
Repaglinide Induces ATF6 Processing and Neuroprotection in Transgenic SOD1G93A Mice
 , , , , and
, , , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
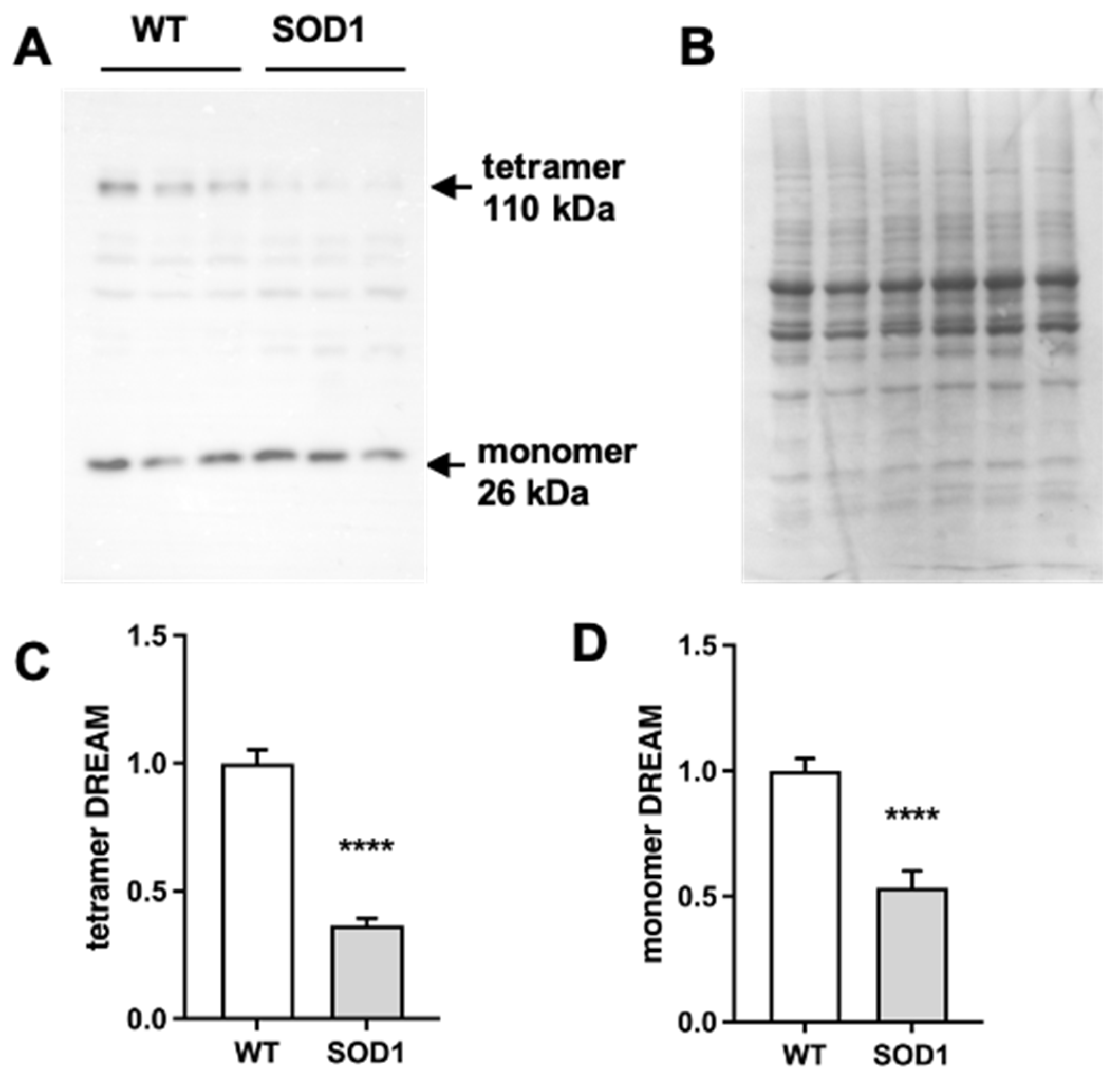
2.1. Expression of DREAM Is Significantly Reduced in the Lumbar Spinal Cord of SOD1G93A Mice
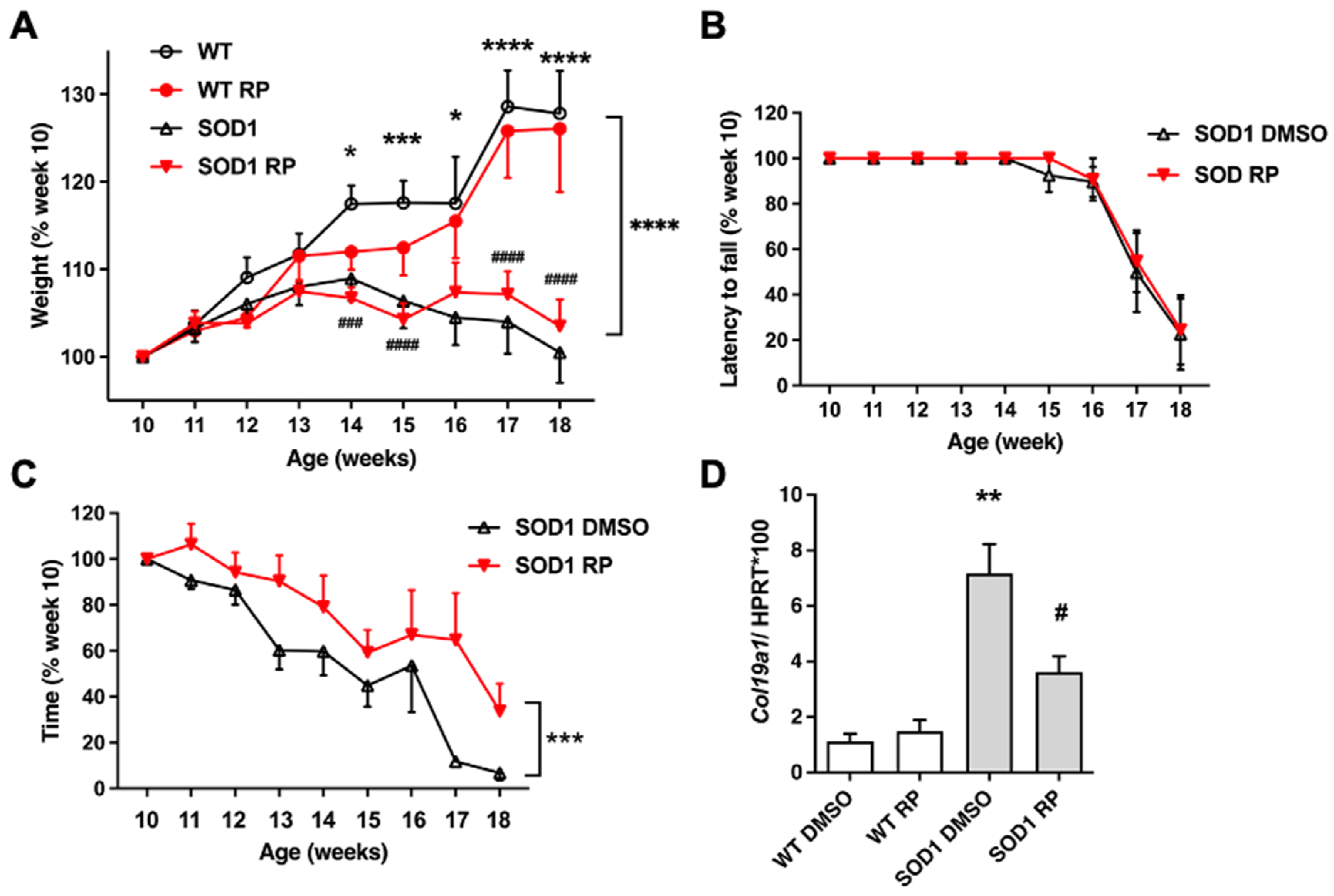
2.2. Repaglinide Treatment Delayed the Loss of Motor Strength in SOD1G93A Mice
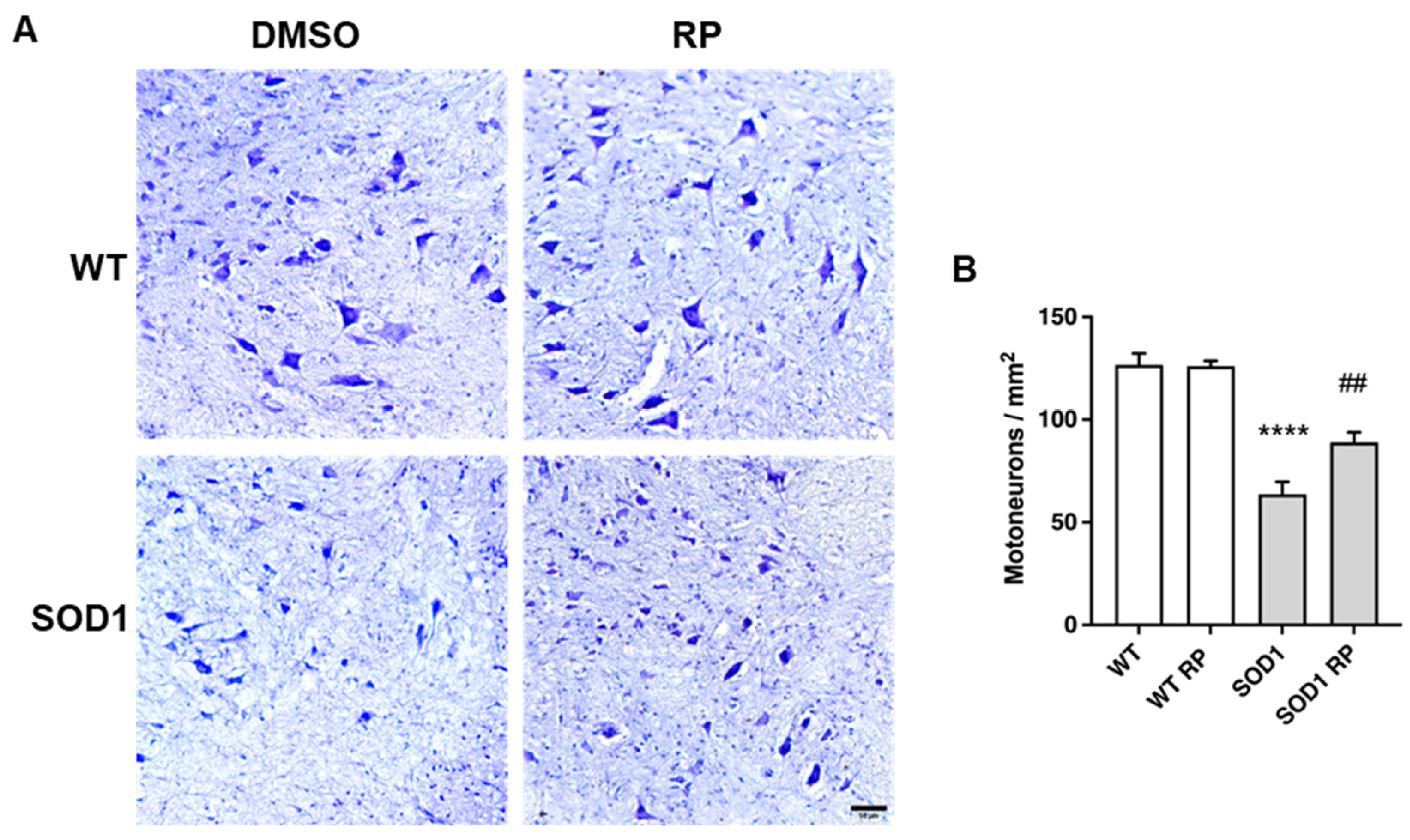
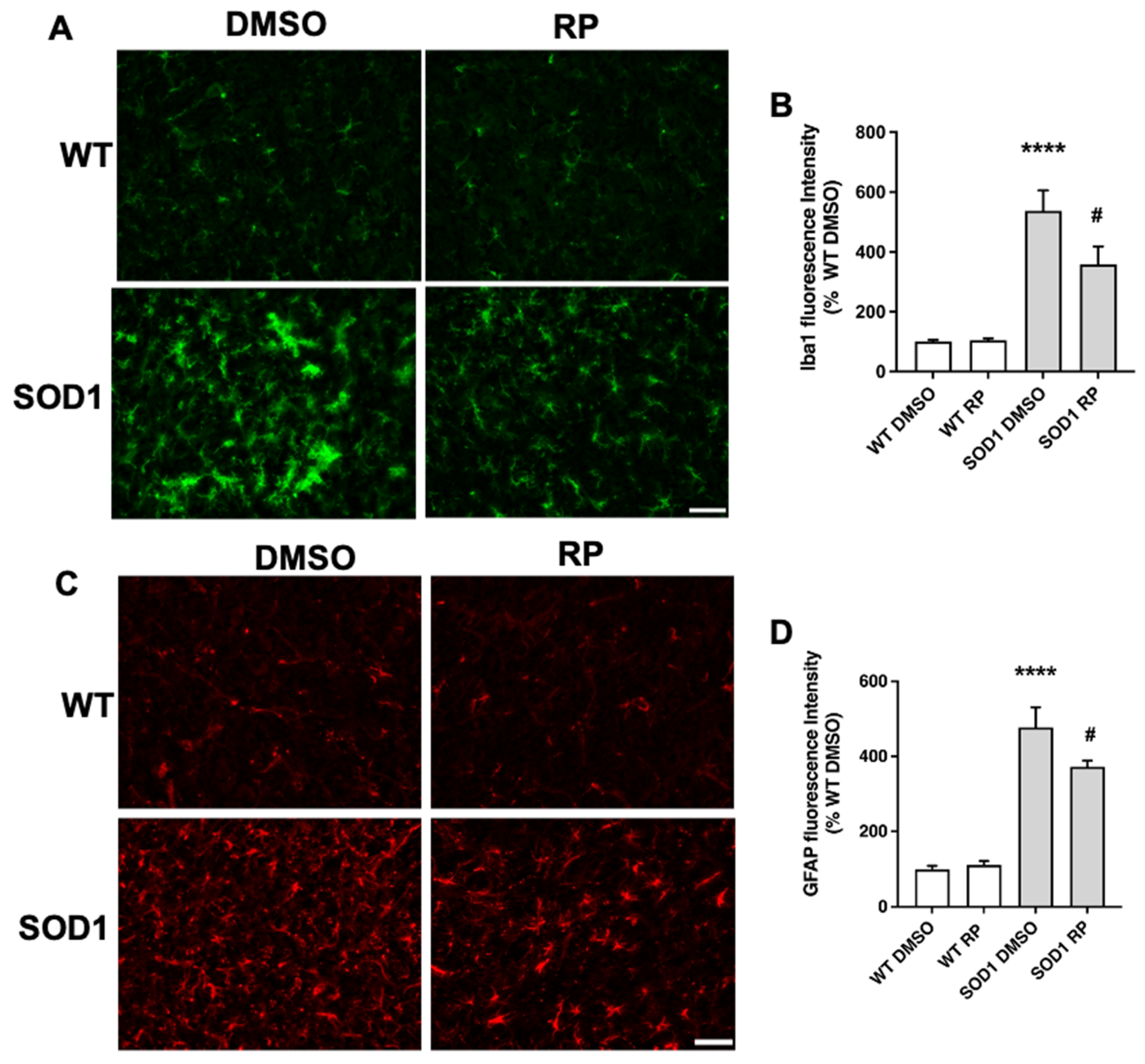
2.3. Treatment with RP Reduces Motoneuron Loss and Improves Gliosis in the Ventral Horn of SOD1G93A Spinal Cord
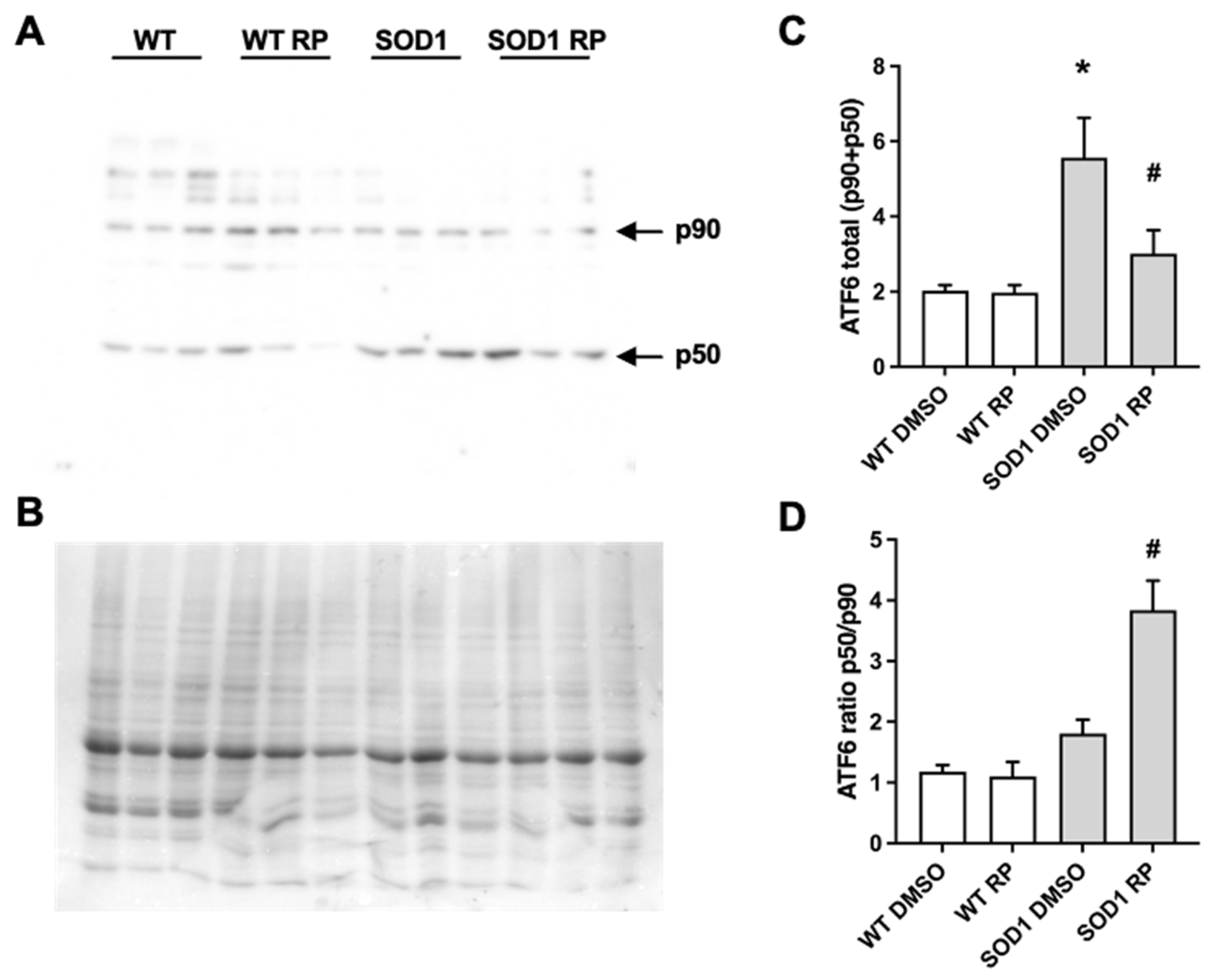
2.4. RP Administration Stimulates ATF6 Processing in the Spinal Cord of SOD1G93A Mice
3. Discussion
4. Materials and Methods
4.1. Animals and Treatments
4.1.1. Animals
4.1.2. Treatments
4.2. Weight Control and Behavioral Tests
4.3. Western Blot
4.4. Real-Time qPCR
4.5. Histology
4.5.1. Tissue Processing
4.5.2. Immunofluorescence, Nissl Staining, and Image Analysis
4.6. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- An, W.F.; Bowlby, M.R.; Betty, M.; Cao, J.; Ling, H.P.; Mendoza, G.; Hinson, J.W.; Mattsson, K.I.; Strassle, B.W.; Trimmer, J.S.; et al. Modulation of A-type potassium channels by a family of calcium sensors. Nature 2000, 403, 553–556. [Google Scholar] [CrossRef] [PubMed]
- Buxbaum, J.D.; Choi, E.K.; Luo, Y.; Lilliehook, C.; Crowley, A.C.; Merriam, D.E.; Wasco, W. Calsenilin: A calcium-binding protein that interacts with the presenilins and regulates the levels of a presenilin fragment. Nat. Med. 1998, 4, 1177–1181. [Google Scholar] [CrossRef] [PubMed]
- Carrion, A.M.; Link, W.A.; Ledo, F.; Mellstrom, B.; Naranjo, J.R. DREAM is a Ca2+-regulated transcriptional repressor. Nature 1999, 398, 80–84. [Google Scholar] [CrossRef]
- Xiong, H.; Kovacs, I.; Zhang, Z. Differential distribution of KChIPs mRNAs in adult mouse brain. Brain Res. Mol. Brain Res. 2004, 128, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Cantero-Recasens, G.; Butnaru, C.M.; Valverde, M.A.; Naranjo, J.R.; Brouwers, N.; Malhotra, V. KChIP3 coupled to Ca(2+) oscillations exerts a tonic brake on baseline mucin release in the colon. Elife 2018, 7, e39729. [Google Scholar] [CrossRef]
- Jerng, H.H.; Pfaffinger, P.J. Modulatory mechanisms and multiple functions of somatodendritic A-type K (+) channel auxiliary subunits. Front. Cell. Neurosci. 2014, 8, 82. [Google Scholar] [CrossRef]
- Venn, N.; Haynes, L.P.; Burgoyne, R.D. Specific effects of KChIP3/calsenilin/DREAM, but not KChIPs 1, 2 and 4, on calcium signalling and regulated secretion in PC12 cells. Biochem. J. 2008, 413, 71–80. [Google Scholar] [CrossRef]
- Molinaro, P.; Sanguigno, L.; Casamassa, A.; Valsecchi, V.; Sirabella, R.; Pignataro, G.; Annunziato, L.; Formisano, L. Emerging Role of DREAM in Healthy Brain and Neurological Diseases. Int. J. Mol. Sci. 2023, 24, 9177. [Google Scholar] [CrossRef]
- Wu, L.Y.; Song, Y.J.; Zhang, C.L.; Liu, J. K(V) Channel-Interacting Proteins in the Neurological and Cardiovascular Systems: An Updated Review. Cells 2023, 12, 1894. [Google Scholar] [CrossRef]
- Rivas, M.; Villar, D.; Gonzalez, P.; Dopazo, X.M.; Mellstrom, B.; Naranjo, J.R. Building the DREAM interactome. Sci. China Life Sci. 2011, 54, 786–792. [Google Scholar] [CrossRef]
- Holmqvist, M.H.; Cao, J.; Knoppers, M.H.; Jurman, M.E.; Distefano, P.S.; Rhodes, K.J.; Xie, Y.; An, W.F. Kinetic modulation of Kv4-mediated A-current by arachidonic acid is dependent on potassium channel interacting proteins. J. Neurosci. 2001, 21, 4154–4161. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Takezawa, D.; Tachibanaki, S.; Kawamura, S.; Tokumitsu, H.; Kobayashi, R. Neuronal calcium sensor proteins are direct targets of the insulinotropic agent repaglinide. Biochem. J. 2003, 375, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Bowlby, M.R.; Chanda, P.; Edris, W.; Hinson, J.; Jow, F.; Katz, A.H.; Kennedy, J.; Krishnamurthy, G.; Pitts, K.; Ryan, K.; et al. Identification and characterization of small molecule modulators of KChIP/Kv4 function. Bioorg. Med. Chem. 2005, 13, 6112–6119. [Google Scholar] [CrossRef] [PubMed]
- Naranjo, J.R.; Zhang, H.; Villar, D.; Gonzalez, P.; Dopazo, X.M.; Moron-Oset, J.; Higueras, E.; Oliveros, J.C.; Arrabal, M.D.; Prieto, A.; et al. Activating transcription factor 6 derepression mediates neuroprotection in Huntington disease. J. Clin. Investig. 2016, 126, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, A.; Chen, A.W.; Varner, J.D. A review of the mammalian unfolded protein response. Biotechnol. Bioeng. 2011, 108, 2777–2793. [Google Scholar] [CrossRef]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers 2017, 3, 17085. [Google Scholar] [CrossRef]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef]
- Larrode, P.; Calvo, A.C.; Moreno-Martinez, L.; de la Torre, M.; Moreno-Garcia, L.; Molina, N.; Castiella, T.; Iniguez, C.; Pascual, L.F.; Mena, F.J.M.; et al. DREAM-Dependent Activation of Astrocytes in Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2018, 55, 1–12. [Google Scholar] [CrossRef]
- Savignac, M.; Mellstrom, B.; Bebin, A.G.; Oliveros, J.C.; Delpy, L.; Pinaud, E.; Naranjo, J.R. Increased B cell proliferation and reduced Ig production in DREAM transgenic mice. J. Immunol. 2010, 185, 7527–7536. [Google Scholar] [CrossRef]
- Calvo, A.C.; Manzano, R.; Atencia-Cibreiro, G.; Olivan, S.; Munoz, M.J.; Zaragoza, P.; Cordero-Vazquez, P.; Esteban-Perez, J.; Garcia-Redondo, A.; Osta, R. Genetic biomarkers for ALS disease in transgenic SOD1(G93A) mice. PLoS ONE 2012, 7, e32632. [Google Scholar] [CrossRef]
- Lev, N.; Barhum, Y.; Lotan, I.; Steiner, I.; Offen, D. DJ-1 knockout augments disease severity and shortens survival in a mouse model of ALS. PLoS ONE 2015, 10, e0117190. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K.E.; Rasmussen, A.L.; Bennett, W.; King, A.; West, A.K.; Chung, R.S.; Chuah, M.I. Microglia and motor neurons during disease progression in the SOD1G93A mouse model of amyotrophic lateral sclerosis: Changes in arginase1 and inducible nitric oxide synthase. J. Neuroinflamm. 2014, 11, 55. [Google Scholar] [CrossRef] [PubMed]
- Rando, A.; de la Torre, M.; Martinez-Muriana, A.; Zaragoza, P.; Musaro, A.; Hernandez, S.; Navarro, X.; Toivonen, J.M.; Osta, R. Chemotherapeutic agent 5-fluorouracil increases survival of SOD1 mouse model of ALS. PLoS ONE 2019, 14, e0210752. [Google Scholar] [CrossRef] [PubMed]
- Ilieva, H.; Polymenidou, M.; Cleveland, D.W. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J. Cell Biol. 2009, 187, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Calatrava-Ferreras, L.; Gonzalo-Gobernado, R.; Herranz, A.S.; Reimers, D.; Montero Vega, T.; Jimenez-Escrig, A.; Richart Lopez, L.A.; Bazan, E. Effects of intravenous administration of human umbilical cord blood stem cells in 3-acetylpyridine-lesioned rats. Stem Cells Int. 2012, 2012, 135187. [Google Scholar] [CrossRef] [PubMed]
- Gonzalo-Gobernado, R.; Perucho, J.; Vallejo-Munoz, M.; Casarejos, M.J.; Reimers, D.; Jimenez-Escrig, A.; Gomez, A.; Ulzurrun de Asanza, G.M.; Bazan, E. Liver Growth Factor “LGF” as a Therapeutic Agent for Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 9201. [Google Scholar] [CrossRef] [PubMed]
- Ferri, A.; Cozzolino, M.; Crosio, C.; Nencini, M.; Casciati, A.; Gralla, E.B.; Rotilio, G.; Valentine, J.S.; Carri, M.T. Familial ALS-superoxide dismutases associate with mitochondria and shift their redox potentials. Proc. Natl. Acad. Sci. USA 2006, 103, 13860–13865. [Google Scholar] [CrossRef]
- Igoudjil, A.; Magrane, J.; Fischer, L.R.; Kim, H.J.; Hervias, I.; Dumont, M.; Cortez, C.; Glass, J.D.; Starkov, A.A.; Manfredi, G. In vivo pathogenic role of mutant SOD1 localized in the mitochondrial intermembrane space. J. Neurosci. 2011, 31, 15826–15837. [Google Scholar] [CrossRef]
- Kawamata, H.; Manfredi, G. Different regulation of wild-type and mutant Cu,Zn superoxide dismutase localization in mammalian mitochondria. Hum. Mol. Genet. 2008, 17, 3303–3317. [Google Scholar] [CrossRef]
- Vijayvergiya, C.; Beal, M.F.; Buck, J.; Manfredi, G. Mutant superoxide dismutase 1 forms aggregates in the brain mitochondrial matrix of amyotrophic lateral sclerosis mice. J. Neurosci. 2005, 25, 2463–2470. [Google Scholar] [CrossRef]
- Nishitoh, H.; Kadowaki, H.; Nagai, A.; Maruyama, T.; Yokota, T.; Fukutomi, H.; Noguchi, T.; Matsuzawa, A.; Takeda, K.; Ichijo, H. ALS-linked mutant SOD1 induces ER stress- and ASK1-dependent motor neuron death by targeting Derlin-1. Genes Dev. 2008, 22, 1451–1464. [Google Scholar] [CrossRef] [PubMed]
- Filezac de L’Etang, A.; Maharjan, N.; Cordeiro Brana, M.; Ruegsegger, C.; Rehmann, R.; Goswami, A.; Roos, A.; Troost, D.; Schneider, B.L.; Weis, J.; et al. Marinesco-Sjogren syndrome protein SIL1 regulates motor neuron subtype-selective ER stress in ALS. Nat. Neurosci. 2015, 18, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Saxena, S.; Cabuy, E.; Caroni, P. A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat. Neurosci. 2009, 12, 627–636. [Google Scholar] [CrossRef]
- Oh, Y.K.; Shin, K.S.; Yuan, J.; Kang, S.J. Superoxide dismutase 1 mutants related to amyotrophic lateral sclerosis induce endoplasmic stress in neuro2a cells. J. Neurochem. 2008, 104, 993–1005. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Thielen, P.; Matus, S.; Nassif, M.; Court, F.; Kiffin, R.; Martinez, G.; Cuervo, A.M.; Brown, R.H.; Glimcher, L.H. XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 2009, 23, 2294–2306. [Google Scholar] [CrossRef]
- Ilieva, E.V.; Ayala, V.; Jove, M.; Dalfo, E.; Cacabelos, D.; Povedano, M.; Bellmunt, M.J.; Ferrer, I.; Pamplona, R.; Portero-Otin, M. Oxidative and endoplasmic reticulum stress interplay in sporadic amyotrophic lateral sclerosis. Brain 2007, 130, 3111–3123. [Google Scholar] [CrossRef]
- Kikuchi, H.; Almer, G.; Yamashita, S.; Guegan, C.; Nagai, M.; Xu, Z.; Sosunov, A.A.; McKhann, G.M., 2nd; Przedborski, S. Spinal cord endoplasmic reticulum stress associated with a microsomal accumulation of mutant superoxide dismutase-1 in an ALS model. Proc. Natl. Acad. Sci. USA 2006, 103, 6025–6030. [Google Scholar] [CrossRef]
- Montibeller, L.; de Belleroche, J. Amyotrophic lateral sclerosis (ALS) and Alzheimer’s disease (AD) are characterised by differential activation of ER stress pathways: Focus on UPR target genes. Cell Stress Chaperones 2018, 23, 897–912. [Google Scholar] [CrossRef]
- Sasaki, S. Endoplasmic reticulum stress in motor neurons of the spinal cord in sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2010, 69, 346–355. [Google Scholar] [CrossRef]
- Kiskinis, E.; Sandoe, J.; Williams, L.A.; Boulting, G.L.; Moccia, R.; Wainger, B.J.; Han, S.; Peng, T.; Thams, S.; Mikkilineni, S.; et al. Pathways disrupted in human ALS motor neurons identified through genetic correction of mutant SOD1. Cell Stem Cell 2014, 14, 781–795. [Google Scholar] [CrossRef]
- Matus, S.; Lopez, E.; Valenzuela, V.; Nassif, M.; Hetz, C. Functional contribution of the transcription factor ATF4 to the pathogenesis of amyotrophic lateral sclerosis. PLoS ONE 2013, 8, e66672. [Google Scholar] [CrossRef]
- Dzhashiashvili, Y.; Monckton, C.P.; Shah, H.S.; Kunjamma, R.B.; Popko, B. The UPR-PERK pathway is not a promising therapeutic target for mutant SOD1-induced ALS. Neurobiol. Dis. 2019, 127, 527–544. [Google Scholar] [CrossRef] [PubMed]
- Charif, S.E.; Vassallu, M.F.; Salvanal, L.; Igaz, L.M. Protein synthesis modulation as a therapeutic approach for amyotrophic lateral sclerosis and frontotemporal dementia. Neural Regen. Res. 2022, 17, 1423–1430. [Google Scholar] [CrossRef]
- Hetz, C.; Axten, J.M.; Patterson, J.B. Pharmacological targeting of the unfolded protein response for disease intervention. Nat. Chem. Biol. 2019, 15, 764–775. [Google Scholar] [CrossRef] [PubMed]
- Medinas, D.B.; Gonzalez, J.V.; Falcon, P.; Hetz, C. Fine-Tuning ER Stress Signal Transducers to Treat Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2017, 10, 216. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Liao, Y.; Rahaman, A.; Kumar, V. Towards Understanding the Relationship Between ER Stress and Unfolded Protein Response in Amyotrophic Lateral Sclerosis. Front. Aging Neurosci. 2022, 14, 892518. [Google Scholar] [CrossRef]
- Halliday, M.; Radford, H.; Sekine, Y.; Moreno, J.; Verity, N.; le Quesne, J.; Ortori, C.A.; Barrett, D.A.; Fromont, C.; Fischer, P.M.; et al. Partial restoration of protein synthesis rates by the small molecule ISRIB prevents neurodegeneration without pancreatic toxicity. Cell Death Dis. 2015, 6, e1672. [Google Scholar] [CrossRef]
- Mercado, G.; Castillo, V.; Soto, P.; Lopez, N.; Axten, J.M.; Sardi, S.P.; Hoozemans, J.J.M.; Hetz, C. Targeting PERK signaling with the small molecule GSK2606414 prevents neurodegeneration in a model of Parkinson’s disease. Neurobiol. Dis. 2018, 112, 136–148. [Google Scholar] [CrossRef]
- Bugallo, R.; Marlin, E.; Baltanas, A.; Toledo, E.; Ferrero, R.; Vinueza-Gavilanes, R.; Larrea, L.; Arrasate, M.; Aragon, T. Fine tuning of the unfolded protein response by ISRIB improves neuronal survival in a model of amyotrophic lateral sclerosis. Cell Death Dis. 2020, 11, 397. [Google Scholar] [CrossRef]
- Halliday, M.; Radford, H.; Zents, K.A.M.; Molloy, C.; Moreno, J.A.; Verity, N.C.; Smith, E.; Ortori, C.A.; Barrett, D.A.; Bushell, M.; et al. Repurposed drugs targeting eIF2α-P-mediated translational repression prevent neurodegeneration in mice. Brain 2017, 140, 1768–1783. [Google Scholar] [CrossRef]
- Sidrauski, C.; McGeachy, A.M.; Ingolia, N.T.; Walter, P. The small molecule ISRIB reverses the effects of eIF2alpha phosphorylation on translation and stress granule assembly. Elife 2015, 4, e05033. [Google Scholar] [CrossRef] [PubMed]
- Sidrauski, C.; Tsai, J.C.; Kampmann, M.; Hearn, B.R.; Vedantham, P.; Jaishankar, P.; Sokabe, M.; Mendez, A.S.; Newton, B.W.; Tang, E.L.; et al. Pharmacological dimerization and activation of the exchange factor eIF2B antagonizes the integrated stress response. Elife 2015, 4, e07314. [Google Scholar] [CrossRef] [PubMed]
- Boyce, M.; Bryant, K.F.; Jousse, C.; Long, K.; Harding, H.P.; Scheuner, D.; Kaufman, R.J.; Ma, D.; Coen, D.M.; Ron, D.; et al. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science 2005, 307, 935–939. [Google Scholar] [CrossRef]
- Dalla Bella, E.; Bersano, E.; Antonini, G.; Borghero, G.; Capasso, M.; Caponnetto, C.; Chio, A.; Corbo, M.; Filosto, M.; Giannini, F.; et al. The unfolded protein response in amyotrophic later sclerosis: Results of a phase 2 trial. Brain 2021, 144, 2635–2647. [Google Scholar] [CrossRef] [PubMed]
- Das, I.; Krzyzosiak, A.; Schneider, K.; Wrabetz, L.; D’Antonio, M.; Barry, N.; Sigurdardottir, A.; Bertolotti, A. Preventing proteostasis diseases by selective inhibition of a phosphatase regulatory subunit. Science 2015, 348, 239–242. [Google Scholar] [CrossRef]
- Tsaytler, P.; Harding, H.P.; Ron, D.; Bertolotti, A. Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science 2011, 332, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Popko, B.; Tixier, E.; Roos, R.P. Guanabenz, which enhances the unfolded protein response, ameliorates mutant SOD1-induced amyotrophic lateral sclerosis. Neurobiol. Dis. 2014, 71, 317–324. [Google Scholar] [CrossRef]
- Cross, B.C.; Bond, P.J.; Sadowski, P.G.; Jha, B.K.; Zak, J.; Goodman, J.M.; Silverman, R.H.; Neubert, T.A.; Baxendale, I.R.; Ron, D.; et al. The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proc. Natl. Acad. Sci. USA 2012, 109, E869–E878. [Google Scholar] [CrossRef]
- Kriss, C.L.; Pinilla-Ibarz, J.A.; Mailloux, A.W.; Powers, J.J.; Tang, C.H.; Kang, C.W.; Zanesi, N.; Epling-Burnette, P.K.; Sotomayor, E.M.; Croce, C.M.; et al. Overexpression of TCL1 activates the endoplasmic reticulum stress response: A novel mechanism of leukemic progression in mice. Blood 2012, 120, 1027–1038. [Google Scholar] [CrossRef]
- Mimura, N.; Fulciniti, M.; Gorgun, G.; Tai, Y.T.; Cirstea, D.; Santo, L.; Hu, Y.; Fabre, C.; Minami, J.; Ohguchi, H.; et al. Blockade of XBP1 splicing by inhibition of IRE1alpha is a promising therapeutic option in multiple myeloma. Blood 2012, 119, 5772–5781. [Google Scholar] [CrossRef]
- Papandreou, I.; Denko, N.C.; Olson, M.; Van Melckebeke, H.; Lust, S.; Tam, A.; Solow-Cordero, D.E.; Bouley, D.M.; Offner, F.; Niwa, M.; et al. Identification of an Ire1alpha endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood 2011, 117, 1311–1314. [Google Scholar] [CrossRef] [PubMed]
- Ri, M.; Tashiro, E.; Oikawa, D.; Shinjo, S.; Tokuda, M.; Yokouchi, Y.; Narita, T.; Masaki, A.; Ito, A.; Ding, J.; et al. Identification of Toyocamycin, an agent cytotoxic for multiple myeloma cells, as a potent inhibitor of ER stress-induced XBP1 mRNA splicing. Blood Cancer J. 2012, 2, e79. [Google Scholar] [CrossRef] [PubMed]
- Volkmann, K.; Lucas, J.L.; Vuga, D.; Wang, X.; Brumm, D.; Stiles, C.; Kriebel, D.; Der-Sarkissian, A.; Krishnan, K.; Schweitzer, C.; et al. Potent and selective inhibitors of the inositol-requiring enzyme 1 endoribonuclease. J. Biol. Chem. 2011, 286, 12743–12755. [Google Scholar] [CrossRef] [PubMed]
- Fribley, A.M.; Cruz, P.G.; Miller, J.R.; Callaghan, M.U.; Cai, P.; Narula, N.; Neubig, R.R.; Showalter, H.D.; Larsen, S.D.; Kirchhoff, P.D.; et al. Complementary cell-based high-throughput screens identify novel modulators of the unfolded protein response. J. Biomol. Screen 2011, 16, 825–835. [Google Scholar] [CrossRef]
- Maher, P. The Potential of Flavonoids for the Treatment of Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 3056. [Google Scholar] [CrossRef]
- Wiseman, R.L.; Zhang, Y.; Lee, K.P.; Harding, H.P.; Haynes, C.M.; Price, J.; Sicheri, F.; Ron, D. Flavonol activation defines an unanticipated ligand-binding site in the kinase-RNase domain of IRE1. Mol. Cell 2010, 38, 291–304. [Google Scholar] [CrossRef]
- Canosa, A.; Calvo, A.; Barberis, M.; Brunetti, M.; Restagno, G.; Cammarosano, S.; Ilardi, A.; Vigliani, M.C.; Chio, A.; Moglia, C. Amyotrophic lateral sclerosis onset after prolonged treatment with a VEGF receptors inhibitor. Amyotroph. Lateral Scler. Front. Degener. 2015, 16, 129–130. [Google Scholar] [CrossRef]
- Fan, F.; Liu, F.; Shen, P.; Tao, L.; Zhang, H.; Wu, H. Salvianolic acid B, a new type I IRE1 kinase inhibitor, abrogates AngII-induced angiogenesis by interacting with IRE1 in its active conformation. Clin. Exp. Pharmacol. Physiol. 2023, 50, 82–95. [Google Scholar] [CrossRef]
- Feldman, H.C.; Vidadala, V.N.; Potter, Z.E.; Papa, F.R.; Backes, B.J.; Maly, D.J. Development of a Chemical Toolset for Studying the Paralog-Specific Function of IRE1. ACS Chem. Biol. 2019, 14, 2595–2605. [Google Scholar] [CrossRef]
- Ghosh, R.; Wang, L.; Wang, E.S.; Perera, B.G.; Igbaria, A.; Morita, S.; Prado, K.; Thamsen, M.; Caswell, D.; Macias, H.; et al. Allosteric inhibition of the IRE1alpha RNase preserves cell viability and function during endoplasmic reticulum stress. Cell 2014, 158, 534–548. [Google Scholar] [CrossRef]
- Wang, Z.; Jiang, C.; Chen, W.; Zhang, G.; Luo, D.; Cao, Y.; Wu, J.; Ding, Y.; Liu, B. Baicalein induces apoptosis and autophagy via endoplasmic reticulum stress in hepatocellular carcinoma cells. Biomed. Res. Int. 2014, 2014, 732516. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.S.; Yen, J.H.; Kou, M.C.; Wu, M.J. Luteolin and Apigenin Attenuate 4-Hydroxy-2-Nonenal-Mediated Cell Death through Modulation of UPR, Nrf2-ARE and MAPK Pathways in PC12 Cells. PLoS ONE 2015, 10, e0130599. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.X.; Conn, P.M. Pharmacoperones as Novel Therapeutics for Diverse Protein Conformational Diseases. Physiol. Rev. 2018, 98, 697–725. [Google Scholar] [CrossRef] [PubMed]
- Kieran, D.; Kalmar, B.; Dick, J.R.; Riddoch-Contreras, J.; Burnstock, G.; Greensmith, L. Treatment with arimoclomol, a coinducer of heat shock proteins, delays disease progression in ALS mice. Nat. Med. 2004, 10, 402–405. [Google Scholar] [CrossRef] [PubMed]
- Engin, F.; Yermalovich, A.; Nguyen, T.; Hummasti, S.; Fu, W.; Eizirik, D.L.; Mathis, D.; Hotamisligil, G.S. Restoration of the unfolded protein response in pancreatic beta cells protects mice against type 1 diabetes. Sci. Transl. Med. 2013, 5, 211ra156. [Google Scholar] [CrossRef]
- Ozcan, U.; Yilmaz, E.; Ozcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Gorgun, C.Z.; Hotamisligil, G.S. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef]
- Batulan, Z.; Taylor, D.M.; Aarons, R.J.; Minotti, S.; Doroudchi, M.M.; Nalbantoglu, J.; Durham, H.D. Induction of multiple heat shock proteins and neuroprotection in a primary culture model of familial amyotrophic lateral sclerosis. Neurobiol. Dis. 2006, 24, 213–225. [Google Scholar] [CrossRef]
- Sittler, A.; Lurz, R.; Lueder, G.; Priller, J.; Lehrach, H.; Hayer-Hartl, M.K.; Hartl, F.U.; Wanker, E.E. Geldanamycin activates a heat shock response and inhibits huntingtin aggregation in a cell culture model of Huntington’s disease. Hum. Mol. Genet 2001, 10, 1307–1315. [Google Scholar] [CrossRef]
- Clinicaltrialls.gov ID: NCT04516096; A Compassionate Use Protocol of AMX0035 for Treatment of Patients with Amyotrophic Lateral Sclerosis (ALS). Available online: https://www.clinicaltrials.gov/study/NCT04516096?term=Amylyx%20AMX0035&rank=1 (accessed on 13 September 2023).
- Martinus, R.D.; Garth, G.P.; Webster, T.L.; Cartwright, P.; Naylor, D.J.; Hoj, P.B.; Hoogenraad, N.J. Selective induction of mitochondrial chaperones in response to loss of the mitochondrial genome. Eur. J. Biochem. 1996, 240, 98–103. [Google Scholar] [CrossRef]
- Zhao, Q.; Wang, J.; Levichkin, I.V.; Stasinopoulos, S.; Ryan, M.T.; Hoogenraad, N.J. A mitochondrial specific stress response in mammalian cells. EMBO J. 2002, 21, 4411–4419. [Google Scholar] [CrossRef]
- Pharaoh, G.; Sataranatarajan, K.; Street, K.; Hill, S.; Gregston, J.; Ahn, B.; Kinter, C.; Kinter, M.; Van Remmen, H. Metabolic and Stress Response Changes Precede Disease Onset in the Spinal Cord of Mutant SOD1 ALS Mice. Front. Neurosci. 2019, 13, 487. [Google Scholar] [CrossRef] [PubMed]
- Riar, A.K.; Burstein, S.R.; Palomo, G.M.; Arreguin, A.; Manfredi, G.; Germain, D. Sex specific activation of the ERalpha axis of the mitochondrial UPR (UPRmt) in the G93A-SOD1 mouse model of familial ALS. Hum. Mol. Genet 2017, 26, 1318–1327. [Google Scholar] [CrossRef] [PubMed]
- Cilleros-Holgado, P.; Gomez-Fernandez, D.; Pinero-Perez, R.; Reche-Lopez, D.; Alvarez-Cordoba, M.; Munuera-Cabeza, M.; Talaveron-Rey, M.; Povea-Cabello, S.; Suarez-Carrillo, A.; Romero-Gonzalez, A.; et al. mtUPR Modulation as a Therapeutic Target for Primary and Secondary Mitochondrial Diseases. Int. J. Mol. Sci. 2023, 24, 1482. [Google Scholar] [CrossRef] [PubMed]
- Pellegrino, M.W.; Nargund, A.M.; Haynes, C.M. Signaling the mitochondrial unfolded protein response. Biochim. Biophys. Acta 2013, 1833, 410–416. [Google Scholar] [CrossRef]
- Siegelin, M.D.; Dohi, T.; Raskett, C.M.; Orlowski, G.M.; Powers, C.M.; Gilbert, C.A.; Ross, A.H.; Plescia, J.; Altieri, D.C. Exploiting the mitochondrial unfolded protein response for cancer therapy in mice and human cells. J. Clin. Investig. 2011, 121, 1349–1360. [Google Scholar] [CrossRef]
- Suarez-Rivero, J.M.; Pastor-Maldonado, C.J.; Povea-Cabello, S.; Alvarez-Cordoba, M.; Villalon-Garcia, I.; Talaveron-Rey, M.; Suarez-Carrillo, A.; Munuera-Cabeza, M.; Reche-Lopez, D.; Cilleros-Holgado, P.; et al. Activation of the Mitochondrial Unfolded Protein Response: A New Therapeutic Target? Biomedicines 2022, 10, 1611. [Google Scholar] [CrossRef]
- Lopez-Hurtado, A.; Burgos, D.F.; Gonzalez, P.; Dopazo, X.M.; Gonzalez, V.; Rabano, A.; Mellstrom, B.; Naranjo, J.R. Inhibition of DREAM-ATF6 interaction delays onset of cognition deficit in a mouse model of Huntington’s disease. Mol. Brain 2018, 11, 13. [Google Scholar] [CrossRef]
- Lopez-Hurtado, A.; Peraza, D.A.; Cercos, P.; Lagartera, L.; Gonzalez, P.; Dopazo, X.M.; Herranz, R.; Gonzalez, T.; Martin-Martinez, M.; Mellstrom, B.; et al. Targeting the neuronal calcium sensor DREAM with small-molecules for Huntington’s disease treatment. Sci. Rep. 2019, 9, 7260. [Google Scholar] [CrossRef]
- Peraza, D.A.; Cercos, P.; Miaja, P.; Merinero, Y.G.; Lagartera, L.; Socuellamos, P.G.; Izquierdo Garcia, C.; Sanchez, S.A.; Lopez-Hurtado, A.; Martin-Martinez, M.; et al. Identification of IQM-266, a Novel DREAM Ligand That Modulates K(V)4 Currents. Front. Mol. Neurosci. 2019, 12, 11. [Google Scholar] [CrossRef]
- Socuellamos, P.G.; Olivos-Ore, L.A.; Barahona, M.V.; Cercos, P.; Perez Pascual, M.; Arribas-Blazquez, M.; Naranjo, J.R.; Valenzuela, C.; Gutierrez-Rodriguez, M.; Artalejo, A.R. IQM-PC332, a Novel DREAM Ligand with Antinociceptive Effect on Peripheral Nerve Injury-Induced Pain. Int. J. Mol. Sci. 2022, 23, 2142. [Google Scholar] [CrossRef]
- Olivan, S.; Calvo, A.C.; Rando, A.; Munoz, M.J.; Zaragoza, P.; Osta, R. Comparative study of behavioural tests in the SOD1G93A mouse model of amyotrophic lateral sclerosis. Exp. Anim. 2015, 64, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Mellstrom, B.; Kastanauskaite, A.; Knafo, S.; Gonzalez, P.; Dopazo, X.M.; Ruiz-Nuno, A.; Jefferys, J.G.; Zhuo, M.; Bliss, T.V.; Naranjo, J.R.; et al. Specific cytoarchitectureal changes in hippocampal subareas in daDREAM mice. Mol. Brain 2016, 9, 22. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonzalo-Gobernado, R.; Moreno-Martínez, L.; González, P.; Dopazo, X.M.; Calvo, A.C.; Pidal-Ladrón de Guevara, I.; Seisdedos, E.; Díaz-Muñoz, R.; Mellström, B.; Osta, R.; et al. Repaglinide Induces ATF6 Processing and Neuroprotection in Transgenic SOD1G93A Mice. Int. J. Mol. Sci. 2023, 24, 15783. https://doi.org/10.3390/ijms242115783
Gonzalo-Gobernado R, Moreno-Martínez L, González P, Dopazo XM, Calvo AC, Pidal-Ladrón de Guevara I, Seisdedos E, Díaz-Muñoz R, Mellström B, Osta R, et al. Repaglinide Induces ATF6 Processing and Neuroprotection in Transgenic SOD1G93A Mice. International Journal of Molecular Sciences. 2023; 24(21):15783. https://doi.org/10.3390/ijms242115783
Chicago/Turabian StyleGonzalo-Gobernado, Rafael, Laura Moreno-Martínez, Paz González, Xose Manuel Dopazo, Ana Cristina Calvo, Isabel Pidal-Ladrón de Guevara, Elisa Seisdedos, Rodrigo Díaz-Muñoz, Britt Mellström, Rosario Osta, and et al. 2023. "Repaglinide Induces ATF6 Processing and Neuroprotection in Transgenic SOD1G93A Mice" International Journal of Molecular Sciences 24, no. 21: 15783. https://doi.org/10.3390/ijms242115783
APA StyleGonzalo-Gobernado, R., Moreno-Martínez, L., González, P., Dopazo, X. M., Calvo, A. C., Pidal-Ladrón de Guevara, I., Seisdedos, E., Díaz-Muñoz, R., Mellström, B., Osta, R., & Naranjo, J. R. (2023). Repaglinide Induces ATF6 Processing and Neuroprotection in Transgenic SOD1G93A Mice. International Journal of Molecular Sciences, 24(21), 15783. https://doi.org/10.3390/ijms242115783