
c-Abl Tyrosine Kinase Is Required for BDNF-Induced Dendritic Branching and Growth


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. c-Abl Is Required for BDNF-Induced Dendritic Arborization
2.2. BDNF Increases c-Abl Activation and the Interaction of TrkB with c-Abl
2.3. BDNF-Induced c-Abl Activation Requires TrkB Activity but Does Not Involve the TrkB Downstream Signaling Pathways Regulated by PI3K, ERK1/2, and PLC-γ Signaling
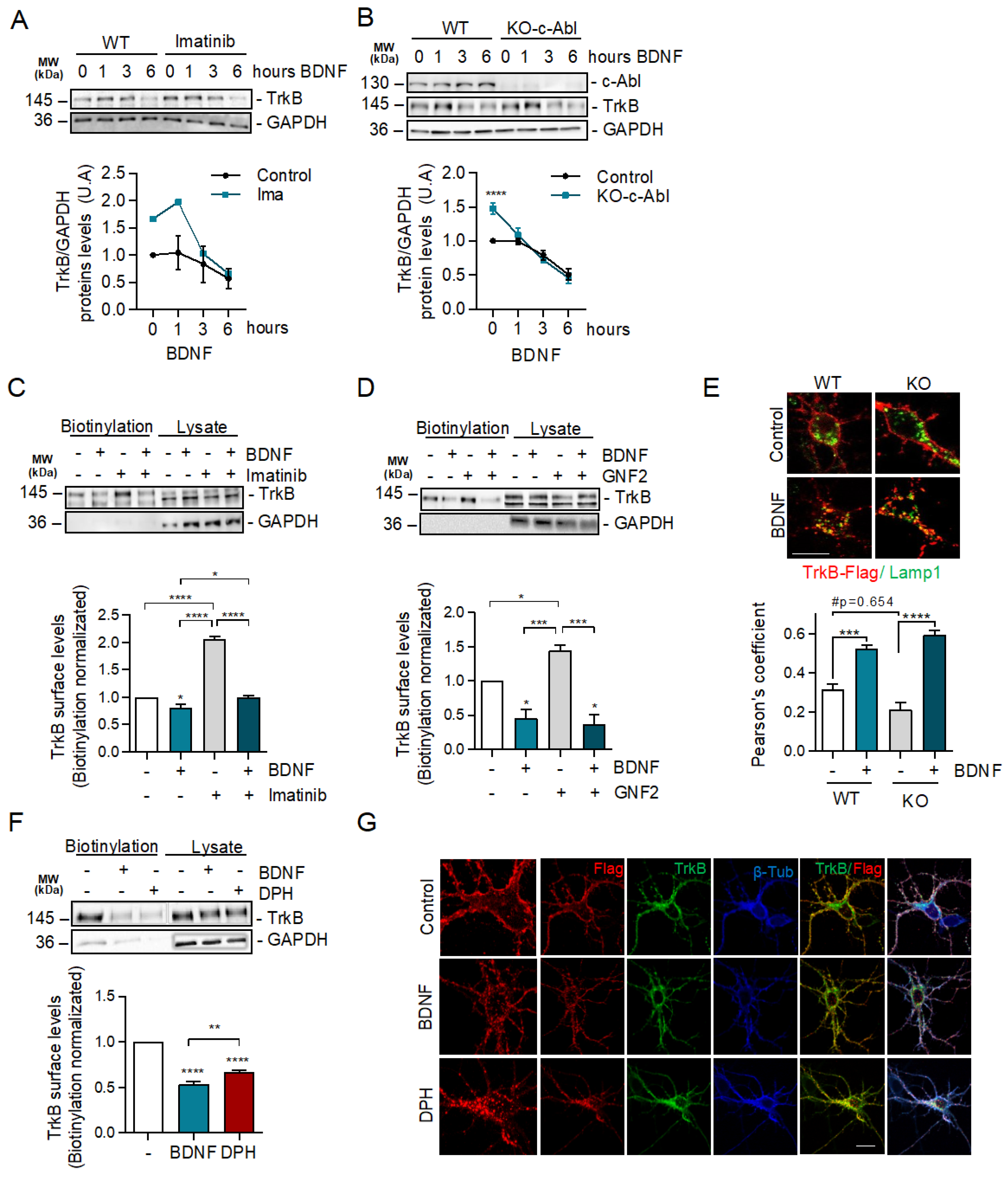
2.4. c-Abl Restricts Basal Levels of TrkB and Downregulates Surface TrkB Levels but Does Not Affect BDNF-Induced Endocytosis
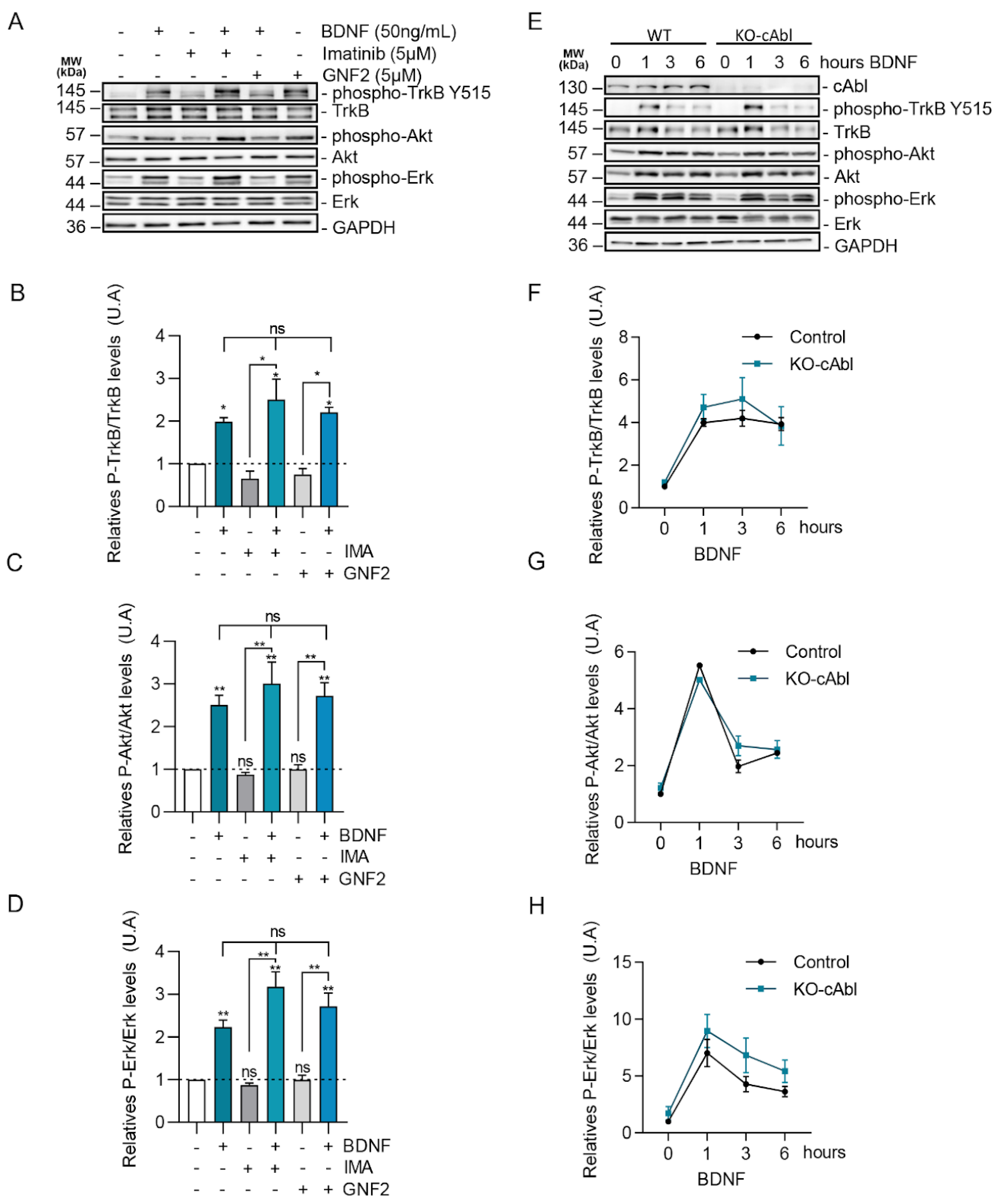
2.5. BDNF Downstream Signaling Is Not Affected by c-Abl Activity
2.6. c-Abl Increases the Retrograde Movement of TrkB Vesicle in Neurites
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Animals
- Abl1-flox forward: 5′-CCT GGC CTC CAA GAG CAC-3′
- Abl1-flox reverse: 5′-AGC CCC AGG GCA TAG ATA GT-3′
- Nes-Cre forward: 5′-TTG CTA AAG CGC TAC ATA GGA-3′ (wild type form)
- Nes-Cre reverse: 5′-GCC TTA TTG TTG AAG GAC TG-3′
- Nes-Cre forward: 5′-CCT TCC TGA AGC AGT AGA GCA-3′ (mutant form)
- Nes-Cre reverse: 5′-GCC TTA TTG TTG AAG GAC TG-3′
- Ntrk2 forward: 5′-AGG GCA AAA GGG TTG CTC-3′ and
- Ntrk2 reverse: 5′-CCA GCA GAA CAC TCG ACT CA-3′
4.3. Antibodies
4.4. Primary Cultures of Hippocampal and Cortical Neurons
4.5. Neuronal Treatments
4.6. Immunofluorescence
4.7. Dendritic Arborization Analysis
4.8. Immunoprecipitation
4.9. Western Blotting
4.10. Live-Cell Imaging of mCherry-TrkB
4.11. Immunoendocytosis of Flag-TrkB
4.12. Surface Biotinylation Assay
4.13. MTT Assay
4.14. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wen, Q.; Chklovskii, D.B. A Cost-Benefit Analysis of Neuronal Morphology. J. Neurophysiol. 2008, 99, 2320–2328. [Google Scholar] [CrossRef] [Green Version]
- Yan, Q.; Rosenfeld, R.D.; Matheson, C.R.; Hawkins, N.; Lopez, O.T.; Bennett, L.; Welcher, A.A. Expression of brain-derived neurotrophic factor protein in the adult rat central nervous system. Neuroscience 1997, 78, 431–448. [Google Scholar] [CrossRef]
- Barde, Y.A.; Edgar, D.; Thoenen, H. Purification of a new neurotrophic factor from mammalian brain. EMBO J. 1982, 1, 549–553. [Google Scholar] [CrossRef]
- Jacobson, M.D.; Weil, M.; Raff, M.C. Programmed cell death in animal development. Cell 1997, 88, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Horch, H.W.; Katz, L.C. BDNF release from single cells elicits local dendritic growth in nearby neurons. Nat. Neurosci. 2002, 5, 1177–1184. [Google Scholar] [CrossRef]
- Tanaka, J.; Horiike, Y.; Matsuzaki, M.; Miyazaki, T.; Ellis-Davies, G.C.; Kasai, H. Protein synthesis and neurotrophin-dependent structural plasticity of single dendritic spines. Science 2008, 319, 1683–1687. [Google Scholar] [CrossRef] [Green Version]
- Rauskolb, S.; Zagrebelsky, M.; Dreznjak, A.; Deogracias, R.; Matsumoto, T.; Wiese, S.; Erne, B.; Sendtner, M.; Schaeren-Wiemers, N.; Korte, M.; et al. Global deprivation of brain-derived neurotrophic factor in the CNS reveals an area-specific requirement for dendritic growth. J. Neurosci. 2010, 30, 1739–1749. [Google Scholar] [CrossRef] [Green Version]
- González-Gutiérrez, A.; Lazo, O.M.; Bronfman, F.C. The Rab5-Rab11 Endosomal Pathway is Required for BDNF-Induced CREB Transcriptional Regulation in Hippocampal Neurons. J. Neurosci. 2020, 40, 8042–8054. [Google Scholar] [CrossRef]
- Haapasalo, A.; Sipola, I.; Larsson, K.; Akerman, K.E.; Stoilov, P.; Stamm, S.; Wong, G.; Castren, E. Regulation of TRKB surface expression by brain-derived neurotrophic factor and truncated TrkB isoforms. J. Biol. Chem. 2002, 277, 43160–43167. [Google Scholar] [CrossRef] [Green Version]
- Ji, Y.; Lu, Y.; Yang, F.; Shen, W.; Tang, T.; Feng, L.; Duan, S.; Lu, B. Acute and gradual increases in BDNF concentration elicit distinct signaling and functions in neurons. Nat. Neurosci. 2010, 13, 302–309. [Google Scholar] [CrossRef]
- Cosker, K.E.; Segal, R.A. Neuronal signaling through endocytosis. Cold Spring Harb. Perspect Biol. 2014, 6, a020669. [Google Scholar] [CrossRef] [PubMed]
- González, A.; Moya-Alvarado, G.; González-Billaut, C.; Bronfman, F.C. Cellular and molecular mechanisms regulating neuronal growth by brain-derived neurotrophic factors. Cytoskeleton 2016, 73, 612–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galic, M.; Tsai, F.C.; Collins, S.R.; Matis, M.; Bandara, S.; Meyer, T. Dynamic recruitment of the curvature-sensitive protein ArhGAP44 to nanoscale membrane deformations limits exploratory filopodia initiation in neurons. Elife 2014, 3, e03116. [Google Scholar] [CrossRef]
- Sainath, R.; Gallo, G. Cytoskeletal and signaling mechanisms of neurite formation. Cell Tissue Res. 2015, 359, 267–278. [Google Scholar] [CrossRef] [Green Version]
- Hedrick, N.; Harward, S.; Hall, C.; Murakoshi, H.; McNamara, J.; Yasuda, R. Rho GTPase complementation underlies BDNF-dependent homo- and heterosynaptic plasticity. Nature 2016, 538, 104–108. [Google Scholar] [CrossRef] [Green Version]
- Cheung, Z.H.; Chin, W.H.; Chen, Y.; Ng, Y.P.; Ip, N.Y. CDK5 is involved in BDNF-stimulated dendritic growth in hippocampal neurons. PLoS Biol. 2007, 5, e63. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, Y.; Yamauchi, J.; Tanoue, A.; Wu, C.; Mobley, W.C. TrkB binds and tyrosine-phosphorylates Tiam1, leading to activation of Rac1 and induction of changes in cellular morphology. Proc. Natl. Acad. Sci. USA 2006, 103, 10444–10449. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.Y. The capable ABL: What is its biological function? Mol. Cell Biol. 2014, 34, 1188–1197. [Google Scholar] [CrossRef] [Green Version]
- Sirvent, A.; Benistant, C.; Roche, S. Cytoplasmic signalling by the c-Abl tyrosine kinase in normal and cancer cells. Biol. Cell 2008, 100, 617–631. [Google Scholar] [CrossRef] [Green Version]
- Tanos, B.; Pendergast, A.M. Abl tyrosine kinase regulate endocytosis of the epidermal growth factor receptor. J. Biol. Chem. 2006, 281, 32714–32723. [Google Scholar] [CrossRef]
- Yu, H.; Zisch, A.; Dodelet, V.; Pasquale, E. Multiple signaling interactions of Abl and Arg kinases with the EphB2 receptor. Oncogene 2001, 20, 3995–4006. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, D.; Kaetzel, D.M.; Plattner, R. Reciprocal regulation of Abl and receptor tyrosine kinases. Cell Signal. 2009, 21, 1143–1150. [Google Scholar] [CrossRef] [Green Version]
- Escalante, M.; Courtney, J.; Chin, W.G.; Teng, K.K.; Kim, J.I.; Fajardo, J.E.; Mayer, B.J.; Hempstead, B.L.; Birge, R.B. Phosphorylation of c-Crk II on the negative regulatory Tyr222 mediates nerve growth factor-induced cell spreading and morphogenesis. J. Biol. Chem. 2000, 275, 24787–24797. [Google Scholar] [CrossRef] [Green Version]
- Woodring, P.J.; Hunter, T.; Wang, J.Y. Regulation of F-actin-dependent processes by the Abl family of tyrosine kinases. J. Cell Sci. 2003, 116, 2613–2626. [Google Scholar] [CrossRef] [Green Version]
- Koleske, A.; Gifford, A.; Scott, M.; Nee, M.; Bronson, R.; Miczek, K.; Baltimore, D. Essential roles for the Abl and Arg tyrosine kinases in neurulation. Neuron 1998, 21, 1259–1272. [Google Scholar] [CrossRef] [Green Version]
- Lanier, L.M.; Gertler, F.B. From Abl to actin: Abl tyrosine kinase and associated proteins in growth cone motility. Curr. Opin. Neurobiol. 2000, 10, 80–87. [Google Scholar] [CrossRef]
- Zukerberg, L.R.; Patrick, G.N.; Nikolic, M.; Humbert, S.; Wu, C.L.; Lanier, L.M.; Gertler, F.B.; Vidal, M.; Van Etten, R.A.; Tsai, L.H. Cables links Cdk5 and c-Abl and facilitates Cdk5 tyrosine phosphorylation, kinase upregulation, and neurite outgrowth. Neuron 2000, 26, 633–646. [Google Scholar] [CrossRef] [Green Version]
- Lu, Q.; Mukhopadhyay, N.K.; Griffin, J.D.; Paredes, M.; Medina, M.; Kosik, K.S. Brain armadillo protein delta-catenin interacts with Abl tyrosine kinase and modulates cellular morphogenesis in response to growth factors. J. Neurosci. Res. 2002, 67, 618–624. [Google Scholar] [CrossRef]
- Moresco, E.M.; Koleske, A.J. Regulation of neuronal morphogenesis and synaptic function by Abl family kinases. Curr. Opin. Neurobiol. 2003, 13, 535–544. [Google Scholar] [CrossRef]
- Woodring, P.J.; Litwack, E.; O’Leary, D.; Lucero, G.; Wang, J.Y.J.; Hunter, T. Modulation of the F-actin cytoskeleton by c-Abl tyrosine kinase in cell spreading and neurite extension. J. Cell Biol. 2002, 156, 879–892. [Google Scholar] [CrossRef]
- Jones, S.B.; Lu, H.Y.; Lu, Q. Abl tyrosine kinase promotes dendrogenesis by inducing actin cytoskeletal rearrangements in cooperation with Rho family small GTPases in hippocampal neurons. J. Neurosci. 2004, 24, 8510–8521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wills, Z.; Emerson, M.; Rusch, J.; Bikoff, J.; Baum, B.; Perrimon, N.; Van Vactor, D. A Drosophila homolog of cyclase-associated proteins collaborate with the Abl tyrosine kinase to control midline axon pathfinding. Neuron 2002, 36, 611–622. [Google Scholar] [CrossRef] [Green Version]
- Tolwani, R.J.; Buckmaster, P.S.; Varma, S.; Cosgaya, J.M.; Wu, Y.; Suri, C.; Shooter, E.M. BDNF overexpression increases dendrite complexity in the hippocampal dentate gyrus. Neuroscience 2002, 114, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Takemoto-Kimura, S.; Ageta-Ishihara, N.; Nonaka, M.; Adachi-Morishima, A.; Mano, T.; Okamura, M.; Fujii, H.; Fuse, T.; Hoshino, M.; Suzuki, S.; et al. Regulation of dendritogenesis via a lipid-raft-associated Ca2+/calmodulin-dependent protein kinase CLICK III/CaMKIgamma. Neuron 2007, 54, 755–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutiérrez, D.A.; Vargas, L.M.; Chandía-Cristi, A.; De la Fuente, C.; Alvarez, A.R. c-Abl Deficiency Provides Synaptic Resiliency Against Aβ-Oligomers. Front. Cell Neurosci. 2019, 26, 526. [Google Scholar] [CrossRef] [Green Version]
- Sholl, D.A. Dendritic organization in the neurons of the visual and motor cortices of the cat. J. Anat. 1953, 87, 387–406. [Google Scholar]
- Lazo, O.M.; González, A.; Ascano, M.; Kuruvilla, R.; Couve, A.; Bronfman, F.C. BDNF regulates Rab11-mediated recycling endosome dynamics to induce dendritic branching. J. Neurosci. 2013, 33, 6112–6122. [Google Scholar] [CrossRef] [Green Version]
- Moya-Alvarado, G.; Gonzalez, A.; Stuardo, N.; Bronfman, F.C. Brain-Derived Neurotrophic Factor (BDNF) Regulates Rab5-Positive Early Endosomes in Hippocampal Neurons to Induce Dendritic Branching. Front. Cell Neurosci. 2018, 12, 493. [Google Scholar] [CrossRef]
- Yano, H.; Cong, F.; Birge, R.B.; Goff, S.P.; Chao, M.W. Association of the Abl tyrosine kinase with the Trk nerve growth factor receptor. J. Neurosci. Res. 2000, 59, 356–364. [Google Scholar] [CrossRef]
- Brasher, B.B.; Van Etten, R.A. c-Abl has high intrinsic tyrosine kinase activity that is stimulated by mutation of the Src homology 3 domain and by autophosphorylation at two distinct regulatory tyrosines. J. Biol. Chem. 2000, 275, 35631–35637. [Google Scholar] [CrossRef] [Green Version]
- Bradley, W.D.; Koleske, A.J. Regulation of cell migration and morphogenesis by Abl family kinases: Emerging mechanisms and physiological contexts. J. Cell Sci. 2009, 122, 3441–3454. [Google Scholar] [CrossRef] [PubMed]
- Arévalo, J.C.; Wu, S.H. Neurotrophin signaling: Many exciting surprises! Cell Mol. Life Sci. 2006, 63, 1523–1537. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Zhang, M.X.; Swank, M.W.; Kunz, J.; Wu, G.Y. Regulation of dendritic morphogenesis by Ras-PI3K-Akt-mTOR and Ras-MAPK signaling pathways. J. Neurosci. 2005, 25, 11288–11299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.H.; Wang, J.; Sui, W.H.; Chen, B.; Zhang, X.Y.; Yan, J.; Geng, Z.; Chen, Z.-Y. BDNF-dependent recycling facilitates TrkB translocation to postsynaptic density during LTP via a Rab11-dependent pathway. J Neurosci. 2013, 33, 9214–9230. [Google Scholar] [CrossRef] [Green Version]
- Song, M.; Giza, J.; Proenca, C.C.; Jing, D.; Elliott, M.; Dincheva, I.; Shmelkov, S.V.; Kim, J.; Schreiner, R.; Huang, S.-H.; et al. Slitrk5 mediates BDNF-dependent TrkB receptor trafficking and signaling. Dev. Cell 2015, 33, 690–702. [Google Scholar] [CrossRef] [Green Version]
- Sui, W.H.; Huang, S.H.; Wang, J.; Chen, Q.; Liu, T.; Chen, Z.Y. Myosin Va mediates BDNF-induced postendocytic recycling of full-length TrkB and its translocation into dendritic spines. J. Cell Sci. 2015, 128, 1108–1122. [Google Scholar] [CrossRef] [Green Version]
- Jan, Y.; Jan, L.Y. Branching out: Mechanisms of dendritic arborization. Nat. Rev. Neurosci. 2010, 11, 316–328. [Google Scholar] [CrossRef]
- Koch, A.; Mancini, A.; Stefan, M.; Niedenthal, R.; Niemann, H.; Tamura, T. Direct interaction of nerve growth factor receptor, TrkA, with non-receptor tyrosine kinase, c-Abl, through the activation loop. FEBS Lett. 2000, 3, 72–76. [Google Scholar] [CrossRef] [Green Version]
- Matsuki, T.; Pramatarova, A.; Howell, B.W. Reduction of Crk and CrkL expression blocks reelin-induced dendritogenesis. J. Cell Sci. 2008, 121, 1869–1875. [Google Scholar] [CrossRef] [Green Version]
- Cao, H.; Schroeder, B.; Chen, J.; Schott, M.B.; McNiven, M.A. The endocytic fate of the transferrin receptor is regulated by c-Abl kinase. J. Biol. Chem. 2016, 291, 16424–16437. [Google Scholar] [CrossRef] [Green Version]
- Jacob, M.; Todd, L.A.; Majumdar, R.S.; Li, Y.; Yamamoto, K.I.; Pure, E. Endogenous c-Abl regulates receptor endocytosis. Cell Signal. 2009, 21, 1308–1316. [Google Scholar] [CrossRef] [PubMed]
- Cancino, G.I.; Perez de Arce, K.; Castro, P.U.; Toledo, E.M.; von Bernhardi, R.; Alvarez, A.R. c-Abl tyrosine kinase modulates tau pathology and Cdk5 phosphorylation in AD transgenic mice. Neurobiol. Aging 2011, 32, 1249–1261. [Google Scholar] [CrossRef]
- Ferreira, A.P.A.; Casamento, A.; Carrillo Roas, S.; Halff, E.F.; Panambalana, J.; Subramaniam, S.; Schützenhofer, K.; Chan, W.; Hak, L.; McGourty, K.; et al. Cdk5 and GSK3β inhibit fast endophilin-mediated endocytosis. Nat. Commun. 2021, 12, 2424. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Fu, X.; Zhu, S.; Liu, J.J. Retrolinkin recruits the WAVE1 protein complex to facilitate BDNF-induced TrkB endocytosis and dendrite outgrowth. Mol. Biol. Cell 2016, 27, 3342–3356. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, D.J.; Blasier, K.R.; Jeffery, E.D.; Ross, M.W.; Pullikuth, A.K.; Suo, D.; Park, J.; Smiley, W.R.; Lo, K.W.; Shabanowitz, J.; et al. Trk activation of the ERK1/2 kinase pathway stimulates intermediate chain phosphorylation and recruits cytoplasmic dynein to signaling endosomes for retrograde axonal transport. J. Neurosci. 2012, 32, 15495–15510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assaife-Lopes, N.; Sousa, V.C.; Pereira, D.B.; Ribeiro, J.A.; Sebastião, A.M. Regulation of TrkB receptor translocation to lipid rafts by adenosine A(2A) receptors and its functional implications for BDNF-induced regulation of synaptic plasticity. Purinergic Signal. 2014, 10, 251–267. [Google Scholar] [CrossRef] [Green Version]
- Bronfman, F.C.; Lazo, O.M.; Flores, C.; Escudero, C.A. Spatiotemporal intracellular dynamics of neurotrophin and its receptors. Implications for neurotrophin signaling and neuronal function. Handb. Exp. Pharmacol. 2014, 220, 33–65. [Google Scholar] [CrossRef]
- Cohen, M.S.; Orth, C.B.; Kim, H.J.; Leon, N.L.; Jaffrey, S.R. Neurotrophin-mediated dendrite-to-nucleus signaling revealed by microfluidic compartmentalization of dendrites. Proc. Natl. Acad. Sci. USA 2011, 108, 11246–11251. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, Q.; Lizarraga, S.F.; Schmidt, M.; Yang, U.; Gong, Y.; Ellisor, D.; Kauer, J.A.; Morrow, E.M. Christianson syndrome protein NHE6 modulates TrkB endosomal signaling required for neuronal circuit development. Neuron 2013, 80, 97–112. [Google Scholar] [CrossRef] [Green Version]
- Peebles, C.L.; Yoo, J.; Thwin, M.T.; Palop, J.J.; Noebels, J.L.; Finkbeiner, S. Arc regulates spine morphology and maintains network stability in vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 18173–18178. [Google Scholar] [CrossRef] [Green Version]
- Morfini, G.; Szebenyi, G.; Brown, H.; Pant, H.C.; Pigino, G.; DeBoer, S.; Beffert, U.; Brady, S.T. A novel CDK5-dependent pathway for regulating GSK3 activity and kinesin-driven motility in neurons. EMBO J. 2004, 23, 2235–2245. [Google Scholar] [CrossRef] [PubMed]
- Kahn, O.I.; Sharma, V.; González-Billault, C.; Baas, P.W. Effects of kinesin-5 inhibition on dendritic architecture and microtubule organization. Mol. Biol. Cell 2015, 26, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, T.; Hirokawa, N. Microtubule Destabilizer KIF2A Undergoes Distinct Site-Specific Phosphorylation Cascades that Differentially Affect Neuronal Morphogenesis. Cell Rep. 2015, 12, 1774–1788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klinman, E.; Holzbaur, E.L. Stress-Induced CDK5 Activation Disrupts Axonal Transport via Lis1/Ndel1/Dynein. Cell Rep. 2015, 12, 462–473. [Google Scholar] [CrossRef] [Green Version]
- Niethammer, M.; Smith, D.S.; Ayala, R.; Peng, J.; Ko, J.; Lee, M.S.; Morabito, M.; Tsai, L.H. NUDEL is a novel Cdk5 substrate that associates with LIS1 and cytoplasmic dynein. Neuron 2000, 28, 697–711. [Google Scholar] [CrossRef] [Green Version]
- Yau, K.W.; Schätzle, P.; Tortosa, E.; Pagès, S.; Holtmaat, A.; Kapitein, L.C.; Hoogenraad, C.C. Dendrites In Vitro and In Vivo Contain Microtubules of Opposite Polarity and Axon Formation Correlates with Uniform Plus-End-Out Microtubule Orientation. J. Neurosci. 2016, 36, 1071–1085. [Google Scholar] [CrossRef] [Green Version]
- Fass, J.; Gehler, S.; Sarmiere, P.; Letourneau, P.; Bamburg, J.R. Regulating filopodial dynamics through actin-depolymerizing factor/cofilin. Anat. Sci. Int. 2004, 79, 173–183. [Google Scholar] [CrossRef]
- Gehler, S.; Gallo, G.; Veien, E.; Letourneau, P.C. p75 neurotrophin receptor signaling regulates growth cone filopodial dynamics through modulating RhoA activity. J. Neurosci. 2004, 24, 4363–4372. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.B.; Jin, M.; Xu, X.; Song, Y.Q.; Wu, C.P.; Poo, M.M.; Duan, S. Signalling and crosstalk of Rho GTPases in mediating axon guidance. Nat. Cell Biol. 2003, 5, 38–45. [Google Scholar] [CrossRef]
- Chen, T.J.; Gehler, S.; Shaw, A.E.; Bamburg, J.R.; Letourneau, P.C. Cdc42 participates in the regulation of ADF/cofilin and retinal growth cone filopodia by brain derived neurotrophic factor. J. Neurobiol. 2006, 66, 103–114. [Google Scholar] [CrossRef]
- Lin, M.Y.; Lin, Y.M.; Kao, T.C.; Chuang, H.H.; Chen, R.H. PDZ-RhoGEF ubiquitination by Cullin3-KLHL20 controls neurotrophin-induced neurite outgrowth. J. Cell Biol. 2011, 193, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Harbott, L.K.; Nobes, C.D. A key role for Abl family kinases in EphA receptor-mediated growth cone collapse. Mol. Cell Neurosci. 2005, 30, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zandy, N.L.; Playford, M.; Pendergast, A.M. Abl tyrosine kinases regulate cell-cell adhesion through Rho GTPases. Proc. Natl. Acad. Sci. USA 2007, 104, 17686–17691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodring, P.J.; Meisenhelder, J.; Johnson, S.A.; Zhou, G.L.; Field, J.; Shah, K.; Bladt, F.; Pawson, T.; Niki, M.; Pandolfi, P.P.; et al. c-Abl phosphorylates Dok1 to promote filopodia during cell spreading. J. Cell Biol. 2004, 165, 493–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sossey-Alaoui, K.; Li, X.; Cowell, J.K. c-Abl-mediated phosphorylation of WAVE3 is required for lamellipodia formation and cell migration. J. Biol. Chem. 2007, 282, 26257–26265. [Google Scholar] [CrossRef] [Green Version]
- Lai, K.O.; Wong, A.S.; Cheung, M.C.; Xu, P.; Liang, Z.; Lok, K.C.; Xie, H.; Palko, M.E.; Yung, W.H.; Tessarollo, L.; et al. TrkB phosphorylation by Cdk5 is required for activity-dependent structural plasticity and spatial memory. Nat. Neurosci. 2012, 15, 1506–1515. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ye, H.; Kuruvilla, R.; Ramanan, N.; Scangos, K.W.; Zhang, C.; Johnson, N.M.; England, P.M.; Shokat, K.M.; Ginty, D.D. A chemical-genetic approach to studying neurotrophin signaling. Neuron 2005, 46, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Kaech, S.; Banker, G. Culturing hippocampal neurons. Nat. Protoc. 2006, 1, 2406–2415. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chandía-Cristi, A.; Stuardo, N.; Trejos, C.; Leal, N.; Urrutia, D.; Bronfman, F.C.; Álvarez Rojas, A. c-Abl Tyrosine Kinase Is Required for BDNF-Induced Dendritic Branching and Growth. Int. J. Mol. Sci. 2023, 24, 1944. https://doi.org/10.3390/ijms24031944
Chandía-Cristi A, Stuardo N, Trejos C, Leal N, Urrutia D, Bronfman FC, Álvarez Rojas A. c-Abl Tyrosine Kinase Is Required for BDNF-Induced Dendritic Branching and Growth. International Journal of Molecular Sciences. 2023; 24(3):1944. https://doi.org/10.3390/ijms24031944
Chicago/Turabian StyleChandía-Cristi, América, Nicolás Stuardo, Cristian Trejos, Nancy Leal, Daniela Urrutia, Francisca C. Bronfman, and Alejandra Álvarez Rojas. 2023. "c-Abl Tyrosine Kinase Is Required for BDNF-Induced Dendritic Branching and Growth" International Journal of Molecular Sciences 24, no. 3: 1944. https://doi.org/10.3390/ijms24031944
APA StyleChandía-Cristi, A., Stuardo, N., Trejos, C., Leal, N., Urrutia, D., Bronfman, F. C., & Álvarez Rojas, A. (2023). c-Abl Tyrosine Kinase Is Required for BDNF-Induced Dendritic Branching and Growth. International Journal of Molecular Sciences, 24(3), 1944. https://doi.org/10.3390/ijms24031944