
The W101C KCNJ5 Mutation Induces Slower Pacing by Constitutively Active GIRK Channels in hiPSC-Derived Cardiomyocytes
 , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Generation and Genetic Correction of hiPSCs from a Patient with the W101C KCNJ5 Variant
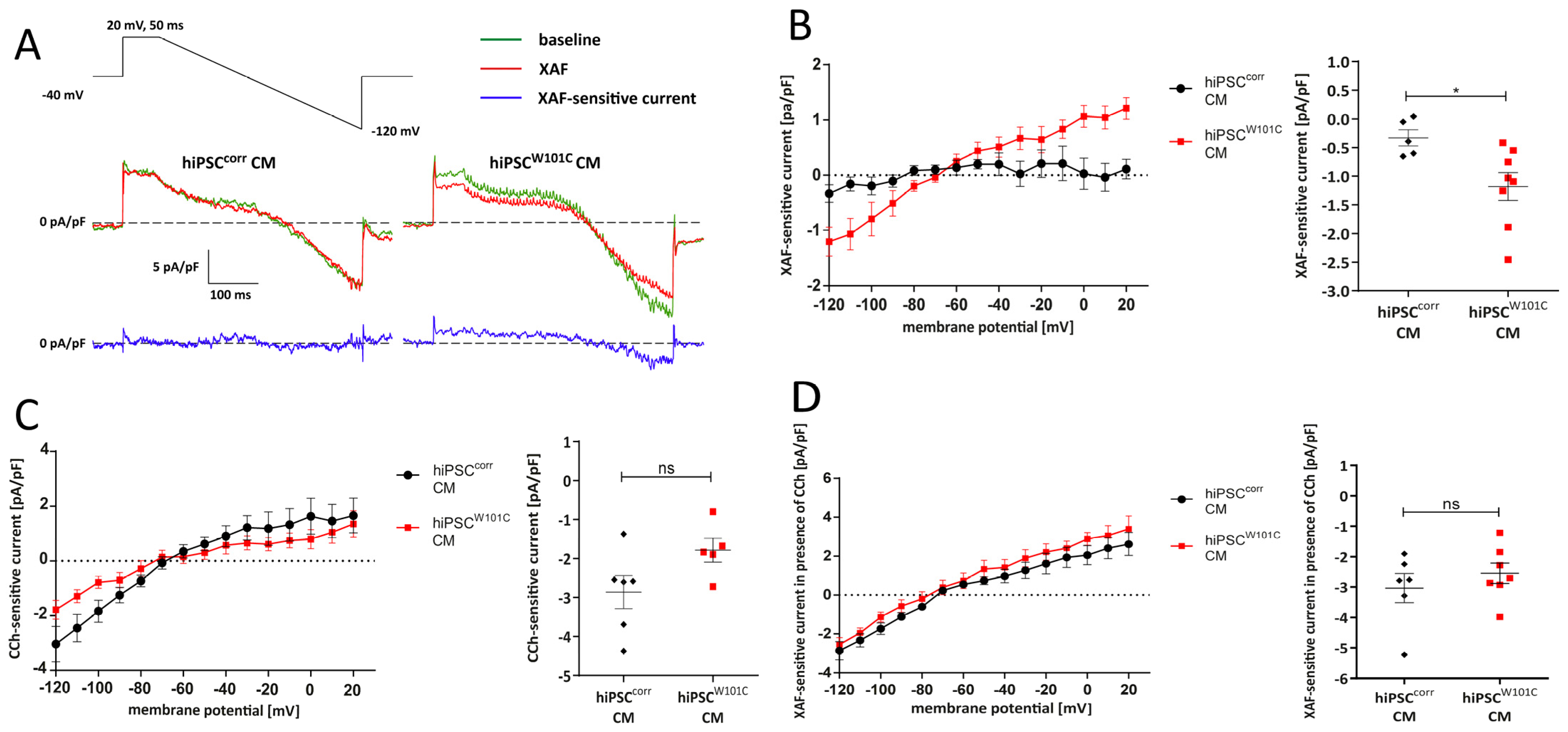
2.2. Variant W101C KCNJ5 Results in a Constitutively Active IK,ACh Current after Retinoic Acid (RA)-Based Differentiation into Atrial-like hiPSCW101C-CMs
2.3. W101C KCNJ5 Results in a Lower Pacemaking Frequency in RA-Treated hiPSC-CMs
2.4. IK,ACh Inhibition Restores Spontaneous Activity in RA-Treated hiPSCW101C-CMs
2.5. hiPSCW101C-Derived Ventricular-like Cardiomyocytes Do Not Show an Electrophysiological Phenotype in Comparison with Controls (hiPSCcorr-CMs)
3. Discussion
4. Materials and Methods
4.1. Human iPSC (hiPSC) Generation
4.2. Human iPSC Culture
4.3. hiPSC Editing Using CRISPR/Cas9 (hiPSCcorr Generation)
4.4. Sequencing Analysis
4.5. Karyotype Analysis
4.6. Cardiac Differentiation of hiPSC
4.7. Immunofluorescence Staining and Imaging
4.8. RT-qPCR
4.9. Cellular Electrophysiology in hiPSC-CMs
4.9.1. Data Acquisition
4.9.2. Voltage Clamp Experiments
4.9.3. Current Clamp Experiments
4.10. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hawks, M.K.; Paul, M.L.B.; Malu, O.O. Sinus Node Dysfunction. Am. Fam. Physician 2021, 104, 179–185. [Google Scholar]
- Monfredi, O.; Boyett, M.R. Sick sinus syndrome and atrial fibrillation in older persons-A view from the sinoatrial nodal myocyte. J. Mol. Cell Cardiol. 2015, 83, 88–100. [Google Scholar] [CrossRef]
- De Ponti, R.; Marazzato, J.; Bagliani, G.; Leonelli, F.M.; Padeletti, L. Sick Sinus Syndrome. Card. Electrophysiol. Clin. 2018, 10, 183–195. [Google Scholar] [CrossRef]
- Manoj, P.; Kim, J.A.; Kim, S.; Li, T.; Sewani, M.; Chelu, M.G.; Li, N. Sinus node dysfunction: Current understanding and future directions. Am. J. Physiol. Heart Circ. Physiol. 2023, 324, H259–H278. [Google Scholar] [CrossRef]
- Sathnur, N.; Ebin, E.; Benditt, D.G. Sinus Node Dysfunction. Cardiol. Clin. 2023, 41, 349–367. [Google Scholar] [CrossRef]
- Porta-Sanchez, A.; Priori, S.G. Genetic Abnormalities of the Sinoatrial Node and Atrioventricular Conduction. Cardiol. Clin. 2023, 41, 333–347. [Google Scholar] [CrossRef]
- Wallace, M.J.; El Refaey, M.; Mesirca, P.; Hund, T.J.; Mangoni, M.E.; Mohler, P.J. Genetic Complexity of Sinoatrial Node Dysfunction. Front. Genet. 2021, 12, 654925. [Google Scholar] [CrossRef]
- Baruscotti, M.; Bottelli, G.; Milanesi, R.; DiFrancesco, J.C.; DiFrancesco, D. HCN-related channelopathies. Pflugers Arch. 2010, 460, 405–415. [Google Scholar] [CrossRef]
- Kuss, J.; Stallmeyer, B.; Goldstein, M.; Rinne, S.; Pees, C.; Zumhagen, S.; Seebohm, G.; Decher, N.; Pott, L.; Kienitz, M.C.; et al. Familial Sinus Node Disease Caused by a Gain of GIRK (G-Protein Activated Inwardly Rectifying K+ Channel) Channel Function. Circ. Genom. Precis. Med. 2019, 12, e002238. [Google Scholar] [CrossRef]
- Krapivinsky, G.; Gordon, E.A.; Wickman, K.; Velimirovic, B.; Krapivinsky, L.; Clapham, D.E. The G-protein-gated atrial K+ channel IKACh is a heteromultimer of two inwardly rectifying K+-channel proteins. Nature 1995, 374, 135–141. [Google Scholar] [CrossRef]
- Ferrer, J.; Nichols, C.G.; Makhina, E.N.; Salkoff, L.; Bernstein, J.; Gerhard, D.; Wasson, J.; Ramanadham, S.; Permutt, A. Pancreatic islet cells express a family of inwardly rectifying K+ channel subunits which interact to form G-protein-activated channels. J. Biol. Chem. 1995, 270, 26086–26091. [Google Scholar] [CrossRef]
- Choi, M.; Scholl, U.I.; Yue, P.; Bjorklund, P.; Zhao, B.; Nelson-Williams, C.; Ji, W.; Cho, Y.; Patel, A.; Men, C.J.; et al. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science 2011, 331, 768–772. [Google Scholar] [CrossRef]
- Wickman, K.; Krapivinsky, G.; Corey, S.; Kennedy, M.; Nemec, J.; Medina, I.; Clapham, D.E. Structure, G protein activation, and functional relevance of the cardiac G protein-gated K+ channel, IKACh. Ann. N. Y. Acad. Sci. 1999, 868, 386–398. [Google Scholar] [CrossRef]
- Ravens, U.; Poulet, C.; Wettwer, E.; Knaut, M. Atrial selectivity of antiarrhythmic drugs. J. Physiol. 2013, 591, 4087–4097. [Google Scholar] [CrossRef]
- Verkerk, A.O.; Geuzebroek, G.S.; Veldkamp, M.W.; Wilders, R. Effects of acetylcholine and noradrenalin on action potentials of isolated rabbit sinoatrial and atrial myocytes. Front. Physiol. 2012, 3, 174. [Google Scholar] [CrossRef]
- DiFrancesco, D.; Ducouret, P.; Robinson, R.B. Muscarinic modulation of cardiac rate at low acetylcholine concentrations. Science 1989, 243, 669–671. [Google Scholar] [CrossRef]
- Choi, H.S.; Wang, D.Y.; Noble, D.; Lee, C.O. Effect of isoprenaline, carbachol, and Cs+ on Na+ activity and pacemaker potential in rabbit SA node cells. Am. J. Physiol. 1999, 276, H205–H214. [Google Scholar] [CrossRef]
- Zaza, A.; Robinson, R.B.; DiFrancesco, D. Basal responses of the L-type Ca2+ and hyperpolarization-activated currents to autonomic agonists in the rabbit sino-atrial node. J. Physiol. 1996, 491 Pt 2, 347–355. [Google Scholar] [CrossRef]
- Campos-Rios, A.; Rueda-Ruzafa, L.; Lamas, J.A. The Relevance of GIRK Channels in Heart Function. Membranes 2022, 12, 1119. [Google Scholar] [CrossRef]
- Wang, J.; Irnaten, M.; Neff, R.A.; Venkatesan, P.; Evans, C.; Loewy, A.D.; Mettenleiter, T.C.; Mendelowitz, D. Synaptic and neurotransmitter activation of cardiac vagal neurons in the nucleus ambiguus. Ann. N. Y. Acad. Sci. 2001, 940, 237–246. [Google Scholar] [CrossRef]
- Marschall, C.; Moscu-Gregor, A.; Klein, H.G. Variant panorama in 1,385 index patients and sensitivity of expanded next-generation sequencing panels in arrhythmogenic disorders. Cardiovasc. Diagn. Ther. 2019, 9, S292–S298. [Google Scholar] [CrossRef] [PubMed]
- Kokunai, Y.; Nakata, T.; Furuta, M.; Sakata, S.; Kimura, H.; Aiba, T.; Yoshinaga, M.; Osaki, Y.; Nakamori, M.; Itoh, H.; et al. A Kir3.4 mutation causes Andersen-Tawil syndrome by an inhibitory effect on Kir2.1. Neurology 2014, 82, 1058–1064. [Google Scholar] [CrossRef] [PubMed]
- Yamada, N.; Asano, Y.; Fujita, M.; Yamazaki, S.; Inanobe, A.; Matsuura, N.; Kobayashi, H.; Ohno, S.; Ebana, Y.; Tsukamoto, O.; et al. Mutant KCNJ3 and KCNJ5 Potassium Channels as Novel Molecular Targets in Bradyarrhythmias and Atrial Fibrillation. Circulation 2019, 139, 2157–2169. [Google Scholar] [CrossRef] [PubMed]
- Proost, D.; Saenen, J.; Vandeweyer, G.; Rotthier, A.; Alaerts, M.; Van Craenenbroeck, E.M.; Van Crombruggen, J.; Mortier, G.; Wuyts, W.; Vrints, C.; et al. Targeted Next-Generation Sequencing of 51 Genes Involved in Primary Electrical Disease. J. Mol. Diagn. 2017, 19, 445–459. [Google Scholar] [CrossRef]
- Akdis, D.; Saguner, A.M.; Medeiros-Domingo, A.; Schaller, A.; Balmer, C.; Steffel, J.; Brunckhorst, C.; Duru, F. Multiple clinical profiles of families with the short QT syndrome. Europace 2018, 20, f113–f121. [Google Scholar] [CrossRef]
- Asatryan, B.; Schaller, A.; Seiler, J.; Servatius, H.; Noti, F.; Baldinger, S.H.; Tanner, H.; Roten, L.; Dillier, R.; Lam, A.; et al. Usefulness of Genetic Testing in Sudden Cardiac Arrest Survivors With or Without Previous Clinical Evidence of Heart Disease. Am. J. Cardiol. 2019, 123, 2031–2038. [Google Scholar] [CrossRef]
- van Lint, F.H.M.; Mook, O.R.F.; Alders, M.; Bikker, H.; Lekanne Dit Deprez, R.H.; Christiaans, I. Large next-generation sequencing gene panels in genetic heart disease: Yield of pathogenic variants and variants of unknown significance. Neth. Heart J. 2019, 27, 304–309. [Google Scholar] [CrossRef]
- Meyer, K.M.; Malhotra, N.; Kwak, J.S.; El Refaey, M. Relevance of KCNJ5 in Pathologies of Heart Disease. Int. J. Mol. Sci. 2023, 24, 10849. [Google Scholar] [CrossRef]
- Duan, S.; Du, J. Sinus node dysfunction and atrial fibrillation-Relationships, clinical phenotypes, new mechanisms, and treatment approaches. Ageing Res. Rev. 2023, 86, 101890. [Google Scholar] [CrossRef]
- Devalla, H.D.; Schwach, V.; Ford, J.W.; Milnes, J.T.; El-Haou, S.; Jackson, C.; Gkatzis, K.; Elliott, D.A.; Chuva de Sousa Lopes, S.M.; Mummery, C.L.; et al. Atrial-like cardiomyocytes from human pluripotent stem cells are a robust preclinical model for assessing atrial-selective pharmacology. EMBO Mol. Med. 2015, 7, 394–410. [Google Scholar] [CrossRef]
- Marczenke, M.; Piccini, I.; Mengarelli, I.; Fell, J.; Ropke, A.; Seebohm, G.; Verkerk, A.O.; Greber, B. Cardiac Subtype-Specific Modeling of K(v)1.5 Ion Channel Deficiency Using Human Pluripotent Stem Cells. Front. Physiol. 2017, 8, 469. [Google Scholar] [CrossRef] [PubMed]
- Veerman, C.C.; Mengarelli, I.; Koopman, C.D.; Wilders, R.; van Amersfoorth, S.C.; Bakker, D.; Wolswinkel, R.; Hababa, M.; de Boer, T.P.; Guan, K.; et al. Genetic variation in GNB5 causes bradycardia by augmenting the cholinergic response via increased acetylcholine-activated potassium current (I(K,ACh)). Dis. Model. Mech. 2019, 12, dmm037994. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wiesinger, A.; Fokkert, L.; Boukens, B.J.; Verkerk, A.O.; Christoffels, V.M.; Boink, G.J.J.; Devalla, H.D. Molecular and electrophysiological evaluation of human cardiomyocyte subtypes to facilitate generation of composite cardiac models. J. Tissue Eng. 2022, 13, 20417314221127908. [Google Scholar] [CrossRef] [PubMed]
- Lemme, M.; Ulmer, B.M.; Lemoine, M.D.; Zech, A.T.L.; Flenner, F.; Ravens, U.; Reichenspurner, H.; Rol-Garcia, M.; Smith, G.; Hansen, A.; et al. Atrial-like Engineered Heart Tissue: An In Vitro Model of the Human Atrium. Stem Cell Rep. 2018, 11, 1378–1390. [Google Scholar] [CrossRef]
- Cyganek, L.; Tiburcy, M.; Sekeres, K.; Gerstenberg, K.; Bohnenberger, H.; Lenz, C.; Henze, S.; Stauske, M.; Salinas, G.; Zimmermann, W.H.; et al. Deep phenotyping of human induced pluripotent stem cell-derived atrial and ventricular cardiomyocytes. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Goldfracht, I.; Protze, S.; Shiti, A.; Setter, N.; Gruber, A.; Shaheen, N.; Nartiss, Y.; Keller, G.; Gepstein, L. Generating ring-shaped engineered heart tissues from ventricular and atrial human pluripotent stem cell-derived cardiomyocytes. Nat. Commun. 2020, 11, 75. [Google Scholar] [CrossRef]
- Fenner, M.F.; Carstensen, H.; Dalgas Nissen, S.; Melis Hesselkilde, E.; Scott Lunddahl, C.; Adler Hess Jensen, M.; Loft-Andersen, A.V.; Sattler, S.M.; Platonov, P.; El-Haou, S.; et al. Effect of selective I(K,ACh) inhibition by XAF-1407 in an equine model of tachypacing-induced persistent atrial fibrillation. Br. J. Pharmacol. 2020, 177, 3778–3794. [Google Scholar] [CrossRef]
- Sobota, V.; Gatta, G.; van Hunnik, A.; van Tuijn, I.; Kuiper, M.; Milnes, J.; Jespersen, T.; Schotten, U.; Verheule, S. The Acetylcholine-Activated Potassium Current Inhibitor XAF-1407 Terminates Persistent Atrial Fibrillation in Goats. Front. Pharmacol. 2020, 11, 608410. [Google Scholar] [CrossRef]
- Linz, B.; Thostrup, A.H.; Saljic, A.; Rombouts, K.; Hertel, J.N.; Hohl, M.; Milnes, J.; Tfelt-Hansen, J.; Linz, D.; Jespersen, T. Pharmacological inhibition of acetylcholine-regulated potassium current (I (K,ACh)) prevents atrial arrhythmogenic changes in a rat model of repetitive obstructive respiratory events. Heart Rhythm O2 2022, 3, 97–104. [Google Scholar] [CrossRef]
- Berecki, G.; Wilders, R.; de Jonge, B.; van Ginneken, A.C.; Verkerk, A.O. Re-evaluation of the action potential upstroke velocity as a measure of the Na+ current in cardiac myocytes at physiological conditions. PLoS ONE 2010, 5, e15772. [Google Scholar] [CrossRef]
- Krishnan, S.C.; Antzelevitch, C. Sodium channel block produces opposite electrophysiological effects in canine ventricular epicardium and endocardium. Circ. Res. 1991, 69, 277–291. [Google Scholar] [CrossRef] [PubMed]
- Horvath, A.; Lemoine, M.D.; Loser, A.; Mannhardt, I.; Flenner, F.; Uzun, A.U.; Neuber, C.; Breckwoldt, K.; Hansen, A.; Girdauskas, E.; et al. Low Resting Membrane Potential and Low Inward Rectifier Potassium Currents Are Not Inherent Features of hiPSC-Derived Cardiomyocytes. Stem Cell Rep. 2018, 10, 822–833. [Google Scholar] [CrossRef] [PubMed]
- Luscher, C.; Slesinger, P.A. Emerging roles for G protein-gated inwardly rectifying potassium (GIRK) channels in health and disease. Nat. Rev. Neurosci. 2010, 11, 301–315. [Google Scholar] [CrossRef] [PubMed]
- Voigt, N.; Abu-Taha, I.; Heijman, J.; Dobrev, D. Constitutive activity of the acetylcholine-activated potassium current IK,ACh in cardiomyocytes. Adv. Pharmacol. 2014, 70, 393–409. [Google Scholar] [CrossRef]
- Charmandari, E.; Sertedaki, A.; Kino, T.; Merakou, C.; Hoffman, D.A.; Hatch, M.M.; Hurt, D.E.; Lin, L.; Xekouki, P.; Stratakis, C.A.; et al. A novel point mutation in the KCNJ5 gene causing primary hyperaldosteronism and early-onset autosomal dominant hypertension. J. Clin. Endocrinol. Metab. 2012, 97, E1532–E1539. [Google Scholar] [CrossRef]
- Cha, T.J.; Ehrlich, J.R.; Chartier, D.; Qi, X.Y.; Xiao, L.; Nattel, S. Kir3-based inward rectifier potassium current: Potential role in atrial tachycardia remodeling effects on atrial repolarization and arrhythmias. Circulation 2006, 113, 1730–1737. [Google Scholar] [CrossRef]
- Makary, S.; Voigt, N.; Maguy, A.; Wakili, R.; Nishida, K.; Harada, M.; Dobrev, D.; Nattel, S. Differential protein kinase C isoform regulation and increased constitutive activity of acetylcholine-regulated potassium channels in atrial remodeling. Circ. Res. 2011, 109, 1031–1043. [Google Scholar] [CrossRef]
- Gada, K.D.; Chang, M.; Chandrashekar, A.; Plant, L.D.; Noujaim, S.F.; Logothetis, D.E. Mechanism of PKCepsilon regulation of cardiac GIRK channel gating. Proc. Natl. Acad. Sci. USA 2023, 120, e2212325120. [Google Scholar] [CrossRef]
- Chan, K.W.; Sui, J.L.; Vivaudou, M.; Logothetis, D.E. Control of channel activity through a unique amino acid residue of a G protein-gated inwardly rectifying K+ channel subunit. Proc. Natl. Acad. Sci. USA 1996, 93, 14193–14198. [Google Scholar] [CrossRef]
- Vivaudou, M.; Chan, K.W.; Sui, J.L.; Jan, L.Y.; Reuveny, E.; Logothetis, D.E. Probing the G-protein regulation of GIRK1 and GIRK4, the two subunits of the KACh channel, using functional homomeric mutants. J. Biol. Chem. 1997, 272, 31553–31560. [Google Scholar] [CrossRef]
- He, C.; Zhang, H.; Mirshahi, T.; Logothetis, D.E. Identification of a potassium channel site that interacts with G protein betagamma subunits to mediate agonist-induced signaling. J. Biol. Chem. 1999, 274, 12517–12524. [Google Scholar] [CrossRef]
- He, C.; Yan, X.; Zhang, H.; Mirshahi, T.; Jin, T.; Huang, A.; Logothetis, D.E. Identification of critical residues controlling G protein-gated inwardly rectifying K+ channel activity through interactions with the beta gamma subunits of G proteins. J. Biol. Chem. 2002, 277, 6088–6096. [Google Scholar] [CrossRef] [PubMed]
- Petersen, J.; Castro, L.; Bengaard, A.K.P.; Pecha, S.; Ismaili, D.; Schulz, C.; Sahni, J.; Steenpass, A.; Meier, C.; Reichenspurner, H.; et al. Muscarinic Receptor Activation Reduces Force and Arrhythmias in Human Atria Independent of IK,ACh. J. Cardiovasc. Pharmacol. 2022, 79, 678–686. [Google Scholar] [CrossRef] [PubMed]
- Verkerk, A.O.; Doszpod, I.J.; Mengarelli, I.; Magyar, T.; Polyak, A.; Paszti, B.; Efimov, I.R.; Wilders, R.; Koncz, I. Acetylcholine Reduces L-Type Calcium Current without Major Changes in Repolarization of Canine and Human Purkinje and Ventricular Tissue. Biomedicines 2022, 10, 2987. [Google Scholar] [CrossRef]
- Koumi, S.; Arentzen, C.E.; Backer, C.L.; Wasserstrom, J.A. Alterations in muscarinic K+ channel response to acetylcholine and to G protein-mediated activation in atrial myocytes isolated from failing human hearts. Circulation 1994, 90, 2213–2224. [Google Scholar] [CrossRef] [PubMed]
- Seibertz, F.; Sutanto, H.; Dulk, R.; Pronto, J.R.D.; Springer, R.; Rapedius, M.; Liutkute, A.; Ritter, M.; Jung, P.; Stelzer, L.; et al. Electrophysiological and calcium-handling development during long-term culture of human-induced pluripotent stem cell-derived cardiomyocytes. Basic. Res. Cardiol. 2023, 118, 14. [Google Scholar] [CrossRef]
- MacDonald, E.A.; Rose, R.A.; Quinn, T.A. Neurohumoral Control of Sinoatrial Node Activity and Heart Rate: Insight From Experimental Models and Findings From Humans. Front. Physiol. 2020, 11, 170. [Google Scholar] [CrossRef]
- Freeman, L.C.; Kass, R.S. Cholinergic inhibition of slow delayed-rectifier K+ current in guinea pig sino-atrial node is not mediated by muscarinic receptors. Mol. Pharmacol. 1995, 47, 1248–1254. [Google Scholar]
- Fleischmann, B.K.; Duan, Y.; Fan, Y.; Schoneberg, T.; Ehlich, A.; Lenka, N.; Viatchenko-Karpinski, S.; Pott, L.; Hescheler, J.; Fakler, B. Differential subunit composition of the G protein-activated inward-rectifier potassium channel during cardiac development. J. Clin. Investig. 2004, 114, 994–1001. [Google Scholar] [CrossRef]
- Frank, S.; Zhang, M.; Scholer, H.R.; Greber, B. Small molecule-assisted, line-independent maintenance of human pluripotent stem cells in defined conditions. PLoS ONE 2012, 7, e41958. [Google Scholar] [CrossRef]
- Zhang, M.; Schulte, J.S.; Heinick, A.; Piccini, I.; Rao, J.; Quaranta, R.; Zeuschner, D.; Malan, D.; Kim, K.P.; Ropke, A.; et al. Universal cardiac induction of human pluripotent stem cells in two and three-dimensional formats: Implications for in vitro maturation. Stem Cells 2015, 33, 1456–1469. [Google Scholar] [CrossRef] [PubMed]
- Haeussler, M.; Schonig, K.; Eckert, H.; Eschstruth, A.; Mianne, J.; Renaud, J.B.; Schneider-Maunoury, S.; Shkumatava, A.; Teboul, L.; Kent, J.; et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016, 17, 148. [Google Scholar] [CrossRef]
- Concordet, J.P.; Haeussler, M. CRISPOR: Intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 2018, 46, W242–W245. [Google Scholar] [CrossRef] [PubMed]
- Li, H.L.; Gee, P.; Ishida, K.; Hotta, A. Efficient genomic correction methods in human iPS cells using CRISPR-Cas9 system. Methods 2016, 101, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Barry, P.H.; Lynch, J.W. Liquid junction potentials and small cell effects in patch-clamp analysis. J. Membr. Biol. 1991, 121, 101–117. [Google Scholar] [CrossRef]
- Lomax, A.E.; Rose, R.A.; Giles, W.R. Electrophysiological evidence for a gradient of G protein-gated K+ current in adult mouse atria. Br. J. Pharmacol. 2003, 140, 576–584. [Google Scholar] [CrossRef]
- Verkerk, A.O.; Wilders, R.; Zegers, J.G.; van Borren, M.M.; Ravesloot, J.H.; Verheijck, E.E. Ca2+-activated Cl− current in rabbit sinoatrial node cells. J. Physiol. 2002, 540, 105–117. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kayser, A.; Dittmann, S.; Šarić, T.; Mearini, G.; Verkerk, A.O.; Schulze-Bahr, E. The W101C KCNJ5 Mutation Induces Slower Pacing by Constitutively Active GIRK Channels in hiPSC-Derived Cardiomyocytes. Int. J. Mol. Sci. 2023, 24, 15290. https://doi.org/10.3390/ijms242015290
Kayser A, Dittmann S, Šarić T, Mearini G, Verkerk AO, Schulze-Bahr E. The W101C KCNJ5 Mutation Induces Slower Pacing by Constitutively Active GIRK Channels in hiPSC-Derived Cardiomyocytes. International Journal of Molecular Sciences. 2023; 24(20):15290. https://doi.org/10.3390/ijms242015290
Chicago/Turabian StyleKayser, Anne, Sven Dittmann, Tomo Šarić, Giulia Mearini, Arie O. Verkerk, and Eric Schulze-Bahr. 2023. "The W101C KCNJ5 Mutation Induces Slower Pacing by Constitutively Active GIRK Channels in hiPSC-Derived Cardiomyocytes" International Journal of Molecular Sciences 24, no. 20: 15290. https://doi.org/10.3390/ijms242015290
APA StyleKayser, A., Dittmann, S., Šarić, T., Mearini, G., Verkerk, A. O., & Schulze-Bahr, E. (2023). The W101C KCNJ5 Mutation Induces Slower Pacing by Constitutively Active GIRK Channels in hiPSC-Derived Cardiomyocytes. International Journal of Molecular Sciences, 24(20), 15290. https://doi.org/10.3390/ijms242015290