
Integrative Metabolomic and Transcriptomic Landscape during Akebia trifoliata Fruit Ripening and Cracking


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
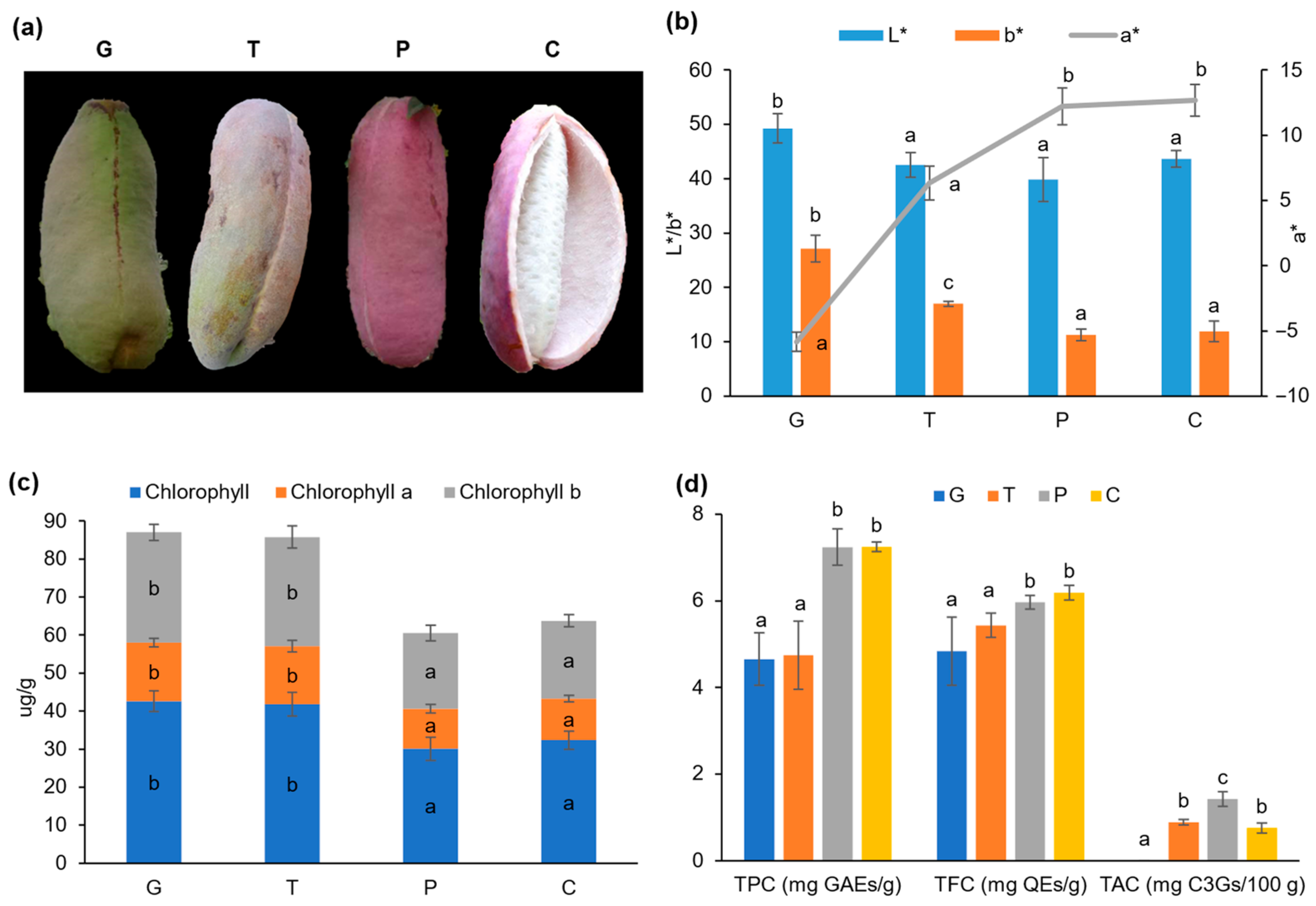
2.1. Alterations in Color, Chlorophyll Content, and Phytochemical Profile in the A. trifoliata Fruit Peels
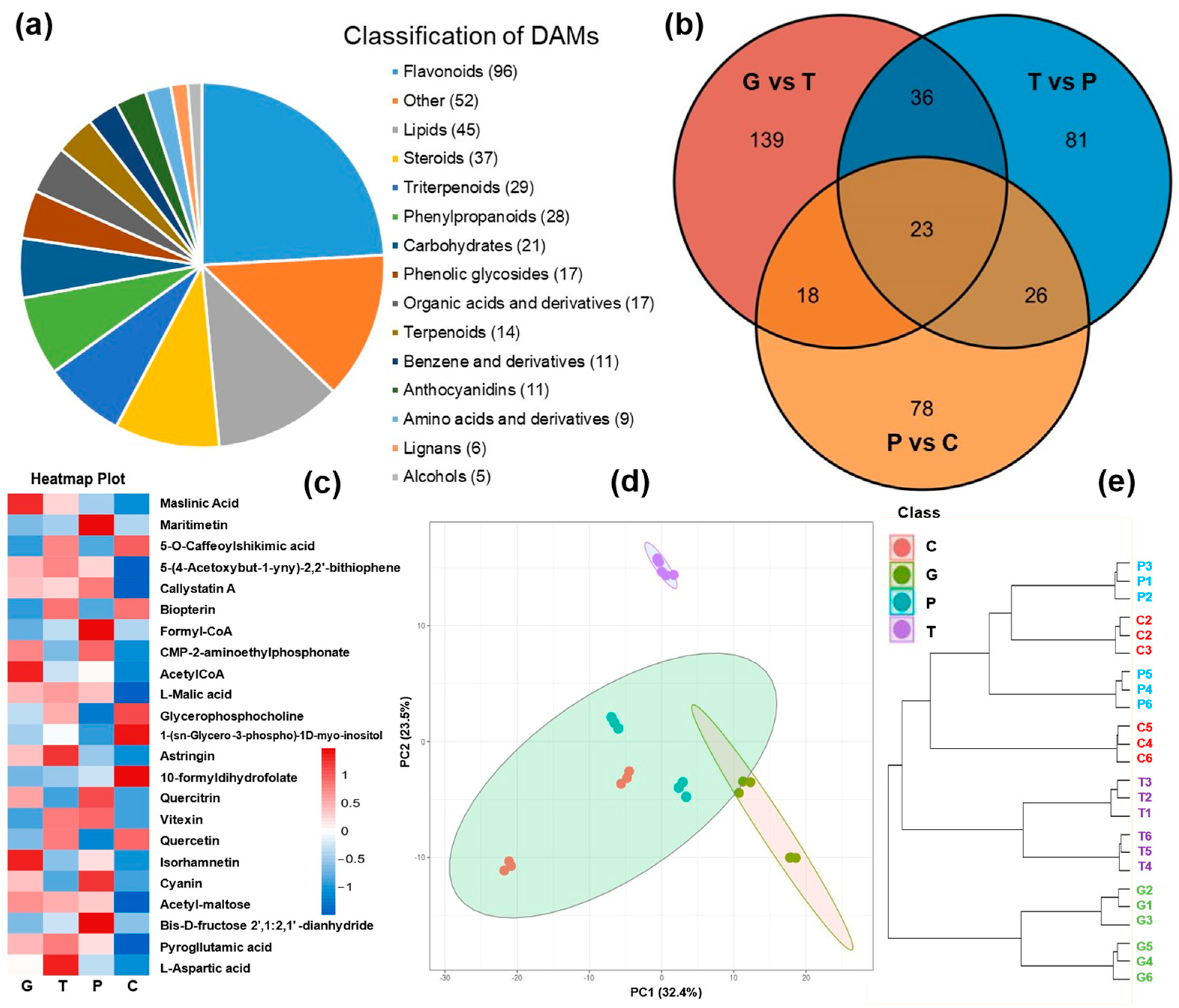
2.2. Metabolome Profiling of A. trifoliata Peel over the Ripening Stages
2.3. Overview of RNA-Seq of A. trifoliata Peel over the Ripening Stages
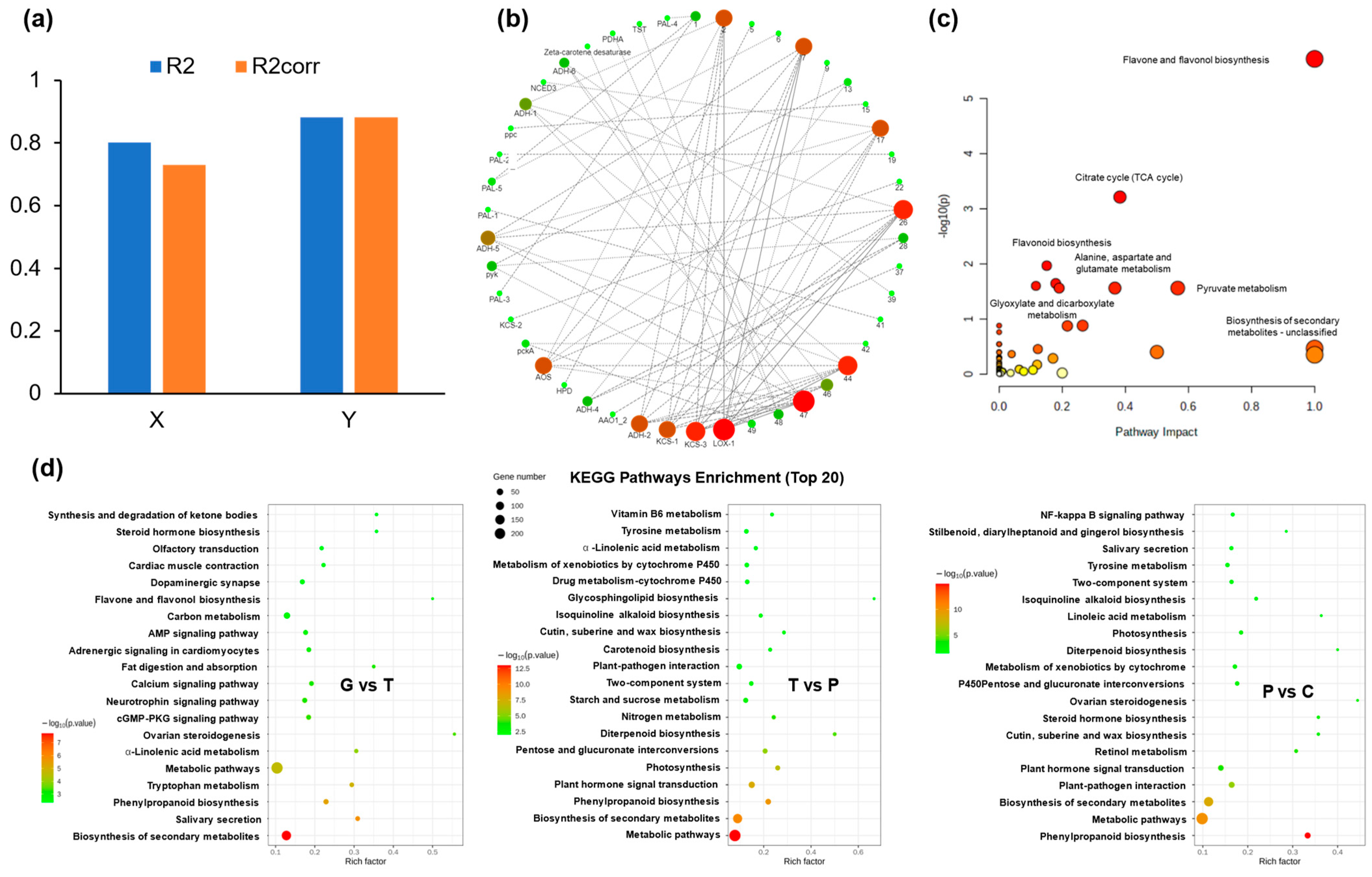
2.4. Identification and Analysis of DEGs
2.5. Joint Analysis of DAMs and DEGs
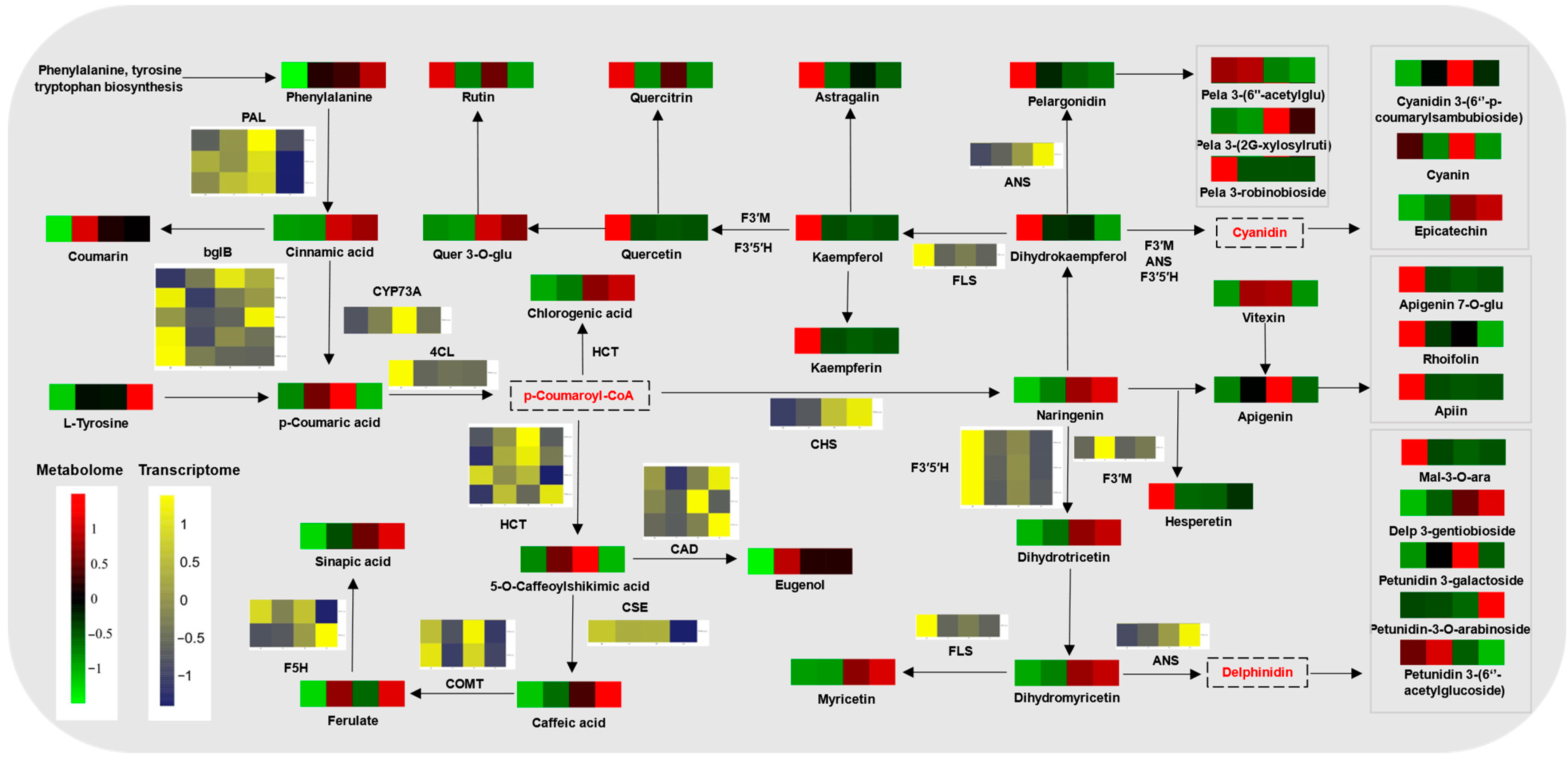
2.6. Phenylpropanoid Metabolism
2.7. Photosynthesis Metabolism
2.8. Plant Hormone Metabolism
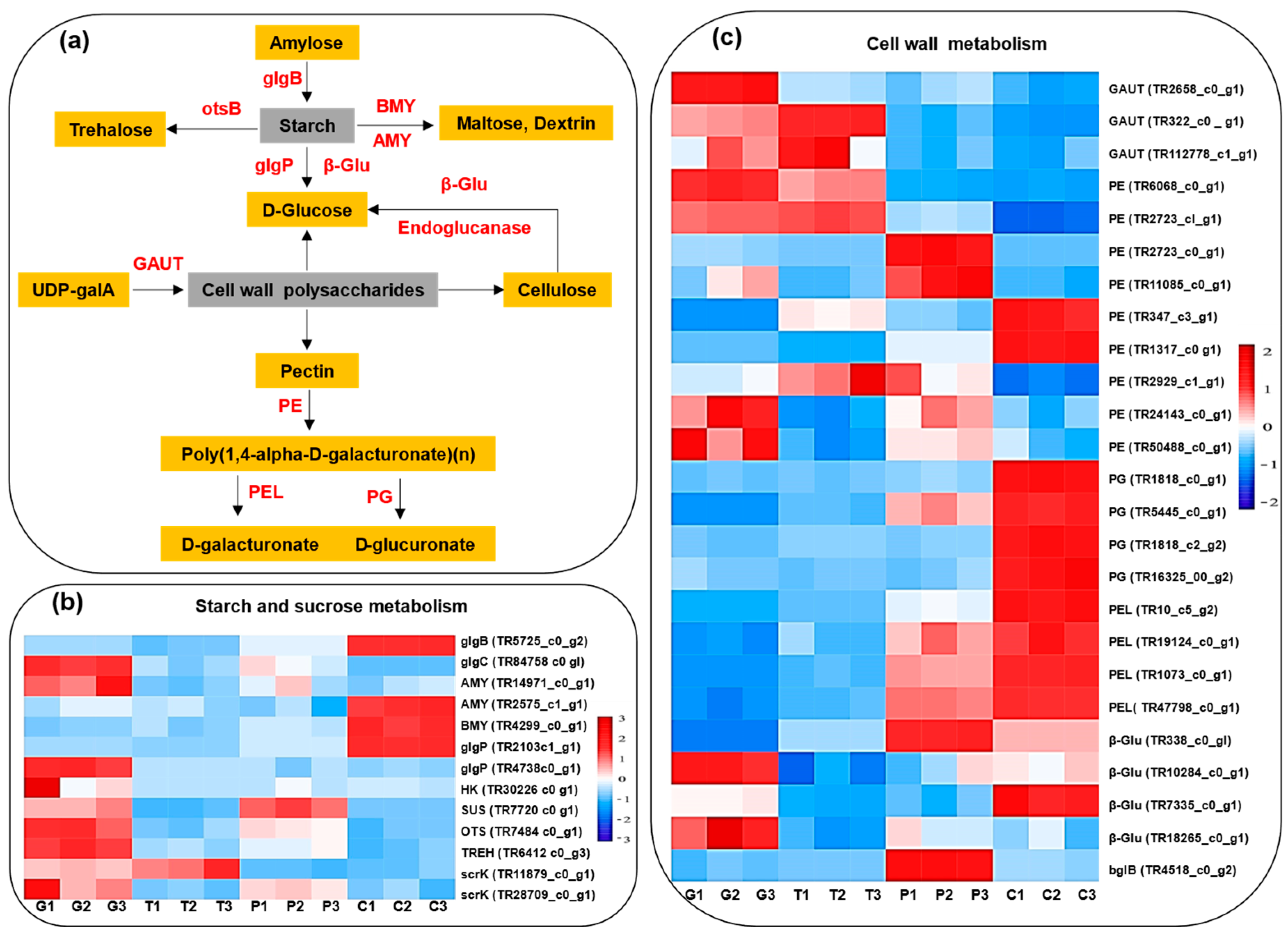
2.9. Cell-Wall Metabolism
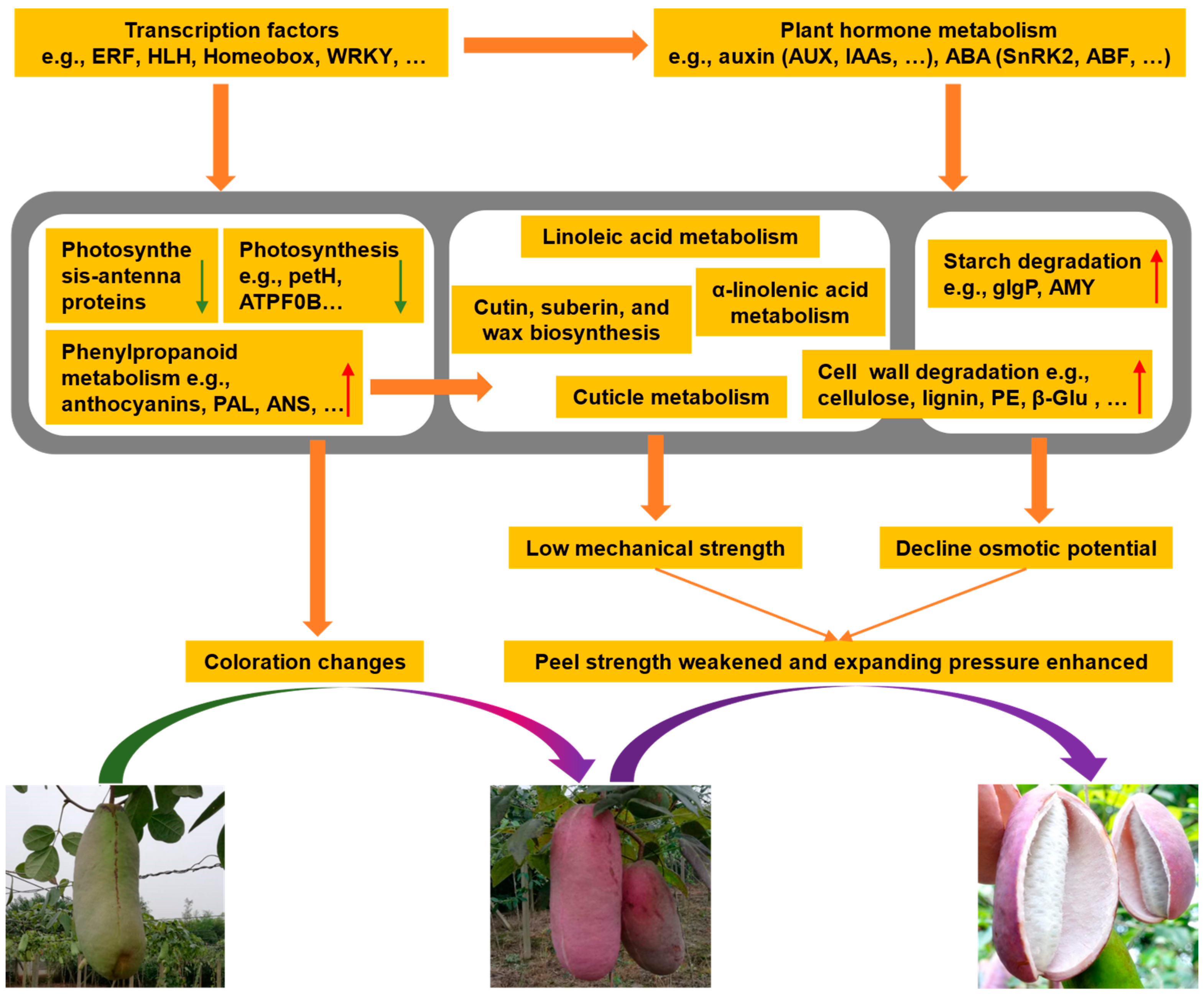
2.10. Possible Explanations for A. trifoliata Fruit Cracking
3. Materials and Methods
3.1. Materials and Reagents
3.2. Color and Chlorophyll Measurements
3.3. Peel Extract Preparations
3.4. Phytochemical Constituent Measurement
3.5. Metabolomics Analysis and Data Processing
3.6. Transcriptome Sequencing and Analysis
3.7. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jiang, Y.; Ding, Y.; Wang, D.; Deng, Y.; Zhao, Y. Radio frequency-assisted enzymatic extraction of anthocyanins from Akebia trifoliata (Thunb.) Koidz. flowers: Process optimization, structure, and bioactivity determination. Ind. Crops Prod. 2020, 149, 112327. [Google Scholar] [CrossRef]
- Zou, S.; Yao, X.; Zhong, C.; Zhao, T.; Huang, H. Effectiveness of recurrent selection in Akebia trifoliata (Lardizabalaceae) breeding. Sci. Hortic. 2019, 246, 79–85. [Google Scholar] [CrossRef]
- Zhou, Y.; Jie, B.; Wu, G.; Cao, Y.; Wu, W.; Meng, H. Research Progress of Akebia trifoliata (Thunb.) Koidz. Agric. Biotechnol. 2017, 6, 1–8. [Google Scholar]
- Niu, J.; Shi, Y.; Huang, K.; Zhong, Y.; Chen, J.; Sun, Z.; Luan, M.; Chen, J. Integrative transcriptome and proteome analyses provide new insights into different stages of Akebia trifoliata fruit cracking during ripening. Biotechnol. Biofuels. 2020, 13, 1–18. [Google Scholar] [CrossRef]
- Li, L.; Yao, X.; Zhong, C.; Chen, X.; Huang, H. Akebia: A potential new fruit crop in China. HortScience 2010, 45, 4–10. [Google Scholar] [CrossRef]
- Knoche, M.; Lang, A. Ongoing Growth Challenges Fruit Skin Integrity. Crit. Rev. Plant. Sci. 2017, 36, 190–215. [Google Scholar] [CrossRef]
- Khadivi-Khub, A. Physiological and genetic factors influencing fruit cracking. Acta. Physiol. Plantarum. 2014, 37, 1718. [Google Scholar] [CrossRef]
- Niu, J.; Wang, Y.; Shi, Y.; Wang, X.; Sun, Z.; Huang, K. Development of SSR markers via de novo transcriptome assembly in Akebia trifoliata (Thunb.) Koidz. Genome 2019, 62, 817–831. [Google Scholar] [CrossRef]
- Yang, H.; Liu, H.; Shi, X.; Ge, F.; Zhao, Z.; Luo, H. Transcriptome analysis of Akebia trifoliate in ripening process. J. Moun. Agr. Biol. 2016, 35, 46–51. [Google Scholar]
- Chen, J.; Duan, Y.; Hu, Y.; Li, W.; Sun, D.; Hu, H.; Xie, J. Transcriptome analysis of atemoya pericarp elucidates the role of polysaccharide metabolism in fruit ripening and cracking after harvest. BMC Plant Biol. 2019, 19, 219. [Google Scholar] [CrossRef]
- Yun, Z.; Li, T.; Gao, H.; Zhu, H.; Gupta, V.K.; Jiang, Y.; Duan, X. Integrated Transcriptomic, Proteomic, and Metabolomics Analysis Reveals Peel Ripening of Harvested Banana under Natural Condition. Biomolecules 2019, 9, 167. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gao, X.; Ma, Z.; Chen, J.; Liu, Y. Analysis of the molecular basis of fruit cracking susceptibility in Litchi chinensis cv. Baitangying by transcriptome and quantitative proteome profiling. J. Plant Physiol. 2019, 234, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Liu, L.; Yu, J.; Chen, R.; Hu, C.; Wang, H.; Li, D.; Wang, Z.; Zhao, Z. Combined transcriptomic and metabolomic analyses reveal the mechanism of debagged ‘Fuji’ apple sunburn. LWT 2023, 181, 114680. [Google Scholar] [CrossRef]
- Li, Y.; Fang, J.; Qi, X.; Lin, M.; Zhong, Y.; Sun, L.; Cui, W. Combined Analysis of the Fruit Metabolome and Transcriptome Reveals Candidate Genes Involved in Flavonoid Biosynthesis in Actinidia arguta. Int. J. Mol. Sci. 2018, 19, 1471. [Google Scholar] [CrossRef]
- Wang, J.; Wu, X.F.; Tang, Y.; Li, J.G.; Zhao, M.L. RNA-Seq Provides New Insights into the Molecular Events Involved in “Ball-Skin versus Bladder Effect” on Fruit Cracking in Litchi. Int. J. Mol. Sci. 2021, 22, 454. [Google Scholar] [CrossRef]
- Zhu, M.; Yu, J.; Zhao, M.; Wang, M.; Yang, G. Transcriptome analysis of metabolisms related to fruit cracking during ripening of a cracking-susceptible grape berry cv. Xiangfei (Vitis vinifera L.). Genes Genom. 2020, 42, 639–650. [Google Scholar] [CrossRef]
- Shen, J.; Zou, Z.; Zhang, X.; Zhou, L.; Wang, Y.; Fang, W.; Zhu, X. Metabolic analyses reveal different mechanisms of leaf color change in two purple-leaf tea plant (Camellia sinensis L.) cultivars. Hortic. Res. 2018, 5, 1. [Google Scholar] [CrossRef]
- Zhang, Q.; Liu, M.; Ruan, J. Metabolomics analysis reveals the metabolic and functional roles of flavonoids in light-sensitive tea leaves. BMC Plant Biol. 2017, 17, 64. [Google Scholar] [CrossRef]
- Correia, S.; Schouten, R.; Silva, A.P.; Gonçalves, B. Sweet cherry fruit cracking mechanisms and prevention strategies: A review. Sci. Hortic. 2018, 240, 369–377. [Google Scholar] [CrossRef]
- Marmon, S.; Sturtevant, D.; Herrfurth, C.; Chapman, K.; Stymne, S.; Feussner, I. Two Acyltransferases Contribute Differently to Linolenic Acid Levels in Seed Oil. Plant Physiol. 2017, 173, 2081–2095. [Google Scholar] [CrossRef]
- Li, W.-C.; Wu, J.-Y.; Zhang, H.-N.; Shi, S.-Y.; Liu, L.-Q.; Shu, B.; Liang, Q.-Z.; Xie, J.-H.; Wei, Y.-Z. De Novo Assembly and Characterization of Pericarp Transcriptome and Identification of Candidate Genes Mediating Fruit Cracking in Litchi chinensis Sonn. Int. J. Mol. Sci. 2014, 15, 17667–17685. [Google Scholar] [CrossRef]
- Forlani, S.; Masiero, S.; Mizzotti, C. Fruit ripening: The role of hormones, cell wall modifications, and their relationship with pathogens. J. Exp. Bot. 2019, 70, 2993–3006. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Chen, X.; Zong, X.; Shu, H.; Gao, D.; Liu, Q. Comparative Transcriptome Analysis of Genes Involved in Anthocyanin Biosynthesis in the Red and Yellow Fruits of Sweet Cherry (Prunus avium L.). PLoS ONE 2015, 10, e0121164. [Google Scholar] [CrossRef] [PubMed]
- Copley, T.R.; Aliferis, K.A.; Kliebenstein, D.J.; Jabaji, S.H. An integrated RNAseq-1H NMR metabolomics approach to understand soybean primary metabolism regulation in response to Rhizoctonia foliar blight disease. BMC Plant Biol. 2017, 17, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wang, L.; Liu, Z.; Zhao, Z.; Zhao, J.; Wang, Z.; Zhou, G.; Liu, P.; Liu, M. Transcriptome and metabolome profiling unveil the mechanisms of Ziziphus jujuba Mill. peel coloration. Food Chem. 2020, 312, 125903. [Google Scholar] [CrossRef]
- Sun, L.; Yuan, B.; Zhang, M.; Wang, L.; Cui, M.; Wang, Q.; Leng, P. Fruit-specific RNAi-mediated suppression of SlNCED1 increases both lycopene and β-carotene contents in tomato fruit. J. Exp. Bot. 2012, 63, 3097–3108. [Google Scholar] [CrossRef] [PubMed]
- Francoz, E.; Ranocha, P.; Nguyen-Kim, H.; Jamet, E.; Burlat, V.; Dunand, C. Roles of cell wall peroxidases in plant development. Phytochemistry 2015, 112, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Joshi, M.; Baghel, R.S.; Fogelman, E.; Stern, R.A.; Ginzberg, I. Identification of candidate genes mediating apple fruit-cracking resistance following the application of gibberellic acids 4 + 7 and the cytokinin 6-benzyladenine. Plant Physiol. Biochem. 2018, 127, 436–445. [Google Scholar] [CrossRef]
- Liu, S.; Meng, Z.; Zhang, H.; Chu, Y.; Qiu, Y.; Jin, B.; Wang, L. Identification and characterization of thirteen gene families involved in flavonoid biosynthesis in Ginkgo biloba. Ind. Crops Prod. 2022, 188, 115576. [Google Scholar] [CrossRef]
- Oliveira, D.M.; Mota, T.R.; Salatta, F.V.; Sinzker, R.C.; Končitíková, R.; Kopečný, D.; Simister, R.; Silva, M.; Goeminne, G.; Morreel, K.; et al. Cell wall remodeling under salt stress: Insights into changes in polysaccharides, feruloylation, lignification, and phenolic metabolism in maize. Plant Cell Environ. 2020, 43, 2172–2191. [Google Scholar] [CrossRef]
- Jiang, Y.; Wu, Y.; Yin, H.; Wang, D.; Zhong, Y.; Deng, Y. Metabolic and bioactive comparative analyses reveal the mechanisms of color changes in Akebia trifoliata (Thunb.) Koidz fruit. Sci. Hortic. 2022, 295, 110819. [Google Scholar] [CrossRef]
- Matas, A.J.; Yeats, T.H.; Buda, G.J.; Zheng, Y.; Chatterjee, S.; Tohge, T.; Ponnala, L.; Adato, A.; Aharoni, A.; Stark, R.; et al. Tissue- and Cell-Type Specific Transcriptome Profiling of Expanding Tomato Fruit Provides Insights into Metabolic and Regulatory Specialization and Cuticle Formation. Plant Cell 2011, 23, 3893–3910. [Google Scholar] [CrossRef] [PubMed]
- Guilfoyle, T.; Hagen, G. Auxin signal transduction. Essays Biochem. 2015, 58, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Yan, J.; Li, W.; Wang, Q.; Wang, C.; Guo, J.; Geng, D.; Guan, Q.; Ma, F. Integrative Analyses of Widely Targeted Metabolic Profiling and Transcriptome Data Reveals Molecular Insight into Metabolomic Variations during Apple (Malus domestica) Fruit Development and Ripening. Int. J. Mol. Sci. 2020, 21, 4797. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, Y.; Du, Y.; Chen, C.; Wang, D.; Zhong, Y.; Deng, Y. Integrative Metabolomic and Transcriptomic Landscape during Akebia trifoliata Fruit Ripening and Cracking. Int. J. Mol. Sci. 2023, 24, 16732. https://doi.org/10.3390/ijms242316732
Jiang Y, Du Y, Chen C, Wang D, Zhong Y, Deng Y. Integrative Metabolomic and Transcriptomic Landscape during Akebia trifoliata Fruit Ripening and Cracking. International Journal of Molecular Sciences. 2023; 24(23):16732. https://doi.org/10.3390/ijms242316732
Chicago/Turabian StyleJiang, Yongli, Yanlin Du, Chongyang Chen, Danfeng Wang, Yu Zhong, and Yun Deng. 2023. "Integrative Metabolomic and Transcriptomic Landscape during Akebia trifoliata Fruit Ripening and Cracking" International Journal of Molecular Sciences 24, no. 23: 16732. https://doi.org/10.3390/ijms242316732
APA StyleJiang, Y., Du, Y., Chen, C., Wang, D., Zhong, Y., & Deng, Y. (2023). Integrative Metabolomic and Transcriptomic Landscape during Akebia trifoliata Fruit Ripening and Cracking. International Journal of Molecular Sciences, 24(23), 16732. https://doi.org/10.3390/ijms242316732