

The Altered Functions of Shelterin Components in ALT Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
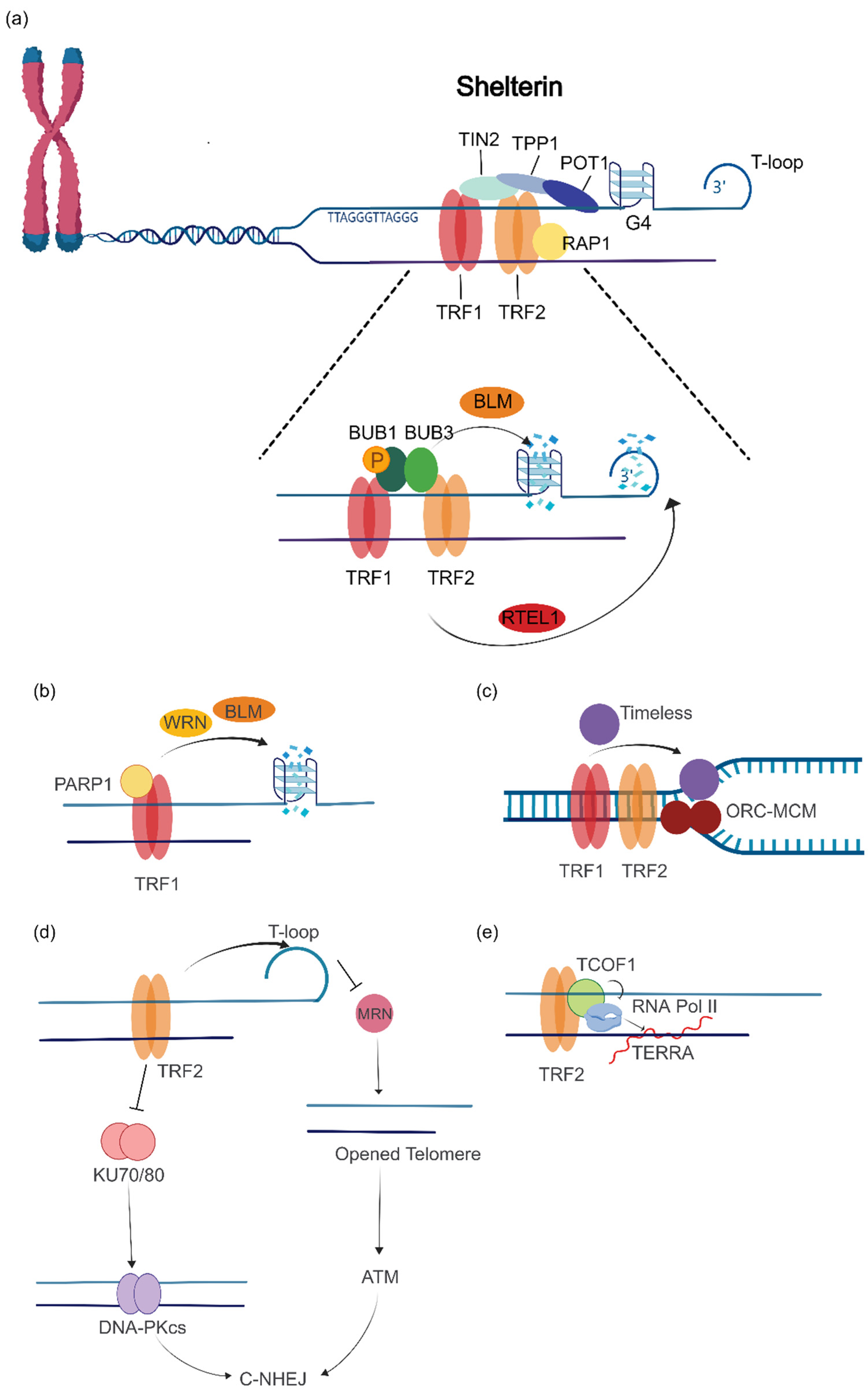
2. TRF1 and TRF2
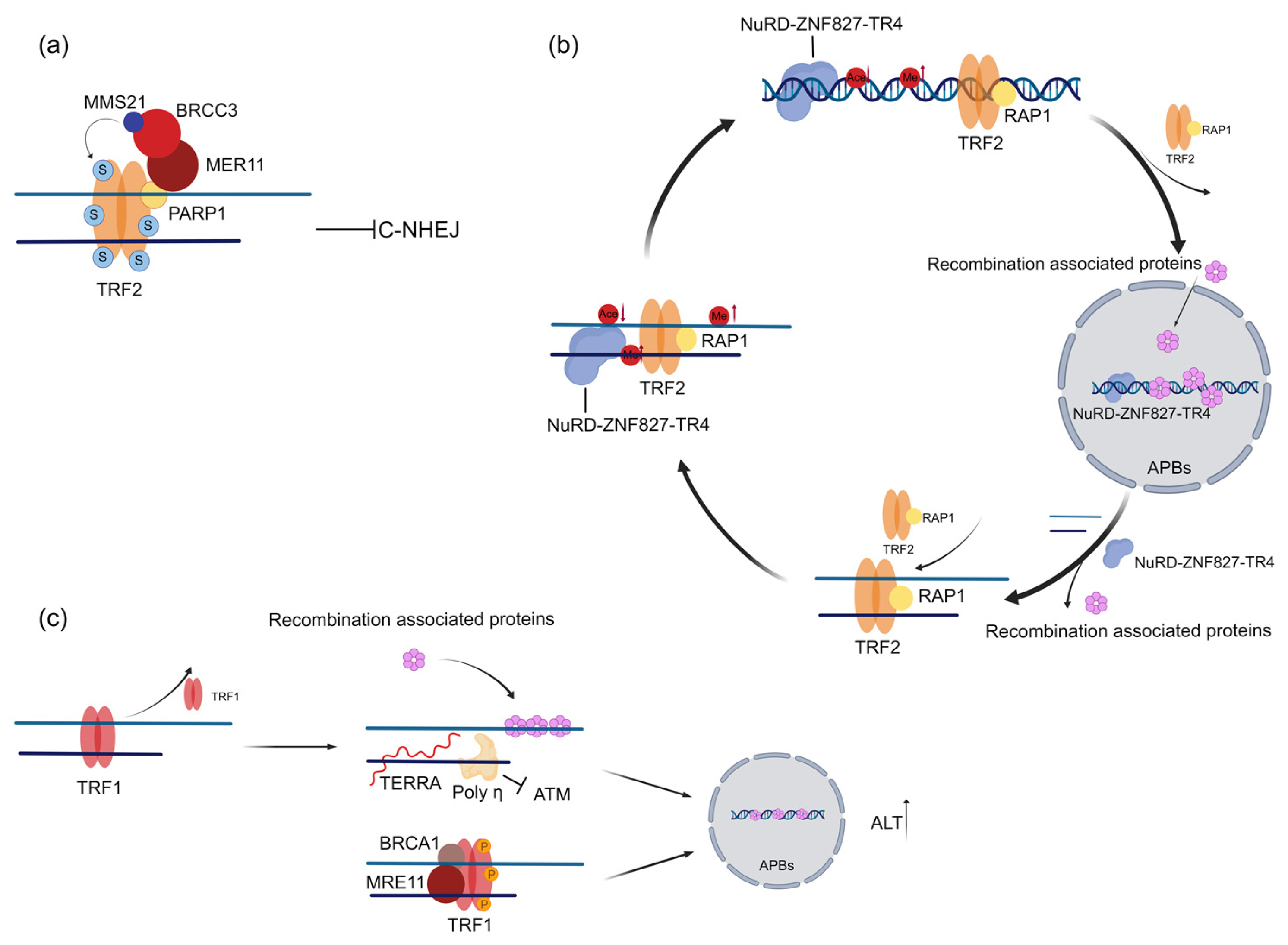
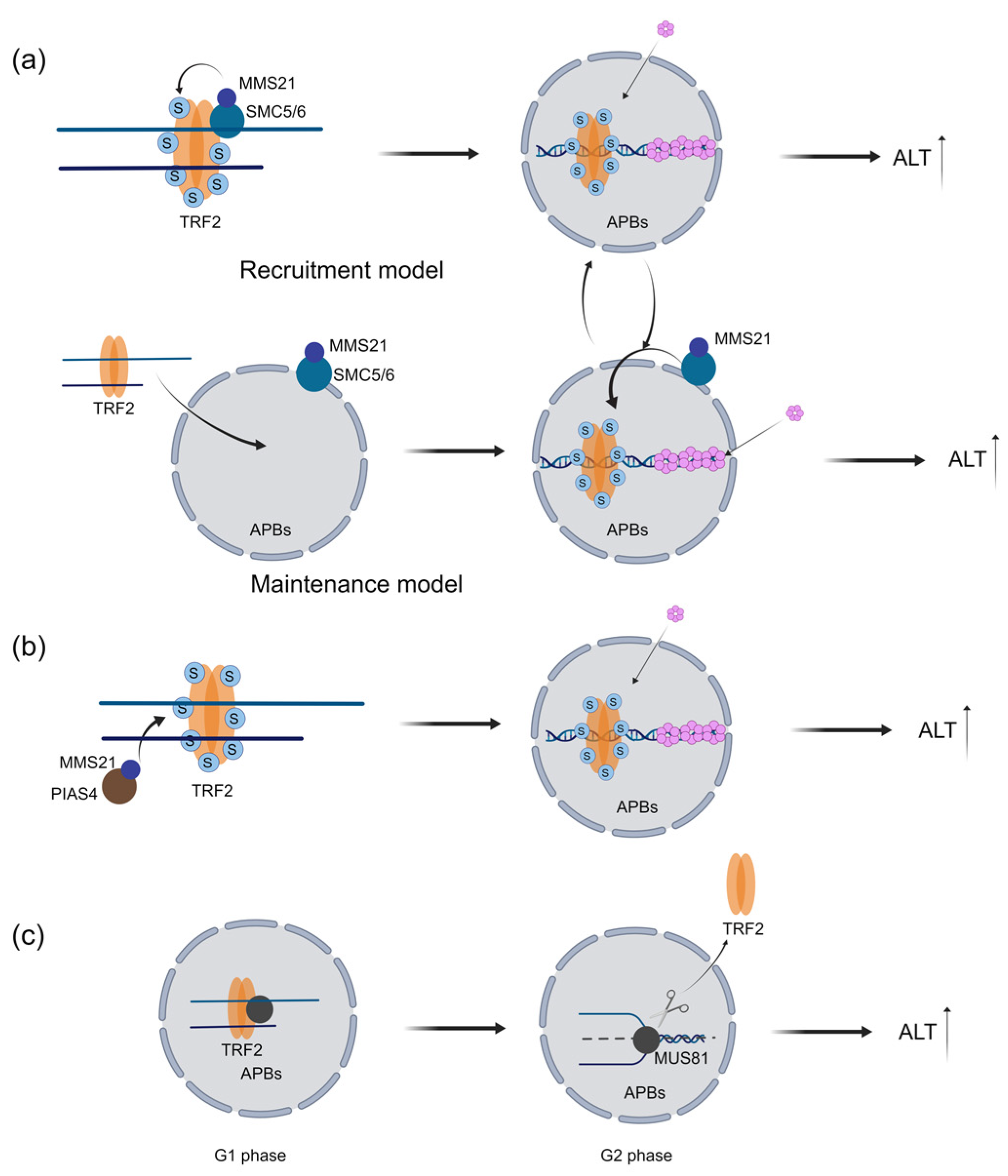
2.1. TRF1 and TRF2 Promote Telomere Maintenance in ALT Cells
2.2. TRF1 and TRF2 Promote Proper Telomere Localization in APBs and Telomere Replication in ALT Cells
3. RAP1
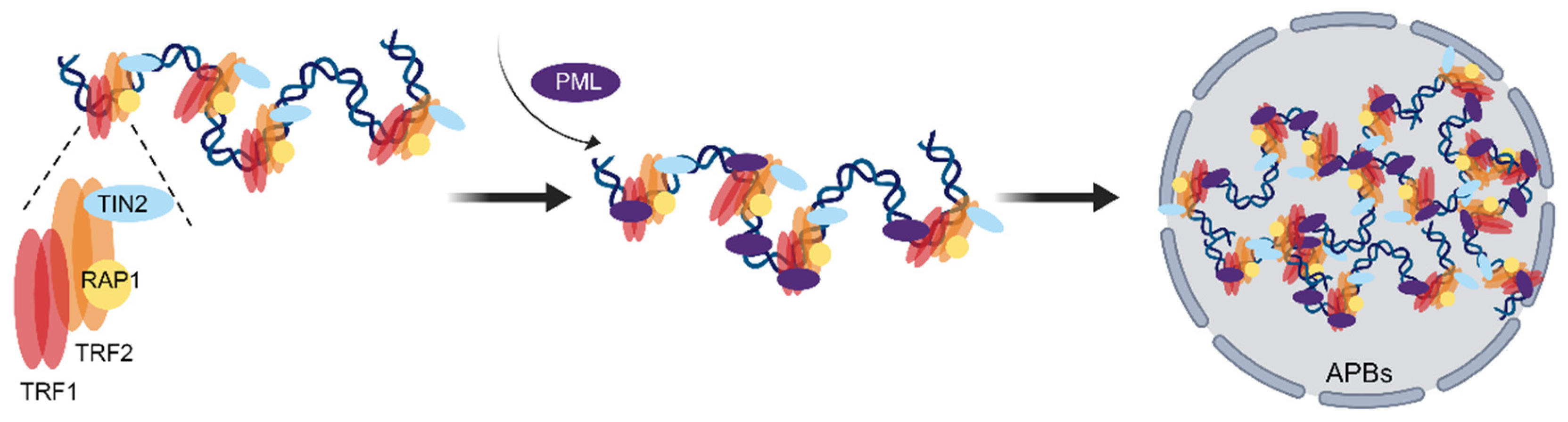
3.1. Depletion of RAP1 Accumulates Replication Stress at Telomeres and Triggers ALT
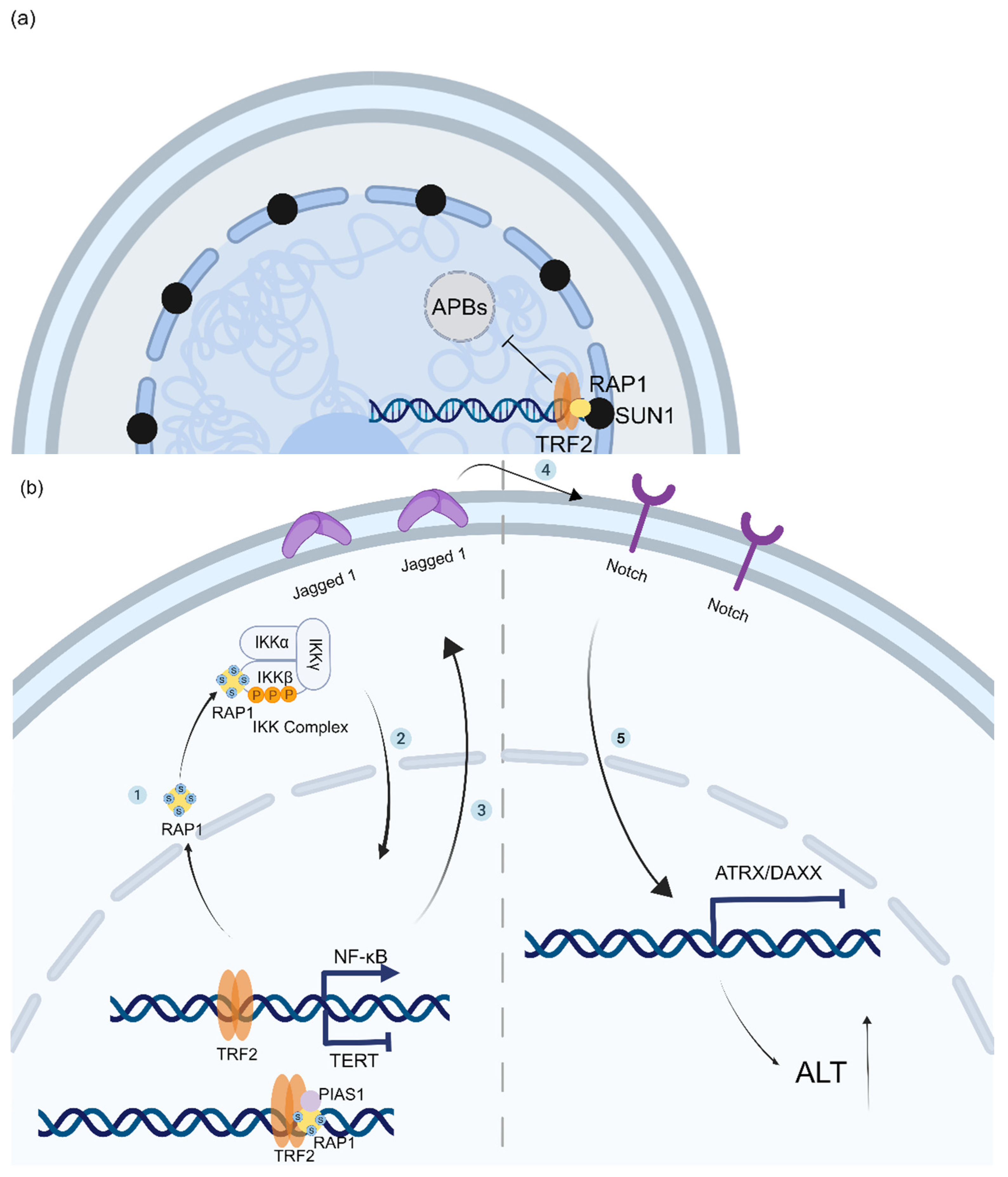
3.2. RAP1 Coordinates the Spatial Localization of Telomeres and the Epigenetic State of Telomere Chromatin to Prevent the Generation of ALT
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- De Lange, T. Shelterin-Mediated Telomere Protection. Annu. Rev. Genet. 2018, 52, 223–247. [Google Scholar] [CrossRef] [PubMed]
- Greider, C.W. Telomerase and telomere-length regulation: Lessons from small eukaryotes to mammals. Cold Spring Harb. Symp. Quant. Biol. 1993, 58, 719–723. [Google Scholar] [CrossRef] [PubMed]
- De Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef] [PubMed]
- Aramburu, T.; Plucinsky, S.; Skordalakes, E. POT1-TPP1 telomere length regulation and disease. Comput. Struct. Biotechnol. J. 2020, 18, 1939–1946. [Google Scholar] [CrossRef] [PubMed]
- Shay, J.W. Role of Telomeres and Telomerase in Aging and Cancer. Cancer Discov. 2016, 6, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Londono-Vallejo, J.A.; Der-Sarkissian, H.; Cazes, L.; Bacchetti, S.; Reddel, R.R. Alternative lengthening of telomeres is characterized by high rates of telomeric exchange. Cancer Res. 2004, 64, 2324–2327. [Google Scholar] [CrossRef] [PubMed]
- Yeager, T.R.; Neumann, A.A.; Englezou, A.; Huschtscha, L.I.; Noble, J.R.; Reddel, R.R. Telomerase-negative immortalized human cells contain a novel type of promyelocytic leukemia (PML) body. Cancer Res. 1999, 59, 4175–4179. [Google Scholar] [PubMed]
- Cesare, A.J.; Kaul, Z.; Cohen, S.B.; Napier, C.E.; Pickett, H.A.; Neumann, A.A.; Reddel, R.R. Spontaneous occurrence of telomeric DNA damage response in the absence of chromosome fusions. Nat. Struct. Mol. Biol. 2009, 16, 1244–1251. [Google Scholar] [CrossRef]
- Daley, J.M.; Niu, H.; Miller, A.S.; Sung, P. Biochemical mechanism of DSB end resection and its regulation. DNA Repair 2015, 32, 66–74. [Google Scholar] [CrossRef]
- Bugreev, D.V.; Huang, F.; Mazina, O.M.; Pezza, R.J.; Voloshin, O.N.; Camerini-Otero, R.D.; Mazin, A.V. HOP2-MND1 modulates RAD51 binding to nucleotides and DNA. Nat. Commun. 2014, 5, 4198. [Google Scholar] [CrossRef]
- Verma, P.; Dilley, R.L.; Zhang, T.; Gyparaki, M.T.; Li, Y.; Greenberg, R.A. RAD52 and SLX4 act nonepistatically to ensure telomere stability during alternative telomere lengthening. Genes. Dev. 2019, 33, 221–235. [Google Scholar] [CrossRef]
- Sobinoff, A.P.; Pickett, H.A. Alternative Lengthening of Telomeres: DNA Repair Pathways Converge. Trends Genet. TIG 2017, 33, 921–932. [Google Scholar] [CrossRef]
- Wilson, J.S.; Tejera, A.M.; Castor, D.; Toth, R.; Blasco, M.A.; Rouse, J. Localization-dependent and -independent roles of SLX4 in regulating telomeres. Cell Rep. 2013, 4, 853–860. [Google Scholar] [CrossRef]
- Zhang, J.M.; Yadav, T.; Ouyang, J.; Lan, L.; Zou, L. Alternative Lengthening of Telomeres through Two Distinct Break-Induced Replication Pathways. Cell Rep. 2019, 26, 955–968.e953. [Google Scholar] [CrossRef]
- Mason-Osann, E.; Terranova, K.; Lupo, N.; Lock, Y.J.; Carson, L.M.; Flynn, R.L. RAD54 promotes alternative lengthening of telomeres by mediating branch migration. EMBO Rep. 2020, 21, e49495. [Google Scholar] [CrossRef]
- Brouwer, A.K.; Schimmel, J.; Wiegant, J.C.; Vertegaal, A.C.; Tanke, H.J.; Dirks, R.W. Telomeric DNA mediates de novo PML body formation. Mol. Biol. Cell 2009, 20, 4804–4815. [Google Scholar] [CrossRef]
- Jiang, W.Q.; Zhong, Z.H.; Henson, J.D.; Reddel, R.R. Identification of candidate alternative lengthening of telomeres genes by methionine restriction and RNA interference. Oncogene 2007, 26, 4635–4647. [Google Scholar] [CrossRef]
- Wang, C.; Songyang, Z.; Huang, Y. TRIM28 inhibits alternative lengthening of telomere phenotypes by protecting SETDB1 from degradation. Cell Biosci. 2021, 11, 149. [Google Scholar] [CrossRef] [PubMed]
- Peuscher, M.H.; Jacobs, J.J. Posttranslational control of telomere maintenance and the telomere damage response. Cell Cycle 2012, 11, 1524–1534. [Google Scholar] [CrossRef] [PubMed]
- Osterwald, S.; Deeg, K.I.; Chung, I.; Parisotto, D.; Wörz, S.; Rohr, K.; Erfle, H.; Rippe, K. PML induces compaction, TRF2 depletion and DNA damage signaling at telomeres and promotes their alternative lengthening. J. Cell Sci. 2015, 128, 1887–1900. [Google Scholar] [CrossRef] [PubMed]
- Gu, P.L.; Jia, S.T.; Takasugi, T.; Tesmer, V.M.; Nandakumar, J.; Chen, Y.; Chang, S. Distinct functions of POT1 proteins contribute to the regulation of telomerase recruitment to telomeres. Nat. Commun. 2021, 12, 5514. [Google Scholar] [CrossRef]
- Iachettini, S.; Ciccarone, F.; Maresca, C.; D’ Angelo, C.; Petti, E.; Di Vito, S.; Ciriolo, M.R.; Zizza, P.; Biroccio, A. The telomeric protein TERF2/TRF2 impairs HMGB1-driven autophagy. Autophagy 2023, 19, 1479–1490. [Google Scholar] [CrossRef]
- Polanska, E.; Dobsakova, Z.; Dvorackova, M.; Fajkus, J.; Stros, M. HMGB1 gene knockout in mouse embryonic fibroblasts results in reduced telomerase activity and telomere dysfunction. Chromosoma 2012, 121, 419–431. [Google Scholar] [CrossRef]
- Li, F.; Kim, H.; Ji, Z.; Zhang, T.; Chen, B.; Ge, Y.; Hu, Y.; Feng, X.; Han, X.; Xu, H.; et al. The BUB3-BUB1 Complex Promotes Telomere DNA Replication. Mol. Cell 2018, 70, 395–407.e394. [Google Scholar] [CrossRef]
- Sarek, G.; Vannier, J.B.; Panier, S.; Petrini, J.H.J.; Boulton, S.J. TRF2 recruits RTEL1 to telomeres in S phase to promote t-loop unwinding. Mol. Cell 2015, 57, 622–635. [Google Scholar] [CrossRef]
- Sfeir, A.; Kosiyatrakul, S.T.; Hockemeyer, D.; MacRae, S.L.; Karlseder, J.; Schildkraut, C.L.; de Lange, T. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell 2009, 138, 90–103. [Google Scholar] [CrossRef]
- Maresca, C.; Dello Stritto, A.; D’Angelo, C.; Petti, E.; Rizzo, A.; Vertecchi, E.; Berardinelli, F.; Bonanni, L.; Sgura, A.; Antoccia, A.; et al. PARP1 allows proper telomere replication through TRF1 poly (ADP-ribosyl)ation and helicase recruitment. Commun. Biol. 2023, 6, 234. [Google Scholar] [CrossRef]
- Leman, A.R.; Dheekollu, J.; Deng, Z.; Lee, S.W.; Das, M.M.; Lieberman, P.M.; Noguchi, E. Timeless preserves telomere length by promoting efficient DNA replication through human telomeres. Cell Cycle 2012, 11, 2337–2347. [Google Scholar] [CrossRef] [PubMed]
- Higa, M.; Matsuda, Y.; Fujii, J.; Sugimoto, N.; Yoshida, K.; Fujita, M. TRF2-mediated ORC recruitment underlies telomere stability upon DNA replication stress. Nucleic Acids Res. 2021, 49, 12234–12251. [Google Scholar] [CrossRef]
- Okamoto, K.; Bartocci, C.; Ouzounov, I.; Diedrich, J.K.; Yates, J.R., 3rd; Denchi, E.L. A two-step mechanism for TRF2-mediated chromosome-end protection. Nature 2013, 494, 502–505. [Google Scholar] [CrossRef] [PubMed]
- Lazzerini-Denchi, E.; Sfeir, A. Stop pulling my strings—What telomeres taught us about the DNA damage response. Nat. Rev. Mol. Cell Biol. 2016, 17, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Griffith, J.D.; Comeau, L.; Rosenfield, S.; Stansel, R.M.; Bianchi, A.; Moss, H.; de Lange, T. Mammalian telomeres end in a large duplex loop. Cell 1999, 97, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Stracker, T.H.; Petrini, J.H. The MRE11 complex: Starting from the ends. Nat. Rev. Mol. Cell Biol. 2011, 12, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Ribes-Zamora, A.; Indiviglio, S.M.; Mihalek, I.; Williams, C.L.; Bertuch, A.A. TRF2 interaction with Ku heterotetramerization interface gives insight into c-NHEJ prevention at human telomeres. Cell Rep. 2013, 5, 194–206. [Google Scholar] [CrossRef]
- Nie, X.; Xiao, D.; Ge, Y.; Xie, Y.; Zhou, H.; Zheng, T.; Li, X.; Liu, H.; Huang, H.; Zhao, Y. TRF2 recruits nucleolar protein TCOF1 to coordinate telomere transcription and replication. Cell Death Differ. 2021, 28, 1062–1075. [Google Scholar] [CrossRef]
- Stagno D’Alcontres, M.; Mendez-Bermudez, A.; Foxon, J.L.; Royle, N.J.; Salomoni, P. Lack of TRF2 in ALT cells causes PML-dependent p53 activation and loss of telomeric DNA. J. Cell Biol. 2007, 179, 855–867. [Google Scholar] [CrossRef] [PubMed]
- Celli, G.B.; de Lange, T. DNA processing is not required for ATM-mediated telomere damage response after TRF2 deletion. Nat. Cell Biol. 2005, 7, 712–718. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, J.; Pandita, A.; Kamalakar, C.; Johannessen, T.C.; Ohba, S.; Tang, Y.; Dalle-Ore, C.L.; Bjerkvig, R.; Pieper, R.O. RETRACTED: A subset of PARP inhibitors induces lethal telomere fusion in ALT-dependent tumor cells. Sci. Transl. Med. 2021, 13, eabc7211. [Google Scholar] [CrossRef]
- Willis, N.A.; Panday, A.; Duffey, E.E.; Scully, R. Rad51 recruitment and exclusion of non-homologous end joining during homologous recombination at a Tus/Ter mammalian replication fork barrier. PLoS Genet. 2018, 14, e1007486. [Google Scholar] [CrossRef]
- Udroiu, I.; Sgura, A. Alternative Lengthening of Telomeres and Chromatin Status. Genes 2019, 11, 45. [Google Scholar] [CrossRef] [PubMed]
- Conomos, D.; Reddel, R.R.; Pickett, H.A. NuRD-ZNF827 recruitment to telomeres creates a molecular scaffold for homologous recombination. Nat. Struct. Mol. Biol. 2014, 21, 760–770. [Google Scholar] [CrossRef] [PubMed]
- Al-Turki, T.M.; Griffith, J.D. Mammalian telomeric RNA (TERRA) can be translated to produce valine-arginine and glycine-leucine dipeptide repeat proteins. Proc. Natl. Acad. Sci. USA 2023, 120, e2221529120. [Google Scholar] [CrossRef]
- Draskovic, I.; Arnoult, N.; Steiner, V.; Bacchetti, S.; Lomonte, P.; Londoño-Vallejo, A. Probing PML body function in ALT cells reveals spatiotemporal requirements for telomere recombination. Proc. Natl. Acad. Sci. USA 2009, 106, 15726–15731. [Google Scholar] [CrossRef] [PubMed]
- Timashev, L.A.; Babcock, H.; Zhuang, X.; de Lange, T. The DDR at telomeres lacking intact shelterin does not require substantial chromatin decompaction. Genes Dev. 2017, 31, 578–589. [Google Scholar] [CrossRef] [PubMed]
- Potts, P.R.; Yu, H. The SMC5/6 complex maintains telomere length in ALT cancer cells through SUMOylation of telomere-binding proteins. Nat. Struct. Mol. Biol. 2007, 14, 581–590. [Google Scholar] [CrossRef]
- Zhang, J.M.; Genois, M.M.; Ouyang, J.; Lan, L.; Zou, L. Alternative lengthening of telomeres is a self-perpetuating process in ALT-associated PML bodies. Mol. Cell 2021, 81, 1027–1042.e1024. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.; Yang, Q. The MUS81 endonuclease is essential for telomerase negative cell proliferation. Cell Cycle 2009, 8, 2157–2160. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.; Xiang, T.; Pandita, T.K.; Gonzalez-Suarez, I.; Gonzalo, S.; Harris, C.C.; Yang, Q. Telomere recombination requires the MUS81 endonuclease. Nat. Cell Biol. 2009, 11, 616–623. [Google Scholar] [CrossRef]
- Deregowska, A.; Wnuk, M. RAP1/TERF2IP-A Multifunctional Player in Cancer Development. Cancers 2021, 13, 5970. [Google Scholar] [CrossRef] [PubMed]
- Sfeir, A.; Kabir, S.; van Overbeek, M.; Celli, G.B.; de Lange, T. Loss of Rap1 induces telomere recombination in the absence of NHEJ or a DNA damage signal. Science 2010, 327, 1657–1661. [Google Scholar] [CrossRef]
- Silva, B.; Arora, R.; Bione, S.; Azzalin, C.M. TERRA transcription destabilizes telomere integrity to initiate break-induced replication in human ALT cells. Nat. Commun. 2021, 12, 3760. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Bennett, H.W.; Liu, N.; Moravec, M.; Williams, J.F.; Azzalin, C.M.; King, M.C. RNA-DNA Hybrids Support Recombination-Based Telomere Maintenance in Fission Yeast. Genetics 2019, 213, 431–447. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, Z.; Liu, X.; Wang, S.; Zhang, Y.; He, X.; Sun, S.; Ma, S.; Shyh-Chang, N.; Liu, F.; et al. Telomere-dependent and telomere-independent roles of RAP1 in regulating human stem cell homeostasis. Protein Cell 2019, 10, 649–667. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, J.; Johannessen, T.C.; Ohba, S.; Chow, T.T.; Jones, L.; Pandita, A.; Pieper, R.O. Mutant IDH1 Cooperates with ATRX Loss to Drive the Alternative Lengthening of Telomere Phenotype in Glioma. Cancer Res. 2018, 78, 2966–2977. [Google Scholar] [CrossRef]
- Arora, R.; Lee, Y.; Wischnewski, H.; Brun, C.M.; Schwarz, T.; Azzalin, C.M. RNaseH1 regulates TERRA-telomeric DNA hybrids and telomere maintenance in ALT tumour cells. Nat. Commun. 2014, 5, 5220. [Google Scholar] [CrossRef] [PubMed]
- Miné-Hattab, J.; Chiolo, I. Complex Chromatin Motions for DNA Repair. Front. Genet. 2020, 11, 800. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Ge, T.; Jian, M.; Chen, L.; Fang, Z.; He, Z.; Huang, C.; An, Y.; Yin, S.; Xiong, Y.; et al. Transmembrane nuclease NUMEN/ENDOD1 regulates DNA repair pathway choice at the nuclear periphery. Nat. Cell Biol. 2023, 25, 1004–1016. [Google Scholar] [CrossRef]
- Yang, C.W.; Hsieh, M.H.; Sun, H.J.; Teng, S.C. Nuclear envelope tethering inhibits the formation of ALT-associated PML bodies in ALT cells. Aging 2021, 13, 10490–10516. [Google Scholar] [CrossRef]
- Lottersberger, F.; Karssemeijer, R.A.; Dimitrova, N.; de Lange, T. 53BP1 and the LINC Complex Promote Microtubule-Dependent DSB Mobility and DNA Repair. Cell 2015, 163, 880–893. [Google Scholar] [CrossRef]
- Robinson, N.J.; Miyagi, M.; Scarborough, J.A.; Scott, J.G.; Taylor, D.J.; Schiemann, W.P. SLX4IP promotes RAP1 SUMOylation by PIAS1 to coordinate telomere maintenance through NF-κB and Notch signaling. Sci. Signal 2021, 14, eabe9613. [Google Scholar] [CrossRef]
- Chen, C.; Gu, P.; Wu, J.; Chen, X.; Niu, S.; Sun, H.; Wu, L.; Li, N.; Peng, J.; Shi, S.; et al. Structural insights into POT1-TPP1 interaction and POT1 C-terminal mutations in human cancer. Nat. Commun. 2017, 8, 14929. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, H.C.; Towbin, B.D.; Jegou, T.; Gasser, S.M. The shelterin protein POT-1 anchors Caenorhabditis elegans telomeres through SUN-1 at the nuclear periphery. J. Cell Biol. 2013, 203, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Manzato, C.; Larini, L.; Pegorar, C.O.; Dello Stritto, M.R.; Jurikova, K.; Jantsch, V.; Cusanelli, E. TERRA expression is regulated by the telomere-binding proteins POT-1 and POT-2 in Caenorhabditis elegans. Nucleic Acids Res. 2023, 51, 10681–10699. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Shi, G.; Zhang, L.; Li, F.; Jiang, Y.; Jiang, S.; Ma, W.; Zhao, Y.; Songyang, Z.; Huang, J. Switch telomerase to ALT mechanism by inducing telomeric DNA damages and dysfunction of ATRX and DAXX. Sci. Rep. 2016, 6, 32280. [Google Scholar] [CrossRef] [PubMed]
- Qiang, W.; Wu, Q.; Zhou, F.; Xie, C.; Wu, C.; Zhou, Y. Suppression of telomere-binding protein TPP1 resulted in telomere dysfunction and enhanced radiation sensitivity in telomerase-negative osteosarcoma cell line. Biochem. Biophys. Res. Commun. 2014, 445, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.; Zhang, F.; Lin, S.; Chen, W.; Weng, K.; Liu, D.; Wang, C.; He, Z.; Chen, Y.; Ma, W.; et al. TIN2 deficiency leads to ALT-associated phenotypes and differentiation defects in embryonic stem cells. Stem Cell Rep. 2022, 17, 1183–1197. [Google Scholar] [CrossRef]
- Storchova, R.; Palek, M.; Palkova, N.; Veverka, P.; Brom, T.; Hofr, C.; Macurek, L. Phosphorylation of TRF2 promotes its interaction with TIN2 and regulates DNA damage response at telomeres. Nucleic Acids Res. 2023, 51, 1154–1172. [Google Scholar] [CrossRef]
- Xu, M.; Qin, J.; Wang, L.; Lee, H.J.; Kao, C.Y.; Liu, D.; Songyang, Z.; Chen, J.; Tsai, M.J.; Tsai, S.Y. Nuclear receptors regulate alternative lengthening of telomeres through a novel noncanonical FANCD2 pathway. Sci. Adv. 2019, 5, eaax6366. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Hou, K.; Tong, J.; Zhang, H.; Xiong, M.; Liu, J.; Jia, S. The Altered Functions of Shelterin Components in ALT Cells. Int. J. Mol. Sci. 2023, 24, 16830. https://doi.org/10.3390/ijms242316830
Zhang Y, Hou K, Tong J, Zhang H, Xiong M, Liu J, Jia S. The Altered Functions of Shelterin Components in ALT Cells. International Journal of Molecular Sciences. 2023; 24(23):16830. https://doi.org/10.3390/ijms242316830
Chicago/Turabian StyleZhang, Yanduo, Kailong Hou, Jinkai Tong, Haonan Zhang, Mengjie Xiong, Jing Liu, and Shuting Jia. 2023. "The Altered Functions of Shelterin Components in ALT Cells" International Journal of Molecular Sciences 24, no. 23: 16830. https://doi.org/10.3390/ijms242316830
APA StyleZhang, Y., Hou, K., Tong, J., Zhang, H., Xiong, M., Liu, J., & Jia, S. (2023). The Altered Functions of Shelterin Components in ALT Cells. International Journal of Molecular Sciences, 24(23), 16830. https://doi.org/10.3390/ijms242316830