). Treg Therapy for the Induction of Immune Tolerance in Transplantation—Not Lost in Translation? International Journal of Molecular Sciences, 24(2), 1752. https://doi.org/10.3390/ijms24021752