

Definition and Characterization of SOX11-Derived T Cell Epitopes towards Immunotherapy of Glioma

 , ,
, , 
Abstract
:1. Introduction
2. Results
2.1. Peptides Derived from SOX11 as Defined by In Silico Prediction
2.2. Experimental HLA-A*02-Binding Affinity of Predicted Peptides on T2 Cells
2.3. In Vitro Generation of CD8+ T Cells Recognizing the SOX11-Derived Peptides
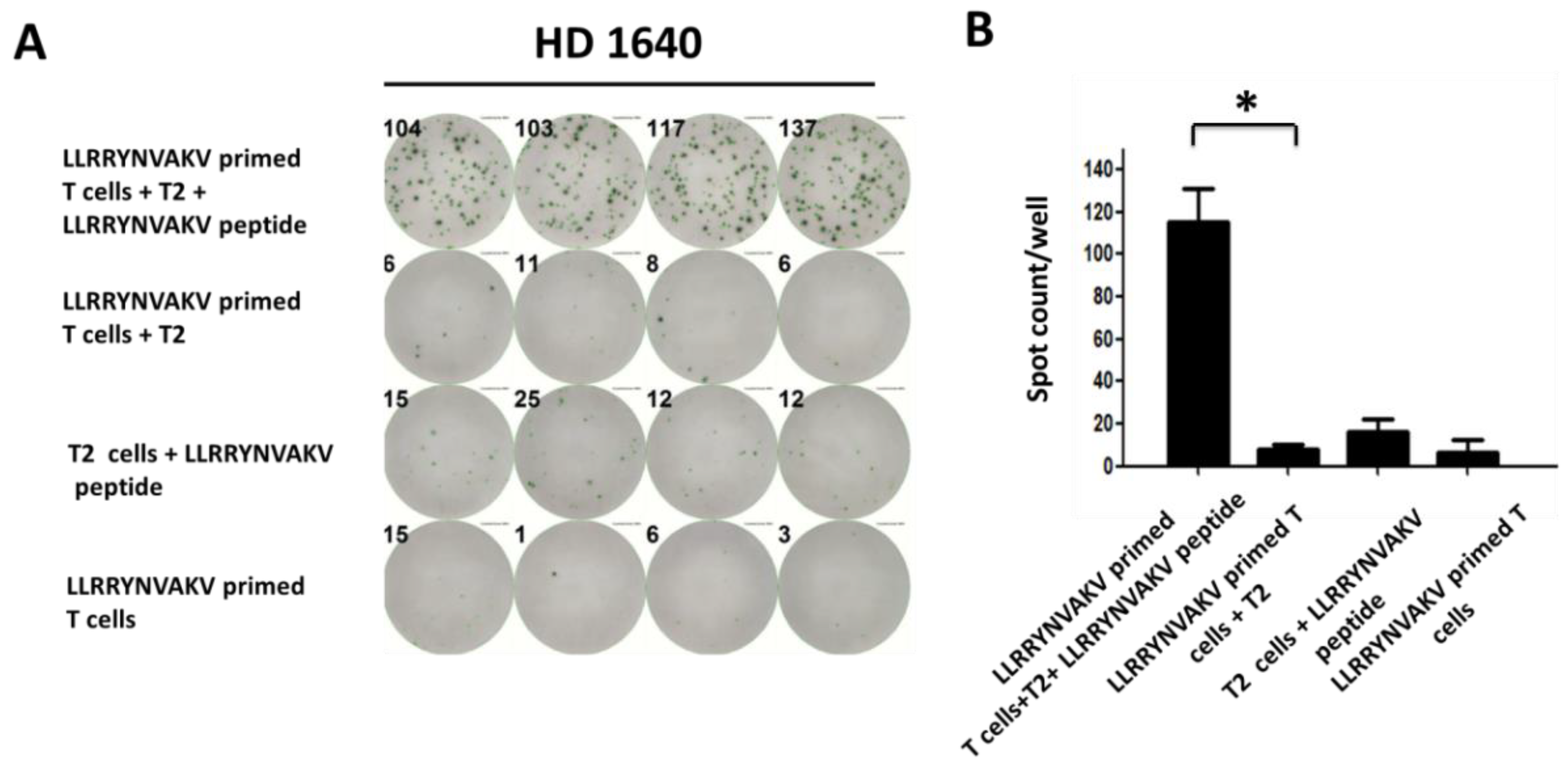
2.4. Recognition of Peptide LLRRYNVAKV by T Cells from a Healthy Donor
2.5. Recognition of Peptides FMACSPVAL, LMFDLSLNF, and SLYDEVRAGA by T Cells from Healthy Donors
2.6. Recognition of Different Peptides by T Cells from Different Healthy Donors
2.7. Evaluation of Cytokine Production and Cytotoxicity of Peptide-Specific CD8+ T Cells
3. Discussion
4. Materials and Methods
4.1. Blood Samples from Healthy Donors
4.2. Cell Line
4.3. Epitope Prediction and Peptide Synthesis
4.4. HLA Typing
4.5. In Vitro Generation of Peptide-Specific CD8+ T Cells with Long Method
4.6. In Vitro Generation of Peptide-Specific CD8+ T Cells with Short Method
4.7. HLA-A*02-Binding Affinity Assay
4.8. IFN-γ ELISPOT Assay
4.9. Intracellular Cytokine Staining
4.10. Cytotoxic Capacitiy of SOX11-Specific CAR T Cells
4.11. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Herholz, K. Brain Tumors: An Update on Clinical PET Research in Gliomas. Semin. Nucl. Med. 2017, 47, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Zhang, L.; Qiu, B.; Meng, L.; Wang, X.; Hou, B.L. Correlation of volume transfer coefficient Ktrans with histopathologic grades of gliomas. J. Magn. Reson. Imaging 2012, 36, 355–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrom, Q.T.; Bauchet, L.; Davis, F.G.; Deltour, I.; Fisher, J.L.; Langer, C.E.; Pekmezci, M.; Schwartzbaum, J.A.; Turner, M.C.; Walsh, K.M.; et al. The epidemiology of glioma in adults: A “state of the science” review. Neuro Oncol. 2014, 16, 896–913. [Google Scholar] [CrossRef] [Green Version]
- Oberoi, R.K.; Parrish, K.E.; Sio, T.T.; Mittapalli, R.K.; Elmquist, W.F.; Sarkaria, J.N. Strategies to improve delivery of anticancer drugs across the blood-brain barrier to treat glioblastoma. Neuro Oncol. 2016, 18, 27–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.; Tang, L.; Li, X.; Fan, F.; Liu, Z. Immunotherapy for glioma: Current management and future application. Cancer Lett. 2020, 476, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Chung, D.S.; Shin, H.J.; Hong, Y.K. A new hope in immunotherapy for malignant gliomas: Adoptive T cell transfer therapy. J. Immunol. Res. 2014, 2014, 326545. [Google Scholar] [CrossRef]
- Leen, A.M.; Rooney, C.M.; Foster, A.E. Improving T cell therapy for cancer. Annu. Rev. Immunol. 2007, 25, 243–265. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, C.J.; Black, K.L.; Liu, G.; Mazer, M.; Zhang, X.X.; Pepkowitz, S.; Goldfinger, D.; Ng, H.; Irvin, D.; Yu, J.S. Vaccination elicits correlated immune and clinical responses in glioblastoma multiforme patients. Cancer Res. 2008, 68, 5955–5964. [Google Scholar] [CrossRef] [Green Version]
- Pevny, L.; Placzek, M. SOX genes and neural progenitor identity. Curr. Opin. Neurobiol. 2005, 15, 7–13. [Google Scholar] [CrossRef]
- Mitchell, D.A.; Fecci, P.E.; Sampson, J.H. Adoptive immunotherapy for malignant glioma. Cancer J. 2003, 9, 157–166. [Google Scholar] [CrossRef]
- Schuler, M.M.; Nastke, M.D.; Stevanovikc, S. SYFPEITHI: Database for searching and T cell epitope prediction. Methods Mol. Biol. 2007, 409, 75–93. [Google Scholar] [PubMed]
- Reynisson, B.; Alvarez, B.; Paul, S.; Peters, B.; Nielsen, M. NetMHCpan-4.1 and NetMHCIIpan-4.0: Improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020, 48, W449–W454. [Google Scholar] [CrossRef] [PubMed]
- Jurtz, V.; Paul, S.; Andreatta, M.; Marcatili, P.; Peters, B.; Nielsen, M. NetMHCpan-4.0: Improved peptide–MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. J. Immunol. 2017, 199, 3360–3368. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, M.; Andreatta, M. NetMHCpan-3.0; improved prediction of binding to MHC class I molecules integrating information from multiple receptor and peptide length datasets. Genome Med. 2016, 8, 33. [Google Scholar] [CrossRef] [Green Version]
- Hoof, I.; Peters, B.; Sidney, J.; Pedersen, L.E.; Sette, A.; Lund, O.; Buus, S.; Nielsen, M. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics 2009, 61, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Andreatta, M.; Nielsen, M. Gapped sequence alignment using artificial neural networks: Application to the MHC class I system. Bioinformatics 2016, 32, 511–517. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, M.; Lundegaard, C.; Worning, P.; Lauemøller, S.L.; Lamberth, K.; Buus, S.; Brunak, S.; Lund, O. Reliable prediction of T cell epitopes using neural networks with novel sequence representations. Protein Sci. 2003, 12, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, A.; Hochedlinger, K. The sox family of transcription factors: Versatile regulators of stem and progenitor cell fate. Cell Stem Cell 2013, 12, 15–30. [Google Scholar] [CrossRef] [Green Version]
- de la Rocha, A.M.; Sampron, N.; Alonso, M.M.; Matheu, A. Role of SOX family of transcription factors in central nervous system tumors. Am. J. Cancer Res. 2014, 4, 312–324. [Google Scholar]
- Weigle, B.; Ebner, R.; Temme, A.; Schwind, S.; Schmitz, M.; Kiessling, A.; Rieger, M.A.; Schackert, G.; Schackert, H.K.; Rieber, E.P. Highly specific overexpression of the transcription factor SOX11 in human malignant gliomas. Oncol. Rep. 2005, 13, 139–144. [Google Scholar] [CrossRef]
- Schmitz, M.; Wehner, R.; Stevanovic, S.; Kiessling, A.; Rieger, M.A.; Temme, A.; Bachmann, M.; Rieber, E.P.; Weigle, B. Identification of a naturally processed T cell epitope derived from the glioma-associated protein SOX11. Cancer Lett. 2007, 245, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Nejo, T.; Yamamichi, A.; Almeida, N.D.; Goretsky, Y.E.; Okada, H. Tumor antigens in glioma. Semin. Immunol. 2020, 47, 101385. [Google Scholar] [CrossRef] [PubMed]
- Bonsack, M.; Hoppe, S.; Winter, J.; Tichy, D.; Zeller, C.; Küpper, M.D.; Schitter, E.C.; Blatnik, R.; Riemer, A.B. Performance evaluation of MHC class-I binding prediction tools based on an experimentally validated MHC–peptide binding data set. Cancer Immunol. Res. 2019, 7, 719–736. [Google Scholar] [CrossRef] [PubMed]
- Valmori, D.; Fonteneau, J.-F.; Lizana, C.M.; Gervois, N.; Liénard, D.; Rimoldi, D.; Jongeneel, V.; Jotereau, F.; Cerottini, J.-C.; Romero, P. Enhanced generation of specific tumor-reactive CTL in vitro by selected Melan-A/MART-1 immunodominant peptide analogues. J. Immunol. 1998, 160, 1750–1758. [Google Scholar] [CrossRef]
- Bonaventura, P.; Shekarian, T.; Alcazer, V.; Valladeau-Guilemond, J.; Valsesia-Wittmann, S.; Amigorena, S.; Caux, C.; Depil, S. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front. Immunol. 2019, 10, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamers, C.H.; Sleijfer, S.; van Steenbergen, S.; van Elzakker, P.; van Krimpen, B.; Groot, C.; Vulto, A.; den Bakker, M.; Oosterwijk, E.; Debets, R.; et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: Clinical evaluation and management of on-target toxicity. Mol. Ther. 2013, 21, 904–912. [Google Scholar] [CrossRef] [PubMed]
- Tsang, S.M.; Oliemuller, E.; Howard, B.A. In Regulatory roles for SOX11 in development, stem cells and cancer. Semin. Cancer Biol. 2020, 67 Pt 1, 3–11. [Google Scholar] [CrossRef]
- Zhang, J.G.; Eguchi, J.; Kruse, C.A.; Gomez, G.G.; Fakhrai, H.; Schroter, S.; Ma, W.; Hoa, N.; Minev, B.; Delgado, C.; et al. Antigenic profiling of glioma cells to generate allogeneic vaccines or dendritic cell-based therapeutics. Clin. Cancer Res. 2007, 13 Pt 1, 566–575. [Google Scholar] [CrossRef] [Green Version]
- Bullain, S.S.; Sahin, A.; Szentirmai, O.; Sanchez, C.; Lin, N.; Baratta, E.; Waterman, P.; Weissleder, R.; Mulligan, R.C.; Carter, B.S. Genetically engineered T cells to target EGFRvIII expressing glioblastoma. J. Neurooncol. 2009, 94, 373–382. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, N.; Salsman, V.S.; Kew, Y.; Shaffer, D.; Powell, S.; Zhang, Y.J.; Grossman, R.G.; Heslop, H.E.; Gottschalk, S. HER2-specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clin. Cancer Res. 2010, 16, 474–485. [Google Scholar] [CrossRef] [Green Version]
- Chow, K.K.; Naik, S.; Kakarla, S.; Brawley, V.S.; Shaffer, D.R.; Yi, Z.; Rainusso, N.; Wu, M.-F.; Liu, H.; Kew, Y. T cells redirected to EphA2 for the immunotherapy of glioblastoma. Mol. Ther. 2013, 21, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, R.; Abe, T.; Yajima, N.; Tsuchiya, N.; Homma, J.; Kobayashi, T.; Narita, M.; Takahashi, M.; Tanaka, R. Vaccination of recurrent glioma patients with tumour lysate-pulsed dendritic cells elicits immune responses: Results of a clinical phase I/II trial. Br. J. Cancer 2003, 89, 1172–1179. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.S.; Wheeler, C.J.; Zeltzer, P.M.; Ying, H.; Finger, D.; Lee, P.; Yong, W.; Incardona, F.; Thompson, R.; Riedinger, M.; et al. Vaccination of malignant glioma patients with peptide-pulsed dendritic cells elicits systemic cytotoxicity and intracranial T cell infiltration. Cancer Res. 2001, 61, 842–847. [Google Scholar] [PubMed]
- Kikuchi, T.; Akasaki, Y.; Irie, M.; Homma, S.; Abe, T.; Ohno, T. Results of a phase I clinical trial of vaccination of glioma patients with fusions of dendritic and glioma cells. Cancer Immunol. Immunother. 2001, 50, 337–344. [Google Scholar] [CrossRef]
- Wu, A.-h.; Xiao, J.; Anker, L.; Hall, W.A.; Gregerson, D.S.; Cavenee, W.K.; Chen, W.; Low, W.C. Identification of EGFRvIII-derived CTL epitopes restricted by HLA A0201 for dendritic cell based immunotherapy of gliomas. J. Neurooncol. 2006, 76, 23–30. [Google Scholar] [CrossRef]
- Morford, L.A.; Dix, A.R.; Brooks, W.H.; Roszman, T.L. Apoptotic elimination of peripheral T lymphocytes in patients with primary intracranial tumors. J. Neurosurg. 1999, 91, 935–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peace, D.J.; Chen, W.; Nelson, H.; Cheever, M. T cell recognition of transforming proteins encoded by mutated ras proto-oncogenes. J. Immunol. 1991, 146, 2059–2065. [Google Scholar] [CrossRef]
- Trolle, T.; McMurtrey, C.P.; Sidney, J.; Bardet, W.; Osborn, S.C.; Kaever, T.; Sette, A.; Hildebrand, W.H.; Nielsen, M.; Peters, B. The length distribution of class I–restricted T cell epitopes is determined by both peptide supply and MHC allele–specific binding preference. J. Immunol. 2016, 196, 1480–1487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mani, J.; Wang, L.; Huckelhoven, A.G.; Schmitt, A.; Gedvilaite, A.; Jin, N.; Kleist, C.; Ho, A.D.; Schmitt, M. Definition and characterization of novel HLA-*A02-restricted CD8+ T cell epitopes derived from JCV polyomavirus with clinical relevance. Oncotarget 2017, 8, 2485–2500. [Google Scholar] [CrossRef] [Green Version]
- Lissina, A.; Briceno, O.; Afonso, G.; Larsen, M.; Gostick, E.; Price, D.A.; Mallone, R.; Appay, V. Priming of Qualitatively Superior Human Effector CD8(+) T Cells Using TLR8 Ligand Combined with FLT3 Ligand. J. Immunol. 2016, 196, 256–263. [Google Scholar] [CrossRef] [Green Version]
- Neuber, B.; Herth, I.; Tolliver, C.; Schoenland, S.; Hegenbart, U.; Hose, D.; Witzens-Harig, M.; Ho, A.D.; Goldschmidt, H.; Klein, B.; et al. Lenalidomide enhances antigen-specific activity and decreases CD45RA expression of T cells from patients with multiple myeloma. J. Immunol. 2011, 187, 1047–1056. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide Sequence | Position a | Length b | NetMHC-4.0 (nM) | Predicted Binding Level c | Number of Other Queried Algorithms That Predicted Binding d |
|---|---|---|---|---|---|
| FMACSPVAL | 26–34 | 9 | 6.02 | strong | 10 |
| EFMACSPVAL | 25–34 | 10 | 34.53 | strong | 1 |
| WLEANFSDLV | 429–438 | 10 | 79.27 | weak | 10 |
| FMACSPVALD | 26–35 | 10 | 117.95 | weak | 9 |
| SLYDEVRAGA | 292–301 | 10 | 155.74 | weak | 10 |
| LMFDLSLNF | 361–369 | 9 | 250.95 | weak | 9 |
| MIAGDWLEA | 424–432 | 9 | 258.82 | weak | 10 |
| LMFDLSLNFS | 361–370 | 10 | 261.92 | weak | 10 |
| KMLKDSEKI | 88–96 | 9 | 316.75 | weak | 10 |
| LLRRYNVAKV | 266–275 | 10 | 1469.73 | none | 10 |
| Positive control: ELAGIGILTV | MART-1 26–35 | 10 | 253.92 | weak | 10 |
| Peptide Sequence | ELISPOT a (long method) | ELISPOT a (short method) | ELISPOT a (short method + long method) | ICS (TNF-α) b (long method) | ICS (IFN-γ) c (long method) |
|---|---|---|---|---|---|
| ELAGIGILTV | 3:4 | 8:18 | 11:22 | 4:10 | 2:10 |
| FMACSPVAL | 6:14 | 3:18 | 9:32 | 1:10 | 1:10 |
| LLRRYNVAKV | 1:14 | 1:8 | 2:22 | - | - |
| LMFDLSLNF | 1:14 | 1:8 | 2:22 | - | - |
| SLYDEVRAGA | 2:4 | 1:8 | 3:12 | - | - |
| EFMACSPVAL | 0:3 | 1:8 | 1:11 | - | - |
| FMACSPVALD | 0:3 | 1:8 | 1:11 | - | - |
| MIAGDWLEA | 0:3 | 0:8 | 0:11 | - | - |
| WLEANFSDLV | 0:3 | 0:8 | 0:11 | - | - |
| KMLKDSEKI | 0:3 | 0:8 | 0:11 | - | - |
| LMFDLSLNFS | 0:3 | 0:8 | 0:11 | - | - |
| Peptide Sequence | Healthy Donor Sample (HD) | Unloaded T2 Cells | Peptide-Loaded T2 Cells | p-Value |
|---|---|---|---|---|
| LLRRYNVAKV | HD 1640 | 7.75 ± 2.00 | 115.25 ± 13.71 | 0.0001 |
| FMACSPVAL | HD 0340 | 7.60 ± 3.21 | 19.00 ± 6.40 | 0.0076 |
| FMACSPVAL | HD 5668 | 12.40 ± 5.21 | 95.20 ± 28.20 | 0.0002 |
| FMACSPVAL | HD 5883 | 8.67 ± 3.50 | 20.00 ± 7.00 | 0.0400 |
| FMACSPVAL | HD 5356 | 2.40 ± 2.90 | 22.00 ± 6.00 | 0.0002 |
| FMACSPVAL | HD 5898 | 9.00 ± 2.70 | 37.40 ± 7.80 | 0.0001 |
| FMACSPVAL | HD 1630 | 15.60 ± 8.00 | 75.60 ± 17.70 | 0.0001 |
| LMFDLSLNF | HD 5896 | 9.33 ± 4.00 | 21.33 ± 3.11 | 0.0148 |
| SLYDEVRAG | HD 9805 | 11.00 ± 1.20 | 51.80 ± 31.80 | 0.0210 |
| SLYDEVRAG | HD 9844 | 25.60 ± 9.21 | 59.60 ± 22.00 | 0.0128 |
| Position Start | Region | Length | Sequence | NetMHCpan 4.1 (EL %rank) | NetMHCpan 4.1 (nM) |
|---|---|---|---|---|---|
| 26 | 26–34 | 9 | FMACSPVAL | 0.309 | 4.45 |
| 25 | 25–34 | 10 | EFMACSPVAL | 7.239 | 152.99 |
| 429 | 429–438 | 10 | WLEANFSDLV | 4.360 | 48.37 |
| 26 | 26–35 | 10 | FMACSPVALD | 7.021 | 166.12 |
| 292 | 292–301 | 10 | SLYDEVRAGA | 0.342 | 91.27 |
| 361 | 361–369 | 9 | LMFDLSLNF | 1.230 | 156.17 |
| 424 | 424–432 | 9 | MIAGDWLEA | 2.829 | 310.33 |
| 361 | 361–370 | 10 | LMFDLSLNFS | 6.528 | 158.03 |
| 88 | 88–96 | 9 | KMLKDSEKI | 0.471 | 234.52 |
| 266 | 266–275 | 10 | LLRRYNVAKV | 12.828 | 2439.73 |
| Short Method | Long Method | |
|---|---|---|
| Cells | Cryopreserved PBMCs | Fresh PBMCs |
| Cell number | 6 × 107 | 4 × 108 |
| Incubation time | 12 days | 21 days |
| Stimulation | One-week stimulation | Two-week stimulation |
| Activation conditions | DCs and PBMCs are cultured together | DCs are matured separately |
| Cytokine cocktail 1 | 800 U/mL human GM-CSF, 500 U/mL IL-4 | 2 mM L-glutamine, 5% human serum, 800 U/mL human GM-CSF, 500 U/mL IL-4 |
| Cytokine cocktail 2 | 10 ng/mL TNF-α, 1 μg/mL PGE2, 1000 U/mL IL-1ß, and 10 ng/mL IL-7 | 10 ng/mL TNF-α, 1 μg/mL PGE2, 1000 U/mL IL-6, and 1000 U/mL IL-1ß |
| Incubation medium of peptide-specific T cells | RPMI1640, 10% FBS, 5 ng/mL IL-7, 25 ng/mL IL-15, 100 U/mL IL-2 | RPMI 1640, 2 mM L-glutamine, 5% human serum, 10 ng/mL IL-7, 50 U/mL IL-2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Keib, A.; Neuber, B.; Wang, L.; Riemer, A.B.; Bonsack, M.; Hückelhoven-Krauss, A.; Schmitt, A.; Müller-Tidow, C.; Schmitt, M. Definition and Characterization of SOX11-Derived T Cell Epitopes towards Immunotherapy of Glioma. Int. J. Mol. Sci. 2023, 24, 1943. https://doi.org/10.3390/ijms24031943
Liu Y, Keib A, Neuber B, Wang L, Riemer AB, Bonsack M, Hückelhoven-Krauss A, Schmitt A, Müller-Tidow C, Schmitt M. Definition and Characterization of SOX11-Derived T Cell Epitopes towards Immunotherapy of Glioma. International Journal of Molecular Sciences. 2023; 24(3):1943. https://doi.org/10.3390/ijms24031943
Chicago/Turabian StyleLiu, Yibin, Anna Keib, Brigitte Neuber, Lei Wang, Angelika B. Riemer, Maria Bonsack, Angela Hückelhoven-Krauss, Anita Schmitt, Carsten Müller-Tidow, and Michael Schmitt. 2023. "Definition and Characterization of SOX11-Derived T Cell Epitopes towards Immunotherapy of Glioma" International Journal of Molecular Sciences 24, no. 3: 1943. https://doi.org/10.3390/ijms24031943
APA StyleLiu, Y., Keib, A., Neuber, B., Wang, L., Riemer, A. B., Bonsack, M., Hückelhoven-Krauss, A., Schmitt, A., Müller-Tidow, C., & Schmitt, M. (2023). Definition and Characterization of SOX11-Derived T Cell Epitopes towards Immunotherapy of Glioma. International Journal of Molecular Sciences, 24(3), 1943. https://doi.org/10.3390/ijms24031943