
Next Generation Therapeutic Strategy for Treatment and Prevention of Alzheimer’s Disease and Aging-Associated Cognitive Decline: Transient, Once-in-a-Lifetime-Only Depletion of Intraneuronal Aβ (iAβ) by Its Targeted Degradation via Augmentation of Intra-iAβ-Cleaving Activities of BACE1 and/or BACE2

Abstract
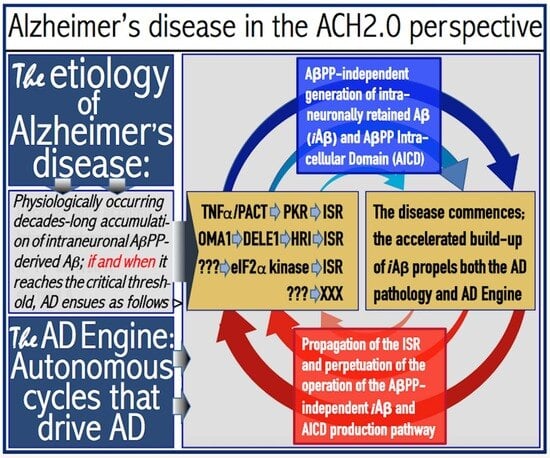
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction: From the ACH to the ACH2.0
1.1. Amyloid Cascade Hypothesis: Extracellular Aβ Causes and Drives AD
1.2. Inconsistencies of the ACH: Extracellular Aβ Can Be Ruled out as the Causative Agent of AD
1.3. Centrality and Causative Role of Aβ Are the Cornerstone of Any Theory of AD: Intraneuronal Aβ, iAβ
1.4. Amyloid Cascade Hypothesis 2.0: The Essence
1.5. The AβPP-Independent Aβ Generation Pathway: The Active Core of AD
2. The Engine That Drives AD
2.1. Life-Long Accumulation of AβPP-Derived iAβ at the First AD Stage: Mechanistic Aspects
2.2. AβPP-Derived iAβ Accumulation Triggers AD but Is a Normal Physiological Process: At the First AD Stage the Difference between Health and Disease Is Quantitative, Not Qualitative
2.3. iAβ-Mediated Activation of the PKR and HRI Kinases Elicits the Integrated Stress Response (ISR) and Triggers Operation of the AβPP-Independent iAβ Production Pathway: Transition to the Second AD Stage
2.4. AβPP-Independent iAβ Production (and Retention) Pathway: Potential Underlying Mechanisms
2.5. Collateral Effect: AβPP-Independent iAβ Production Pathway also Generates AβPP Intracellular Domain, AICD
2.6. The AD Engine: Continuous Cycles of the iAβ-Stimulated Propagation of Its Own AβPP-Independent Production
3. Dynamics of AβPP-Derived iAβ Accumulation in Health and Disease: Alzheimer’s Disorder and Aging-Associated Cognitive Decline and the Conditionality of the “First Stage of AD”
3.1. Dynamics of AβPP-Derived iAβ Accumulation in Health and Disease: Alzheimer’s Disease
3.2. Conditionality of the “First Stage of AD”
3.3. Dynamics of AβPP-Derived iAβ Accumulation in Health and Disease: Aging-Associated Cognitive Decline
4. ACH-Based AD Drugs: A Definition
5. In the ACH2.0 Paradigm, Conceptually, ACH-Based Drugs Cannot Be Effective at Symptomatic Stages of AD
6. In the ACH2.0 Paradigm, Conceptually, ACH-Based Drugs Can Be Effective in Prevention of AD
6.1. Long-Term Treatment with ACH-Based Drugs: Two Possible Modes of Action
6.2. Inefficiency of Short-Duration Treatment with ACH-Based AD Drugs
7. Effect of Lecanemab and Donanemab in Early AD: A Dual Proof of Concepts for Inefficiency of ACH-Based Drugs in Symptomatic AD and for Their Applicability in Prevention of AD
7.1. Effect of Lecanemab and Donanemab in Early AD: Mechanistic Interpretation in the ACH2.0 Perspective
7.2. Outcomes of Lecanemab and Donanemab Clinical Trials Constitute Dual Proof of Concepts: ACH-Based Drugs Are Inefficient in Symptomatic AD but Can Be Effective Preventively
8. ACH-Based AD Drugs: Complications with the Long-Term Treatment for Prevention of AD
8.1. Long-Term Treatment with Current ACH-Based Drugs Is Inconceivable in Low-Risk Individuals
8.2. Long-Term ACH-Based Drugs Treatment for Prevention of Sporadic AD Would Be Inefficient in High-Risk Individuals
9. In the ACH2.0 Paradigm, ACH-Based Drugs Should Be Effective in Treatment of AACD
10. ACH-Based Drugs Were Spectacularly Successful in Transgenic AD Models for the Same Reason They Would Be in AACD: Mechanistic Interpretation in the ACH2.0 Perspective
11. ACH-Based Drugs in Treatment of Different Stages of AACD
12. Symptoms of AACD-Related Cognitive Impairment Overlap with and Are Indistinguishable from Those of AD-Associated MCI: Ramifications for ACH-Based Drugs Therapy
13. Clinical Trials of ACH-Based Drugs in Prevention of AD with All Participants at Sub-T1 Levels of iAβ: Apparently Straightforward but Impractical
14. Clinical Trials of ACH-Based Drugs in Prevention of AD with Participants Representing a Cross-Section of the General Population Aged 65 and Over: Results Could Be Grossly Misleading
14.1. The Overall Picture
14.2. Detailed Analysis: Duration of Trials Could Be Prohibitively Excessive Prior to the Divergence of the Outcomes in Medicated and Placebo Groups
15. Clinical Trials of ACH-Based Drugs in Prevention of AD with High-Risk Participants: Practically Impossible and Self-Defeating
16. Clinical Trials of ACH-Based Drugs in Treatment of AACD
16.1. Clinical Trials of ACH-Based Drugs in Treatment of AACD with Subjects including Late-Stage Patients: Potential Complications
16.2. Clinical Trials of ACH-Based Drugs in Treatment of Early AACD: The Only Unproblematic Clinical Trials of ACH-Based Drugs
17. The ACH2.0-Guided Next Generation Therapeutic Strategy for AD and AACD: The Depletion of iAβ via Its Targeted Degradation
17.1. Effective AD Therapy Requires Both the Reduction of the Levels of iAβ and Cessation of the Operation of the AβPP-Independent Pathway of Its Production
17.2. A Sufficient Depletion of iAβ Would Cease the Activity of the AβPP-Independent iAβ Production Pathway
17.3. Inhibition of the ISR Is Not a Viable Option for AD
In a Fraction of AD Cases, the Effective Therapy May Require Anti-Inflammatory Treatment in Addition to the Depletion of iAβ
18. ACH2.0-Based Drugs for Prevention and Treatment of AD and AACD: A Definition
19. Implementation of ACH2.0-Based Drugs in Prevention of AD by Depletion of iAβ via Its Transient, Short-Duration, Targeted Degradation
19.1. Prevention of AD by Transient Depletion of iAβ via Its Targeted Degradation in Asymptomatic Low-Risk Individuals with the Entire Neuronal Population Sub-T1
19.2. Prevention of AD by Transient Depletion of iAβ via Its Targeted Degradation in Asymptomatic High-Risk Individuals with Over-T1 Neuronal Subpopulation
20. Implementation of ACH2.0-Based Drugs in Prevention and Treatment of AACD
20.1. Transient iAβ Depletion Treatment in Prevention of AACD
20.2. Transient iAβ Depletion Treatment in Early AACD

20.3. Transient iAβ Depletion Treatment in Late AACD
21. The Proverbial “Holy Grail” of the Alzheimer’s Research Attained: ACH2.0-Based Drugs Are Capable of Treatment of AD at Its Symptomatic Stages
22. Clinical Trials of ACH2.0-Based Drugs in Prevention of AD: Unambiguous Evaluation Even with High-Risk Over-T1 Participants
23. Clinical Trials of ACH2.0-Based Drugs in Treatment of AACD: No Complications Even with Late-Stage Participants
24. Clinical Trials of ACH2.0-Based Drugs in Treatment of Symptomatic AD: Lucidly Interpretable Outcomes at All Stages of the Disease
25. The Uniform Mechanistic Effect of the ACH2.0-Based Drugs Elicits Distinctly Stage-Specific Outcomes
26. Activators of Physiologically Occurring Intra-iAβ-Cleaving Capabilities of BACE1 and/or BACE2: Highly Plausible ACH2.0-Based Drugs for Prevention and Treatment of AD and AACD
27. Conclusions
Author Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Volloch, V.; Rits-Volloch, S. The Amyloid Cascade Hypothesis 2.0: On the possibility of once-in-a-lifetime-only treatment for prevention of Alzheimer’s disease and for its potential cure at symptomatic stages. J. Alz Dis. Rep. 2022, 6, 369–399. [Google Scholar] [CrossRef]
- Volloch, V.; Rits-Volloch, S. The Amyloid Cascade Hypothesis 2.0: Generalization of the Concept. J. Alz Dis. Rep. 2023, 7, 21–35. [Google Scholar] [CrossRef]
- Volloch, V.; Rits-Volloch, S. Effect of Lecanemab and Donanemab in Early Alzheimer’s Disease: Mechanistic Interpretation in the Amyloid Cascade Hypothesis 2.0 Perspective. J. Alz Dis. 2023, 93, 1277–1284. [Google Scholar] [CrossRef]
- Volloch, V.; Rits-Volloch, S. The Amyloid Cascade Hypothesis 2.0 for Alzheimer’s Disease and Aging-Associated Cognitive Decline: From Molecular Basis to Effective Therapy. Int. J. Mol. Sci. 2023, 24, 12246. [Google Scholar] [CrossRef]
- Volloch, V.; Rits-Volloch, S. Principles of Design of Clinical Trials for Prevention and Treatment of Alzheimer’s Disease and Aging-Associated Cognitive Decline in the ACH2.0 Perspective: Potential Outcomes, Challenges and Solutions. J. Alz Dis. Rep. 2023, 7, 921–955. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Goate, A.; Chartier-Harlin, M.C.; Mullan, M.; Brown, J.; Crawford, F.; Fidani, L.; Giuffra, L.; Haynes, A.; Irving, N.; James, L.; et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 1991, 349, 704–706. [Google Scholar] [CrossRef]
- Keskin, A.D.; Kekuš, M.; Adelsberger, H.; Neumann, U.; Shimshek, D.R.; Song, B.; Zott, B.; Peng, T.; Förstl, H.; Staufenbiel, M.; et al. BACE inhibition-dependent repair of Alzheimer’s pathophysiology. Proc. Natl. Acad. Sci. USA 2017, 114, 8631–8636. [Google Scholar] [CrossRef]
- Hu, X.; Das, B.; Hou, H.; He, W.; Yan, R. BACE1 deletion in the adult mouse reverses preformed amyloid deposition and improves cognitive functions. J. Exp. Med. 2018, 10, 1084. [Google Scholar] [CrossRef]
- Kennedy, M.E.; Stamford, A.W.; Chen, X.; Cox, K.; Cumming, J.N.; Dockendorf, M.F.; Egan, M.; Ereshefsky, L.; Hodgson, R.A.; Hyde, L.A.; et al. The BACE1 inhibitor verubecestat (MK-8931) reduces CNS β-amyloid in animal models and in Alzheimer’s disease patients. Sci. Transl. Med. 2016, 8, 363ra150. [Google Scholar] [CrossRef]
- Egan, M.F.; Kost, J.; Tariot, P.N.; Aisen, P.S.; Cummings, J.L.; Vellas, B.; Sur, C.; Mukai, Y.; Voss, T.; Furtek, C.; et al. Randomized trials of verubecestat for mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 2018, 378, 1691–1703. [Google Scholar] [CrossRef]
- Egan, M.F.; Kost, J.; Voss, T.; Mukai, Y.; Aisen, P.S.; Cummings, J.L.; Tariot, P.N.; Vellas, B.; van Dyck, C.H.; Boada, M.; et al. Randomized trial of verubecestat for prodromal Alzheimer’s disease. N. Engl. J. Med. 2019, 380, 1408–1420. [Google Scholar] [CrossRef]
- Katzman, R.; Terry, R.; DeTeresa, R.; Brown, T.; Davies, P.; Fuld, P.; Rebing, X.; Peck, A. Clinical, pathological, and neurochemical changes in dementia: A subgroup with preserved mental status and numerous neocortical plaques. Ann. Neurol. 1988, 23, 138–144. [Google Scholar] [CrossRef]
- Delaere, P.; Duyckaerts, C.; Masters, C.; Beyreuther, K.; Piette, F.; Hauw, J. Large amounts of neocortical beta A4 deposits without neuritic plaques nor tangles in a psychometrically assessed, non-demented person. Neurosci. Lett. 1990, 116, 87–93. [Google Scholar] [CrossRef]
- Dickson, D.; Crystal, H.; Mattiace, L.; Masur, D.; Blau, A.; Davies, P.; Yen, S.; Aronson, M. Identification of normal and pathological aging in prospectively studied nondemented elderly humans. Neurobiol. Aging 1992, 13, 178–189. [Google Scholar] [CrossRef]
- Aizenstein, H.; Nebes, R.; Saxton, J.; Price, J.; Mathis, C.; Tsopelas, N.; Ziolko, S.; James, J.; Snitz, B.; Houck, P.; et al. Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch. Neurol. 2008, 65, 1509–1517. [Google Scholar] [CrossRef]
- Klunk, W.; Mathis, C.; Price, J.; DeKosky, S.; Lopresti, B.; Tsopelas, N.; Saxton, J.; Nebes, R. Amyloid imaging with PET in Alzheimer’s disease, mild cognitive impairment, and clinically unimpaired subjects. In PET in the Evaluation of Alzheimer’s Disease and Related Disorders; Silverman, D., Ed.; Springer: New York, NY, USA, 2009; pp. 119–147. [Google Scholar]
- Villemagne, V.; Pike, K.; Chételat, G.; Ellis, K.; Mulligan, R.; Bourgeat, P.; Ackermann, U.; Jones, G.; Szoeke, C.; Salvado, O.; et al. Longitudinal assessment of Abeta and cognition in aging and Alzheimer disease. Ann. Neurol. 2011, 69, 181–192. [Google Scholar] [CrossRef]
- Seto, M.; Weiner, R.L.; Dumitrescu, L.; Hohman, T.J. Protective genes and pathways in Alzheimer’s disease: Moving towards precision interventions. Mol. Neurodegen 2021, 16, 29. [Google Scholar] [CrossRef]
- Makin, S. The amyloid hypothesis on trial. Nature 2018, 559, S4–S7. Available online: https://media.nature.com/original/magazine-assets/d41586-018-05719-4/d41586-018-05719-4.pdf (accessed on 1 March 2021). [CrossRef]
- Jonsson, T.; Atwal, J.K.; Steinberg, S.; Snaedal, J.; Jonsson, P.V.; Bjornsson, S.; Stefansson, H.; Sulem, P.; Gudbjartsson, D.; Maloney, J.; et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 2012, 488, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Harper, A.R.; Nayee, S.; Topol, E.J. Protective alleles and modifier variants in human health and disease. Nat. Rev. Genet. 2015, 16, 689–701. [Google Scholar] [CrossRef]
- Kinoshita, A.; Fukumoto, H.; Shah, T.; Whelan, C.M.; Irizarry, M.C.; Hyman, B.T. Demonstration by FRET of BACE interaction with the amyloid precursor protein at the cell surface and in early endosomes. J. Cell Sci. 2003, 116, 3339–3346. [Google Scholar] [CrossRef]
- Xu, H.; Greengard, P.; Gandy, S. Regulated formation of Golgi secretory vesicles containing Alzheimer β-amyloid precursor protein. J. Biol. Chem. 1995, 270, 23243–23245. [Google Scholar] [CrossRef] [PubMed]
- Mizuguchi, M.; Ikeda, K.; Kim, S. Differential distribution of cellular forms of β-amyloid precursor protein in murine glial cell cultures. Brain Res. 1992, 584, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Cook, D.G.; Forman, M.S.; Sung, J.C.; Leight, S.; Kolson, D.L.; Iwatsubo, T.; Lee, V.M.; Doms, R.W. Alzheimer’s Aβ-42 is generated in the endoplasmic reticulum/intermediate compartment of NT2N cells. Nature Med. 1997, 3, 1021–1023. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, T.; Bieger, S.C.; Brühl, B.; Tienari, P.J.; Ida, N.; Allsop, D.; Roberts, G.W.; Masters, C.L.; Dotti, C.G.; Unsicker, K.; et al. Distinct sites of intracellular production for Alzheimer’s disease Aβ-40/42 amyloid peptides. Nature Med. 1997, 3, 1016–1020. [Google Scholar] [CrossRef] [PubMed]
- Wild-Bode, C.; Yamazaki, T.; Capell, A.; Leimer, U.; Steiner, H.; Ihara, Y.; Haass, C. Intracellular generation and accumulation of amyloid beta-peptide terminating at amino acid 42. J. Biol. Chem. 1997, 272, 16085–16088. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Liyanage, U.; Bickel, P.E.; Xia, W.; Lansbury, P.T., Jr.; Kosik, K.S. A detergent-insoluble membrane compartment contains Aβ in vivo. Nature Med. 1998, 4, 730–734. [Google Scholar] [CrossRef]
- Skovronsky, D.; Doms, R.; Lee, V. Detection of a novel intraneuronal pool of insoluble amyloid β protein that accumulates with time in culture. J. Cell Biol. 1998, 141, 1031–1039. [Google Scholar] [CrossRef]
- Manczak, M.; Anekonda, T.S.; Henson, E.; Park, B.S.; Quinn, J.; Reddy, P.H. Mitochondria are a direct site of Aβ accumulation in Alzheimer’s disease neurons: Implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet 2006, 15, 1437–1449. [Google Scholar] [CrossRef]
- Chafekar, S.; Baas, F.; Scheper, W. Oligomer-specific amyloid-beta toxicity in cell models is mediated by selective uptake. Biochem. Biophys. Acta 2008, 9, 523–531. [Google Scholar]
- Wesen, E.; Jeffries, G.; Dzebo, M.; Esbjorner, M. Endocytic uptake of monomeric amyloid-β peptides is clathrin- and dynamin-independent and results in selective accumulation of Aβ(1–42) compared to Aβ(1–40). Sci. Rep. 2017, 7, 2021. [Google Scholar] [CrossRef] [PubMed]
- Kumar-Singh, S.; Theuns, J.; Van Broeck, B.; Pirici, D.; Vennekens, K.; Corsmit, E.; Cruts, M.; Dermaut, B.; Wang, R.; Van Broeckhoven, C. Mean age-of-onset of familial Alzheimer disease caused by presenilin mutations correlates with both increased Abeta42 and decreased Abeta40. Hum. Mutat. 2006, 27, 686–695. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Crick, S.L.; Bu, G.; Frieden, C.; Pappu, R.V.; Lee, J.M. Amyloid seeds formed by cellular uptake, concentration, and aggregation of the amyloid-beta peptide. Proc. Natl. Acad. Sci. USA 2009, 106, 20324–20329. [Google Scholar] [CrossRef]
- Yajima, R.; Tokutake, T.; Koyama, A.; Kasuga, K.; Tezuka, T.; Nishizawa, M.; Ikeuchi, T. ApoE-isoform-dependent cellular uptake of amyloid-β is mediated by lipoprotein receptor LR11/SorLA. Biochem. Biophys. Res. Comm. 2015, 456, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Omtri, R.S.; Davidson, M.W.; Arumugam, B.; Poduslo, J.F.; Kandimalla, K.K. Differences in the cellular uptake and intracellular itineraries of amyloid beta proteins 40 and 42: Ramifications for the Alzheimer’s drug discovery. Mol. Pharm. 2012, 9, 1887. [Google Scholar] [CrossRef]
- Bu, G.; Cam, J.; Zerbinatti, C. LRP in amyloid-β production and metabolism. Ann. N. Y. Acad. Sci. 2006, 1086, 35–53. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Lee, D.H.; D’Andrea, M.R.; Peterson, P.A.; Shank, R.P.; Reitz, A.B. β-Amyloid1–42 binds to α7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer’s disease pathology. J. Biol. Chem. 2000, 275, 5626–5632. [Google Scholar] [CrossRef]
- Nagele, R.; D’Andrea, M.; Anderson, W.; Wang, H. Intracellular accumulation of Aβ42 in neurons is facilitated by the α7 nicotinic acetylcholine receptor in Alzheimer’s disease. Neuroscience 2002, 110, 199–211. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Green, K.N.; Liang, K.; Tran, L.; Chen, Y.; Leslie, F.M.; LaFerla, F.M. Chronic nicotine administration exacerbates tau pathology in a transgenic model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2005, 102, 3046–3051. [Google Scholar] [CrossRef]
- Yan, S.D.; Chen, X.; Fu, J.; Chen, M.; Zhu, H.; Roher, A.; Slattery, T.; Zhao, L.; Nagashima, M.; Morser, J.; et al. RAGE and amyloid-β peptide neurotoxicity in Alzheimer’s disease. Nature 1996, 382, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, N.; Toki, S.; Chowei, H.; Saito, T.; Nakano, N.; Hayashi, Y.; Takeuchi, M.; Makita, Z. Immunohistochemical distribution of the receptor for advanced glycation end products in neurons and astrocytes in Alzheimer’s disease. Brain Res. 2001, 888, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Deane, R.; Du Yan, S.; Submamaryan, R.K.; LaRue, B.; Jovanovic, S.; Hogg, E.; Welch, D.; Manness, L.; Lin, C.; Yu, J.; et al. RAGE mediates amyloid-β peptide transport across the blood–brain barrier and accumulation in brain. Nat. Med. 2003, 9, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Iribarren, P.; Zhou, Y.; Hu, J.; Le, Y.; Wang, J. Role of formyl peptide receptor-like 1 (FPRL1/FPR2) in mononuclear phagocyte responses in Alzheimer disease. Immunol. Res. 2005, 31, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Snyder, E.M.; Nong, Y.; Almeida, C.G.; Paul, S.; Moran, T.; Choi, E.Y.; Nairn, A.C.; Salter, M.W.; Lombroso, P.J.; Gouras, G.K.; et al. Regulation of NMDA receptor trafficking by amyloid-β. Nat. Neurosci. 2005, 8, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- Casas, C.; Sergeant, N.; Itier, J.M.; Blanchard, V.; Wirths, O.; van der Kolk, N.; Vingtdeux, V.; van de Steeg, E.; Ret, G.; Canton, T.; et al. Massive CA1/2 neuronal loss with intraneuronal and N-terminal truncated Abeta42 accumulation in a novel Alzheimer transgenic model. Am. J. Pathol. 2004, 165, 1289–1300. [Google Scholar] [CrossRef]
- Bayer, T.A.; Wirths, O. Review on the APP/PS1KI mouse model: Intraneuronal Abeta accumulation triggers axonopathy, neuron loss and working memory impairment. Genes Brain Behav. 2008, 1, 6–11. [Google Scholar] [CrossRef]
- Bayer, T.A.; Breyhan, H.; Duan, K.; Rettig, J.; Wirths, O. Intraneuronal beta-amyloid is a major risk factor--novel evidence from the APP/PS1KI mouse model. Neurodegener. Dis. 2008, 5, 140–142. [Google Scholar] [CrossRef]
- Wirths, O.; Breyhan, H.; Cynis, H.; Schilling, S.; Demuth, H.U.; Bayer, T.A. Intraneuronal pyroglutamate-Abeta 3-42 triggers neurodegeneration and lethal neurological deficits in a transgenic mouse model. Acta Neuropathol. 2009, 118, 487–496. [Google Scholar] [CrossRef]
- Christensen, D.Z.; Bayer, T.A.; Wirths, O. Intracellular Aβ triggers neuron loss in the cholinergic system of the APP/PS1KI mouse model of Alzheimer’s disease. Neurobiol. Aging 2010, 31, 1153–1163. [Google Scholar] [CrossRef]
- Christensen, D.Z.; Schneider-Axmann, T.; Lucassen, P.J.; Bayer, T.A.; Wirths, O. Accumulation of intraneuronal Abeta correlates with ApoE4 genotype. Acta Neuropathol. 2010, 119, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Bayer, T.A.; Wirths, O. Intracellular accumulation of amyloid-Beta—A predictor for synaptic dysfunction and neuron loss in Alzheimer’s disease. Front. Aging Neurosci. 2010, 2, 8. [Google Scholar] [CrossRef] [PubMed]
- Bayer, T.A.; Wirths, O. Intraneuronal Aβ as a trigger for neuron loss: Can this be translated into human pathology? Biochem. Soc. Trans. 2011, 39, 857–861. [Google Scholar] [CrossRef] [PubMed]
- Wirths, O.; Bayer, T.A. Intraneuronal Aβ accumulation and neurodegeneration: Lessons from transgenic models. Life Sci. 2012, 91, 1148–1152. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Wirths, O.; Theil, S.; Gerth, J.; Bayer, T.A.; Walter, J. Early intraneuronal accumulation and increased aggregation of phosphorylated Abeta in a mouse model of Alzheimer’s disease. Acta Neuropathol. 2013, 125, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Ripoli, C.; Cocco, S.; Li Puma, D.D.; Piacentini, R.; Mastrodonato, A.; Scala, F.; Puzzo, D.; D’Ascenzo, M.; Grassi, C. Intracellular accumulation of amyloid-β (Aβ) protein plays a major role in Aβ-induced alterations of glutamatergic synaptic transmission and plasticity. J. Neurosci. 2014, 34, 12893–12903. [Google Scholar] [CrossRef]
- Scala, F.; Fusco, S.; Ripoli, C.; Piacentini, R.; Li Puma, D.D.; Spinelli, M.; Laezza, F.; Grassi, C.; D’Ascenzo, M. Intraneuronal Aβ accumulation induces hippocampal neuron hyperexcitability through A-type K+ current inhibition mediated by activation of caspases and GSK-3. Neurobiol. Aging 2015, 36, 886–900. [Google Scholar] [CrossRef]
- Christensen, D.Z.; Kraus, S.L.; Flohr, A.; Cotel, M.C.; Wirths, O.; Bayer, T.A. Transient intraneuronal Abeta rather than extracellular plaque pathology correlates with neuron loss in the frontal cortex of APP/PS1KI mice. Acta Neuropathol. 2008, 116, 647–655. [Google Scholar] [CrossRef]
- Martin, B.; Schrader-Fischer, G.; Busciglio, J.; Duke, M.; Paganetti, P.; Yankner, B. Intracellular accumulation of beta-amyloid in cells expressing the Swedish mutant amyloid precursor protein. J. Biol. Chem. 1995, 270, 26727–26730. [Google Scholar] [CrossRef]
- Sannerud, R.; Esselens, C.; Ejsmont, P.; Mattera, R.; Rochin, L.; Tharkeshwar, A.K.; De Baets, G.; De Wever, V.; Habets, R.; Baert, V.; et al. Restricted Location of PSEN2/gamma-secretase determines substrate specificity and generates an intracellular Abeta pool. Cell 2016, 166, 193–208. [Google Scholar] [CrossRef]
- De Jonghe, C.; Zehr, C.; Yager, D.; Prada, C.M.; Younkin, S.; Hendriks, L.; Van Broeckhoven, C.; Eckman, C.B. Flemish and Dutch mutations in amyloid beta precursor protein have different effects on amyloid beta secretion. Neurobiol. Dis. 1998, 5, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.C.; Suen, K.C.; Ma, C.H.; Elyaman, W.; Ng, H.K.; Hugon, J. Involvement of double-stranded RNA-dependent protein kinase and phosphorylation of eukaryotic initiation factor-2alpha in neuronal degeneration. J. Neurochem. 2002, 83, 1215–1225. [Google Scholar] [CrossRef] [PubMed]
- Peel, A.L. PKR activation in neurodegenerative disease. J. Neuropathol. Exp. Neurol. 2004, 63, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Peel, A.L.; Bredesen, D.E. Activation of the cell stress kinase PKR in Alzheimer’s disease and human amyloid precursor protein transgenic mice. Neurobiol. Dis. 2003, 14, 52–62. [Google Scholar] [CrossRef]
- Chang, R.C.; Wong, A.K.; Ng, H.K.; Hugon, J. Phosphorylation of eukaryotic initiation factor-2alpha (eIF2alpha) is associated with neuronal degeneration in Alzheimer’s disease. Neuroreport 2002, 13, 2429–2432. [Google Scholar] [CrossRef]
- Onuki, R.; Bando, Y.; Suyama, E.; Katayama, T.; Kawasaki, H.; Baba, T.; Tohyama, M.; Taira, K. An RNA-dependent protein kinase is involved in tunicamycin-induced apoptosis and Alzheimer’s disease. EMBO J. 2004, 23, 959–968. [Google Scholar] [CrossRef] [PubMed]
- Lourenco, M.V.; Clarke, J.R.; Frozza, R.L.; Bomfim, T.R.; Forny-Germano, L.; Batista, A.F.; Sathler, L.B.; Brito-Moreira, J.; Amaral, O.B.; Silva, C.A.; et al. TNF-alpha mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimer’s beta-amyloid oligomers in mice and monkeys. Cell Metab. 2013, 18, 831–843. [Google Scholar] [CrossRef]
- Paquet, C.; Mouton-Liger, F.; Meurs, E.F.; Mazot, P.; Bouras, C.; Pradier, L.; Gray, F.; Hugon, J. The PKR activator PACT is induced by Abeta: Involvement in Alzheimer’s disease. Brain Pathol. 2012, 22, 219–229. [Google Scholar] [CrossRef]
- Zhu, X.; Perry, G.; Moreira, P.I.; Aliev, G.; Cash, A.D.; Hirai, K.; Smith, M.A. Mitochondrial abnormalities and oxidative imbalance in Alzheimer disease. J. Alzheimers Dis. 2006, 9, 147–153. [Google Scholar] [CrossRef]
- Blass, J.P. The mitochondrial spiral. An adequate cause of dementia in the Alzheimer’s syndrome. Ann. N. Y. Acad. Sci. 2000, 924, 170–183. [Google Scholar] [CrossRef]
- Manczak, M.; Park, B.S.; Jung, Y.; Reddy, P.H. Differential expression of oxidative phosphorylation genes in patients with Alzheimer’s disease: Implications for early mitochondrial dysfunction and oxidative damage. Neuromolecular Med. 2004, 5, 147–162. [Google Scholar] [CrossRef]
- Qin, W.; Haroutunian, V.; Katsel, P.; Cardozo, C.P.; Ho, L.; Buxbaum, J.D.; Pasinetti, G.M. PGC-1alpha expression decreases in the Alzheimer disease brain as a function of dementia. Arch. Neurol. 2009, 66, 352–361. [Google Scholar] [CrossRef]
- Du, H.; Guo, L.; Yan, S.; Sosunov, A.A.; McKhann, G.M.; Yan, S.S. Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc. Natl. Acad. Sci. USA 2010, 107, 18670–18675. [Google Scholar] [CrossRef]
- Lin, M.T.; Simon, D.K.; Ahn, C.H.; Kim, L.M.; Beal, M.F. High aggregate burden of somatic mtDNA point mutations in aging and Alzheimer’s disease brain. Hum. Mol. Genet. 2002, 11, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Calkins, M.; Manczak, M.; Mao, P.; Shirendeb, U.; Reddy, P.H. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2011, 20, 4515–4529. [Google Scholar] [CrossRef] [PubMed]
- Anandatheerthavarada, H.K.; Biswas, G.; Robin, M.A.; Avadhani, N.G. Mitochondrial targeting and a novel transmembrane arrest of Alzheimer’s amyloid precursor protein impairs mitochondrial function in neuronal cells. J. Cell Biol. 2003, 161, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Caspersen, C.; Wang, N.; Yao, J.; Sosunov, A.; Chen, X.; Lustbader, J.W.; Xu, H.W.; Stern, D.; McKhann, G.; Yan, S.D. Mitochondrial Abeta: A potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 2005, 19, 2040–2041. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.X.; Yan, S.S. Role of mitochondrial amyloid-beta in Alzheimer’s disease. J. Alzheimers Dis. 2010, 20, S569–S578. [Google Scholar] [CrossRef] [PubMed]
- Hansson Petersen, C.A.; Alikhani, N.; Behbahani, H.; Wiehager, B.; Pavlov, P.F.; Alafuzoff, I.; Leinonen, V.; Ito, A.; Winblad, B.; Glaser, E.; et al. The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. USA 2008, 105, 13145–13150. [Google Scholar] [CrossRef] [PubMed]
- de la Monte, S.M.; Luong, T.; Neely, T.R.; Robinson, D.; Wands, J.R. Mitochondrial DNA damage as a mechanism of cell loss in Alzheimer’s disease. Lab. Investig. 2000, 80, 1323–1335. [Google Scholar] [CrossRef]
- Brooks, W.M.; Lynch, P.J.; Ingle, C.C.; Hatton, A.; Emson, P.C.; Faull, R.L.; Starkey, M.P. Gene expression profiles of metabolic enzyme transcripts in Alzheimer’s disease. Brain Res. 2007, 1127, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Perry, G.; Smith, M.A.; Zhu, X. Amyloid-beta-derived diffusible ligands cause impaired axonal transport of mitochondria in neurons. Neurodegener. Dis. 2007, 7, 56–59. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Su, B.; Fujioka, H.; Zhu, X. Dynamin-like protein 1 reduction underlies mitochondrial morphology and distribution abnormalities in fibroblasts from sporadic Alzheimer’s disease patients. Am. J. Pathol. 2008, 173, 470–482. [Google Scholar] [CrossRef]
- Wang, X.; Su, B.; Lee, H.G.; Li, X.; Perry, G.; Smith, M.A.; Zhu, X. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 2009, 29, 9090–9103. [Google Scholar] [CrossRef]
- Wang, X.; Su, B.; Siedlak, S.L.; Moreira, P.I.; Fujioka, H.; Wang, Y.; Casadesus, G.; Zhu, X. Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 19318–19323. [Google Scholar] [CrossRef]
- Brewer, G.; Herrera, R.; Philipp, S.; Sosna, J.; Reyes-Ruiz, J.; Glabe, C. Age-Related Intraneuronal Aggregation of Amyloid-β in Endosomes, Mitochondria, Autophagosomes, and Lysosomes. J. Alzheimers Dis. 2020, 73, 229–246. [Google Scholar] [CrossRef]
- Guo, X.; Aviles, G.; Liu, Y.; Tian, R.; Unger, B.A.; Lin, Y.T.; Wiita, A.P.; Xu, K.; Correia, M.A.; Kampmann, M. Mitochondrial stress is relayed to the cytosol by an OMA1-DELE1-HRI pathway. Nature 2020, 579, 427–432. [Google Scholar] [CrossRef]
- Fessler, E.; Eckl, E.M.; Schmitt, S.; Mancilla, I.A.; Meyer-Bender, M.F.; Hanf, M.; Philippou-Massier, J.; Krebs, S.; Zischka, H.; Jae, L.T. A pathway coordinated by DELE1 relays mitochondrial stress to the cytosol. Nature 2020, 579, 433–437. [Google Scholar] [CrossRef]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef]
- Ron, D. Translational control in the endoplasmic reticulum stress response. J. Clin. Investig. 2002, 110, 1383–1388. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Brostrom, C.O.; Prostko, C.R.; Kaufman, R.J.; Brostrom, M.A. Inhibition of translational initiation by activators of the glucose-regulated stress protein and heat shock protein stress response systems. Role of the interferon-inducible double-stranded RNA-activated eukaryotic initiation factor 2alpha kinase. J. Biol. Chem. 1996, 271, 24995–25002. [Google Scholar] [CrossRef] [PubMed]
- Dever, T.E.; Feng, L.; Wek, R.C.; Hinnebusch, A.G. Phosphorylation of initiation factor 2 alpha by protein kinase GCN2 mediates gene-specific translational control of GCN4 in yeast. Cell 1992, 68, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Wek, R.C.; Jiang, H.Y.; Anthony, T.G. Coping with stress: eIF2 kinases and translational control. Biochem. Soc. Trans. 2006, 34, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Rzymski, T.; Milani, M.; Pike, L.; Buffa, F.; Mellor, H.R.; Winchester, L.; Pires, I.; Hammond, E.; Ragoussis, I.; Harris, A.L. Regulation of autophagy by ATF4 in response to severe hypoxia. Oncogene 2010, 29, 4424–4435. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Kumanova, M.; Hart, L.S.; Sloane, K.; Zhang, H.; De Panis, D.N.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Ron, D.; Koumenis, C. The GCN2-ATF4 pathway is critical for tumour cell survival and proliferation in response to nutrient deprivation. EMBO J. 2010, 29, 2082–2096. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.A.; Meurs, E.F.; Esteban, M. The dsRNA protein kinase PKR: Virus and cell control. Biochimie 2007, 89, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef]
- Breimer, L.; Denny, P. Alzheimer amyloid aspects. Nature 1987, 326, 749–750. [Google Scholar] [CrossRef]
- Kang, J.; Lemaire, H.G.; Unterbeck, A.; Salbaum, J.M.; Masters, C.L.; Grzeschik, K.H.; Multhaup, G.; Beyreuther, K.; Müller-Hill, B. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 1987, 325, 733–736. [Google Scholar] [CrossRef]
- Robakis, N.K.; Ramakrishna, N.; Wolfe, G.; Wisniewski, H.M. Molecular cloning and characterization of a cDNA encoding the cerebrovascular and the neuritic plaque amyloid peptides. Proc. Natl. Acad. Sci. USA 1987, 84, 4190–4194. [Google Scholar] [CrossRef] [PubMed]
- Tanzi, R.E.; Gusella, J.F.; Watkins, P.C.; Bruns, G.A.; St George-Hyslop, P.; Van Keuren, M.L.; Patterson, D.; Pagan, S.; Kurnit, D.M.; Neve, R.L. Amyloid beta protein gene: cDNA, mRNA distribution, and genetic linkage near the Alzheimer locus. Science 1987, 235, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Citron, M.; Haass, C.; Selkoe, D. Production of amyloid beta peptide by cultured cells: No evidence for internal initiation of translation at Met596. Neurobiol. Aging 1993, 14, 571–573. [Google Scholar] [CrossRef]
- Macq, A.; Philippe, B.; Octave, N. The amyloid peptide of Alzheimer’s disease is not produced by internal initiation of translation generating C-terminal amyloidogenic fragments of its precursor. Neurosci. Lett. 1994, 182, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Frottin, F.; Martinez, A.; Peinot, P.; Mitra, S.; Holz, R.; Giglione, C.; Meinnel, T. The proteomics of N-terminal methionine cleavage. Mol. Cell Proteom. MCP 2006, 5, 2336–2349. [Google Scholar] [CrossRef]
- Xiao, Q.; Zhang, F.; Nacev, B.; Liu, J.; Pei, D. Protein N-Terminal Processing: Substrate Specificity of Escherichia coli and Human Methionine Aminopeptidases. Biochemistry 2010, 49, 5588–5599. [Google Scholar] [CrossRef] [PubMed]
- Varshavsky, A. The N-end rule pathway and regulation by proteolysis. Protein Sci. 2011, 20, 1298–1345. [Google Scholar] [CrossRef] [PubMed]
- Wingfield, P. N-Terminal Methionine Processing. Curr. Protoc. Protein Sci. 2017, 88, 6.14.1–6.14.3. [Google Scholar] [CrossRef]
- Varland, S.; Osberg, C.; Arnesen, T. N-terminal modifications of cellular proteins: The enzymes involved, their substrate specificities and biological effects. Proteomics 2015, 15, 2385–2401. [Google Scholar] [CrossRef]
- Polevoda, B.; Sherman, F. N-terminal acetyltransferases and sequence requirements for N-terminal acetylation of eukaryotic proteins. J. Mol. Biol. 2003, 325, 595–622. [Google Scholar] [CrossRef]
- Pardossi-Piquard, R.; Checler, F. The physiology of the β-amyloid precursor protein intracellular domain AICD. J. Neurochem. 2012, 120 (Suppl. S1), 109–124. [Google Scholar] [CrossRef]
- Szögi, T.; Schuster, I.; Borbély, E.; Gyebrovszki, A.; Bozsó, Z.; Gera, J.; Rajkó, R.; Sántha, M.; Penke, B.; Fülöp, L. Effects of the Pentapeptide P33 on Memory and Synaptic Plasticity in APP/PS1 Transgenic Mice: A Novel Mechanism Presenting the Protein Fe65 as a Target. Int. J. Mol. Sci. 2019, 20, 3050. [Google Scholar] [CrossRef] [PubMed]
- Borg, J.-P.; Yang, Y.; De Taddeo-Borg, M.; Margolis, B.; Turner, R.S. The X11α protein slows cellular amyloid precursor protein processing and reduces Aβ40 and Aβ42 secretion. J. Biol. Chem. 1998, 273, 14761–14766. [Google Scholar] [CrossRef] [PubMed]
- Dunning, C.J.R.; Black, H.L.; Andrews, K.L.; Davenport, E.C.; Conboy, M.; Chawla, S.; Dowle, A.A.; Ashford, D.; Thomas, J.R.; Evans, G.J.O. Multisite tyrosine phosphorylation of the N-terminus of Mint1/X11α by Src kinase regulates the trafficking of amyloid precursor protein. J. Neurochem. 2016, 137, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.C.J.; McLoughlin, D.M.; Lau, K.-F.; Tennant, M.E.; Rogelj, B. The X11 proteins, Aβ production and Alzheimer’s disease. Trends Neurosci. 2006, 29, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Tamayev, R.; Zhou, D.; D’Adamio, L. The interactome of the amyloid β precursor protein family members is shaped by phosphorylation of their intracellular domains. Mol. Neurodegener. 2009, 4, 28. [Google Scholar] [CrossRef]
- Hoe, H.-S.; Tran, T.S.; Matsuoka, Y.; Howell, B.W.; Rebeck, G.W. DAB1 and Reelin Effects on Amyloid Precursor Protein and ApoE Receptor 2 Trafficking and Processing. J. Biol. Chem. 2006, 281, 35176–35185. [Google Scholar] [CrossRef]
- Russo, C.; Dolcini, V.; Salis, S.; Venezia, V.; Zambrano, N.; Russo, T.; Schettini, G. Signal Transduction through Tyrosine-phosphorylated C-terminal Fragments of Amyloid Precursor Protein via an Enhanced Interaction with Shc/Grb2 Adaptor Proteins in Reactive Astrocytes of Alzheimer’s Disease Brain. J. Biol. Chem. 2002, 277, 35282–35288. [Google Scholar] [CrossRef]
- Tarr, P.E.; Roncarati, R.; Pelicci, G.; Pelicci, P.G.; D’Adamio, L. Tyrosine phosphorylation of the β-amyloid precursor protein cytoplasmic tail promotes interaction with Shc. J. Biol. Chem. 2002, 277, 16798–16804. [Google Scholar] [CrossRef]
- Bukhari, H.; Glotzbach, A.; Kolbe, K.; Leonhardt, G.; Loosse, C.; Mueller, T. Small things matter: Implications of APP intracellular domain AICD nuclear signaling in the progression and pathogenesis of Alzheimer’s disease. Prog. Neurobiol. 2017, 156, 189–213. [Google Scholar] [CrossRef]
- Chang, K.-A.; Kim, H.-S.; Ha, T.-Y.; Ha, J.-W.; Shin, K.Y.; Jeong, Y.H.; Lee, J.-P.; Park, C.-H.; Kim, S.; Baik, T.-K.; et al. Phosphorylation of amyloid precursor protein (APP) at Thr668 regulates the nuclear translocation of the APP intracellular domain and induces neurodegeneration. Mol. Cell. Biol. 2006, 26, 4327–4338. [Google Scholar] [CrossRef] [PubMed]
- Ando, K.; Iijima, K.-I.; Elliott, J.I.; Kirino, Y.; Suzuki, T. Phosphorylation-dependent regulation of the interaction of amyloid precursor protein with Fe65 affects the production of β-amyloid. J. Biol. Chem. 2001, 276, 40353–40361. [Google Scholar] [CrossRef] [PubMed]
- Müller, T.; Meyer, H.E.; Egensperger, R.; Marcus, K. The amyloid precursor protein intracellular domain (AICD) as modulator of gene expression, apoptosis, and cytoskeletal dynamics-relevance for Alzheimer’s disease. Prog. Neurobiol. 2008, 85, 393–406. [Google Scholar] [CrossRef]
- Shu, R.; Wong, W.; Ma, Q.H.; Yang, Z.Z.; Zhu, H.; Liu, F.J.; Wang, P.; Ma, J.; Yan, S.; Polo, J.M.; et al. APP intracellular domain acts as a transcriptional regulator of miR-663 suppressing neuronal differentiation. Cell Death Dis. 2015, 6, e1651. [Google Scholar] [CrossRef]
- Grimm, M.O.; Mett, J.; Stahlmann, C.P.; Grösgen, S.; Haupenthal, V.J.; Blümel, T.; Hundsdörfer, B.; Zimmer, V.C.; Mylonas, N.T.; Tanila, H.; et al. APP intracellular domain derived from amyloidogenic β- and γ-secretase cleavage regulates neprilysin expression. Front. Aging Neurosci. 2015, 7, 77. [Google Scholar] [CrossRef]
- Pousinha, P.A.; Mouska, X.; Bianchi, D.; Temido-Ferreira, M.; Rajão-Saraiva, J.; Gomes, R.; Fernandez, S.P.; Salgueiro-Pereira, A.R.; Gandin, C.; Raymond, E.F.; et al. The Amyloid Precursor Protein C-Terminal Domain Alters CA1 Neuron Firing, Modifying Hippocampus Oscillations and Impairing Spatial Memory Encoding. Cell Rep. 2019, 29, 317–331.e5. [Google Scholar] [CrossRef]
- Almeida, C.; Takahashi, R.; Gouras, G. β-amyloid accumulation impairs multivesicular body sorting by inhibiting the ubiquitin–proteasome system. J. Neurosci. 2006, 26, 4277–4288. [Google Scholar] [CrossRef]
- Gregori, L.; Fuchs, C.; Figueiredo-Pereira, M.; Van Nostrand, W.; Goldgaber, D. Amyloid β-protein inhibits ubiquitin-dependent protein degradation in vitro. J. Biol. Chem. 1995, 270, 19702–19708. [Google Scholar] [CrossRef]
- Oh, S.; Hong, H.S.; Hwang, E.; Sim, H.J.; Lee, W.; Shin, S.J.; Mook-Jung, I. Amyloid peptide attenuates the proteasome activity in neuronal cells. Mech. Ageing Dev. 2005, 126, 1292–1299. [Google Scholar] [CrossRef]
- Tseng, B.; Green, K.; Chan, J.; Blurton-Jones, M.; LaFerla, F. Aβ inhibits the proteasome and enhances amyloid and tau accumulation. Neurobiol. Aging 2008, 29, 1607–1618. [Google Scholar] [CrossRef]
- Tucker-Drob, E.M. Cognitive Aging and Dementia: A Life Span Perspective. Annu. Rev. Dev. Psychol. 2019, 1, 177–196. [Google Scholar] [CrossRef] [PubMed]
- Matthews, F.E.; Arthur, A.; Barnes, L.E.; Bond, J.; Jagger, C.; Robinson, L.; Brayne, C. Medical Research Council Cognitive Function and Ageing Collaboration. A two-decade comparison of prevalence of dementia in individuals aged 65 years and older from three geographical areas of England: Results of the Cognitive Function and Ageing Study I and II. Lancet. 2013, 382, 1405–1412. [Google Scholar] [PubMed]
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Mintun, M.; Lo, A.; Duggan-Evans, C.; Wessels, A.; Ardayfio, P.; Andersen, S.; Shcherbinin, S.; Sparks, J.; Sims, J.; Brys, M.; et al. Donanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2021, 384, 1691–1704. [Google Scholar] [CrossRef] [PubMed]
- Lilly, E. Donanemab Significantly Slowed Cognitive and Functional Decline in Phase 3 Study of Early Alzheimer’s Disease. Pressrlease. 2023. Available online: https://investor.lilly.com/news-releases/news-release-details/lillys-donanemab-significantly-slowed-cognitive-and-functional (accessed on 1 October 2023).
- Sims, J.; Zimmer, J.; Evans, C.; Lu, M.; Ardayfio, P.; Sparks, J.; Wessels, A.; Sncherbinin, S.; Wang, H.; Nery, E.; et al. Donanemab in Early Symptomatic Alzheimer Disease: The TRAILBLAZER-ALZ 2 Randomized Clinical Trial. JAMA 2023, 330, 512–527. [Google Scholar] [CrossRef]
- Abbott, A. The next generation of Alzheimer’s treatments. Nature 2023, 616, 26–28. [Google Scholar] [CrossRef]
- Huse, J.; Liu, K.; Pijak, D.; Carlin, D.; Lee VDoms, R. Beta-secretase processing in the trans-Golgi network preferentially generates truncated amyloid species that accumulate in Alzheimer’s disease brain. J. Biol. Chem. 2002, 277, 16278–16284. [Google Scholar] [CrossRef]
- Liu, K.; Doms, R.; Lee, M. Glu11 site cleavage and N-terminally truncated A beta production upon BACE overexpression. Biochemistry 2002, 41, 3128–3136. [Google Scholar] [CrossRef]
- Lee, E.B.; Zhang, B.; Liu, K.; Greenbaum, E.A.; Doms, R.W.; Trojanowski, J.Q.; Lee, V.M. BACE overexpression alters the subcellular processing of APP and inhibits Aβ deposition in vivo. J. Cell Biol. 2005, 168, 291–302. [Google Scholar] [CrossRef]
- Kimura, A.; Hata, S.; Suzuki, T. Alternative selection of beta-site APP-cleaving enzyme 1 (BACE1) cleavage sites in amyloid beta-protein precursor (APP) harboring protective and pathogenic mutations within the Abeta sequence. J. Biol. Chem. 2016, 291, 24041–24053. [Google Scholar] [CrossRef]
- Fluhrer, R.; Multhaup, G.; Schlicksupp, A.; Okochi, M.; Takeda, M.; Lammich, S.; Willem, M.; Westmeyer, G.; Bode, W.; Walter, J.; et al. Identification of a beta-secretase activity, which truncates amyloid beta-peptide after its presenilin-dependent generation. J. Biol. Chem. 2003, 278, 5531–5538. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.P.; Tugusheva, K.; Bruce, J.E.; Lucka, A.; Wu, G.X.; Chen-Dodson, E.; Price, E.; Li, Y.; Xu, M.; Huang, Q.; et al. Beta-secretase cleavage at amino acid residue 34 in the amyloid beta peptide is dependent upon gamma-secretase activity. J. Biol. Chem. 2003, 278, 21286–21294. [Google Scholar] [CrossRef] [PubMed]
- Liebsch, F.; Kulic, L.; Teunissen, C.; Shobo, A.; Ulku, I.; Engelschalt, V.; Hancock, M.A.; van der Flier, W.M.; Kunach, P.; Rosa-Neto, P.; et al. Abeta34 is a BACE1-derived degradation intermediate associated with amyloid clearance and Alzheimer’s disease progression. Nat. Commun. 2019, 10, 2240. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gonzales, L.; Pilat, D.; Rivera, S. Emerging Alternative Proteinases in APP Metabolism and Alzheimer’s Disease Pathogenesis. Front. Aging Neurosci. 2019, 11, 244. [Google Scholar] [CrossRef]
- Farzan, M.; Schnitzler, C.E.; Vasilieva, N.; Leung, D.; Choe, H. BACE2, a β-secretase homolog, cleaves at the β site and within the amyloid-β region of the amyloid-β precursor protein. Proc. Natl. Acad. Sci. USA 2000, 97, 9712–9717. [Google Scholar] [CrossRef]
- Yan, R.; Munzner, J.; Shuck, M.; Bienkowski, M. BACE2 functions as an alternative alpha secretase in cells. J. Biol. Chem. 2001, 276, 34019–34027. [Google Scholar] [CrossRef]
- Luo, J.; Zou, H.; Guo, Y.; Huang, K.; Ngan, E.S.; Li, P. BACE2 variant identified from HSCR patient causes AD-like phenotypes in hPSC-derived brain organoids. Cell Death Discov. 2022, 8, 47. [Google Scholar] [CrossRef]
- Basi, G.; Frigon, N.; Barbour, R.; Doan, T.; Gordon, G.; McConlogue, L.; Sinha, S.; Zeller, M. Antagonistic effects of beta-site amyloid precursor protein-cleaving enzymes 1 and 2 on beta-amyloid peptide production in cells. J. Biol. Chem. 2003, 278, 31512–31520. [Google Scholar] [CrossRef]




















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Volloch, V.; Rits-Volloch, S. Next Generation Therapeutic Strategy for Treatment and Prevention of Alzheimer’s Disease and Aging-Associated Cognitive Decline: Transient, Once-in-a-Lifetime-Only Depletion of Intraneuronal Aβ (iAβ) by Its Targeted Degradation via Augmentation of Intra-iAβ-Cleaving Activities of BACE1 and/or BACE2. Int. J. Mol. Sci. 2023, 24, 17586. https://doi.org/10.3390/ijms242417586
Volloch V, Rits-Volloch S. Next Generation Therapeutic Strategy for Treatment and Prevention of Alzheimer’s Disease and Aging-Associated Cognitive Decline: Transient, Once-in-a-Lifetime-Only Depletion of Intraneuronal Aβ (iAβ) by Its Targeted Degradation via Augmentation of Intra-iAβ-Cleaving Activities of BACE1 and/or BACE2. International Journal of Molecular Sciences. 2023; 24(24):17586. https://doi.org/10.3390/ijms242417586
Chicago/Turabian StyleVolloch, Vladimir, and Sophia Rits-Volloch. 2023. "Next Generation Therapeutic Strategy for Treatment and Prevention of Alzheimer’s Disease and Aging-Associated Cognitive Decline: Transient, Once-in-a-Lifetime-Only Depletion of Intraneuronal Aβ (iAβ) by Its Targeted Degradation via Augmentation of Intra-iAβ-Cleaving Activities of BACE1 and/or BACE2" International Journal of Molecular Sciences 24, no. 24: 17586. https://doi.org/10.3390/ijms242417586
APA StyleVolloch, V., & Rits-Volloch, S. (2023). Next Generation Therapeutic Strategy for Treatment and Prevention of Alzheimer’s Disease and Aging-Associated Cognitive Decline: Transient, Once-in-a-Lifetime-Only Depletion of Intraneuronal Aβ (iAβ) by Its Targeted Degradation via Augmentation of Intra-iAβ-Cleaving Activities of BACE1 and/or BACE2. International Journal of Molecular Sciences, 24(24), 17586. https://doi.org/10.3390/ijms242417586