

Meta-Analysis and Multivariate GWAS Analyses in 80,950 Individuals of African Ancestry Identify Novel Variants Associated with Blood Pressure Traits

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Results
2.1. Results Overview
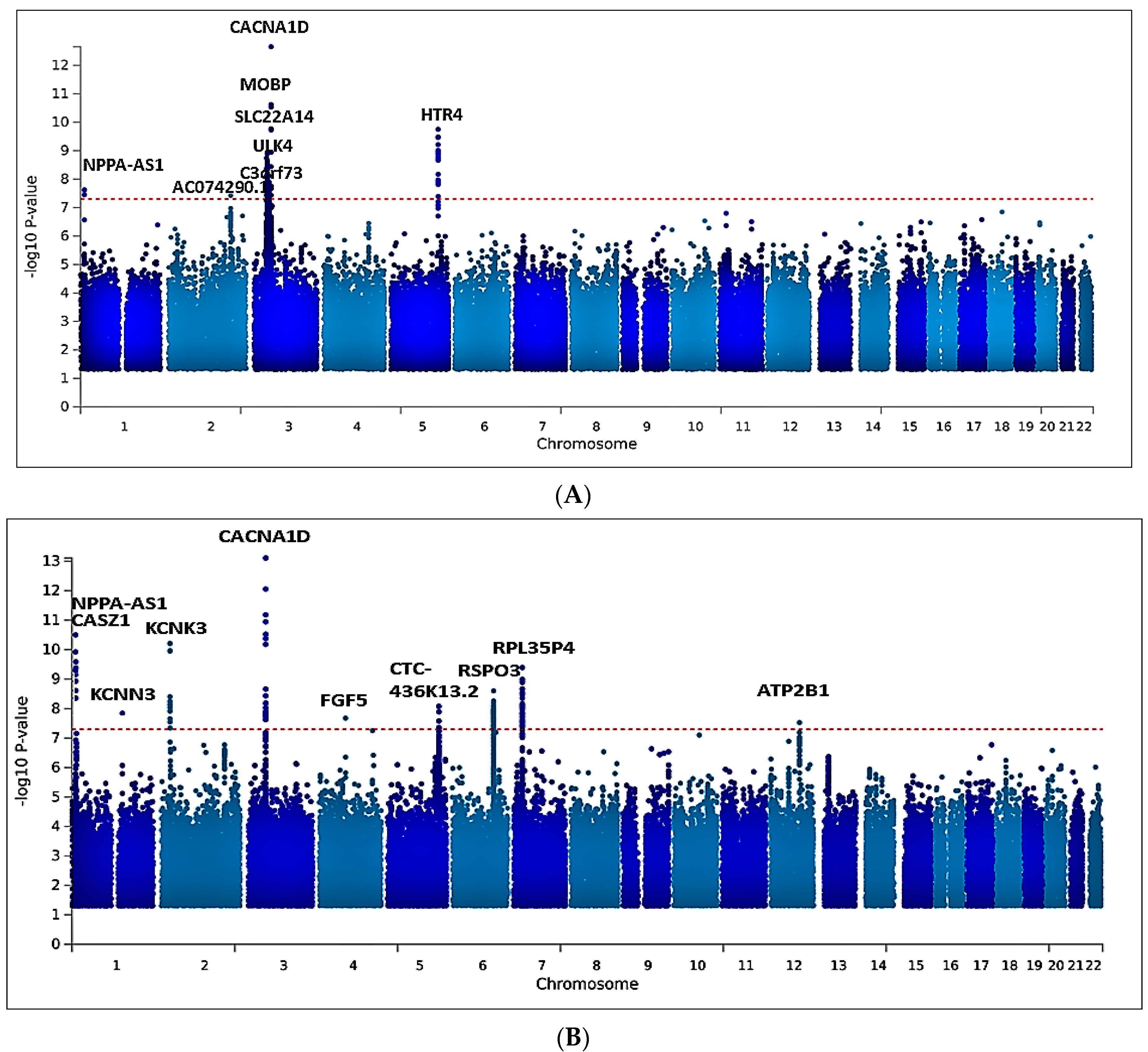
2.2. Univariate GWAS Meta-Analysis
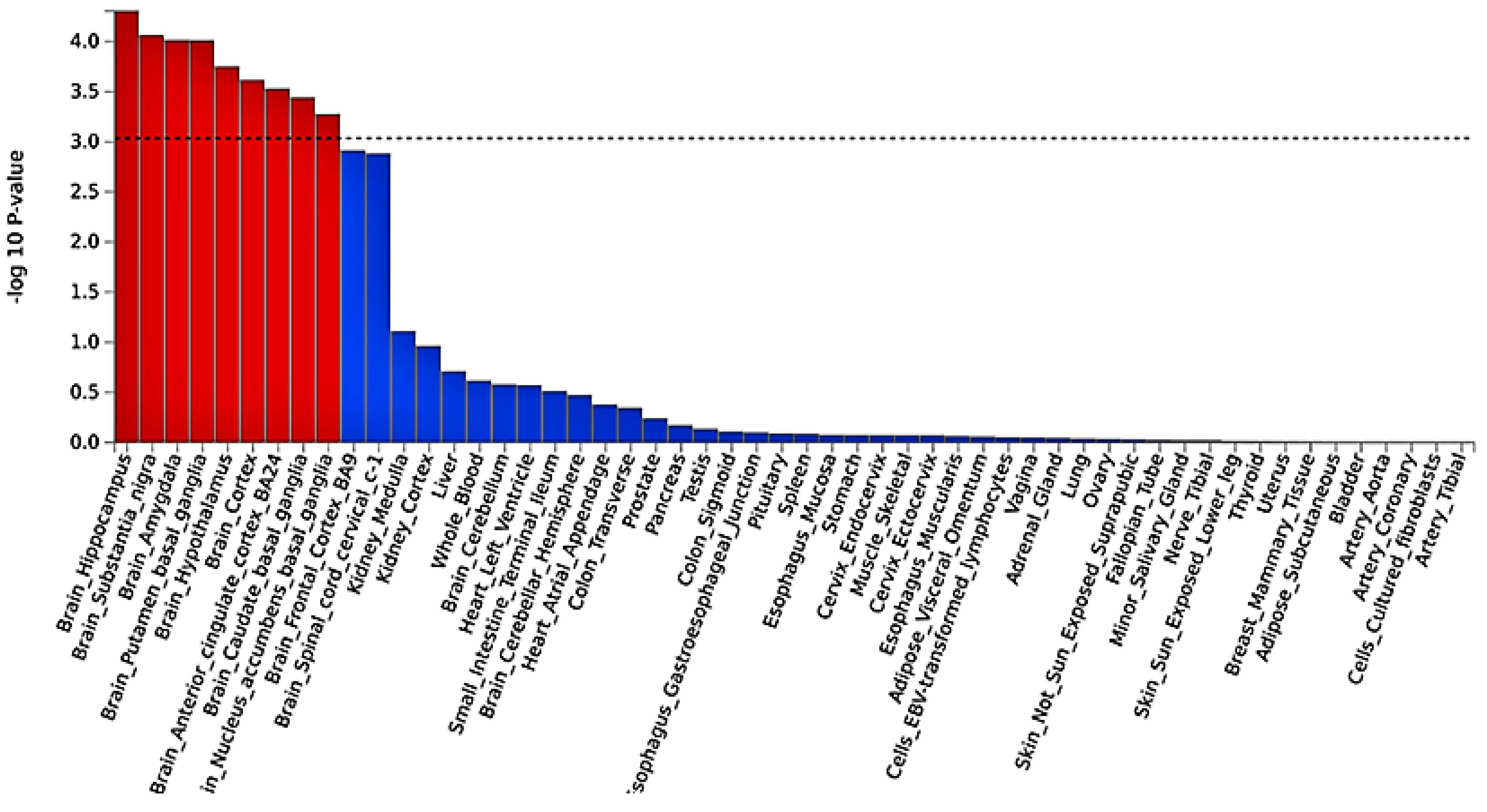
2.3. Functional Mapping and Annotation Analyses from FUMA of the Meta-Analysis
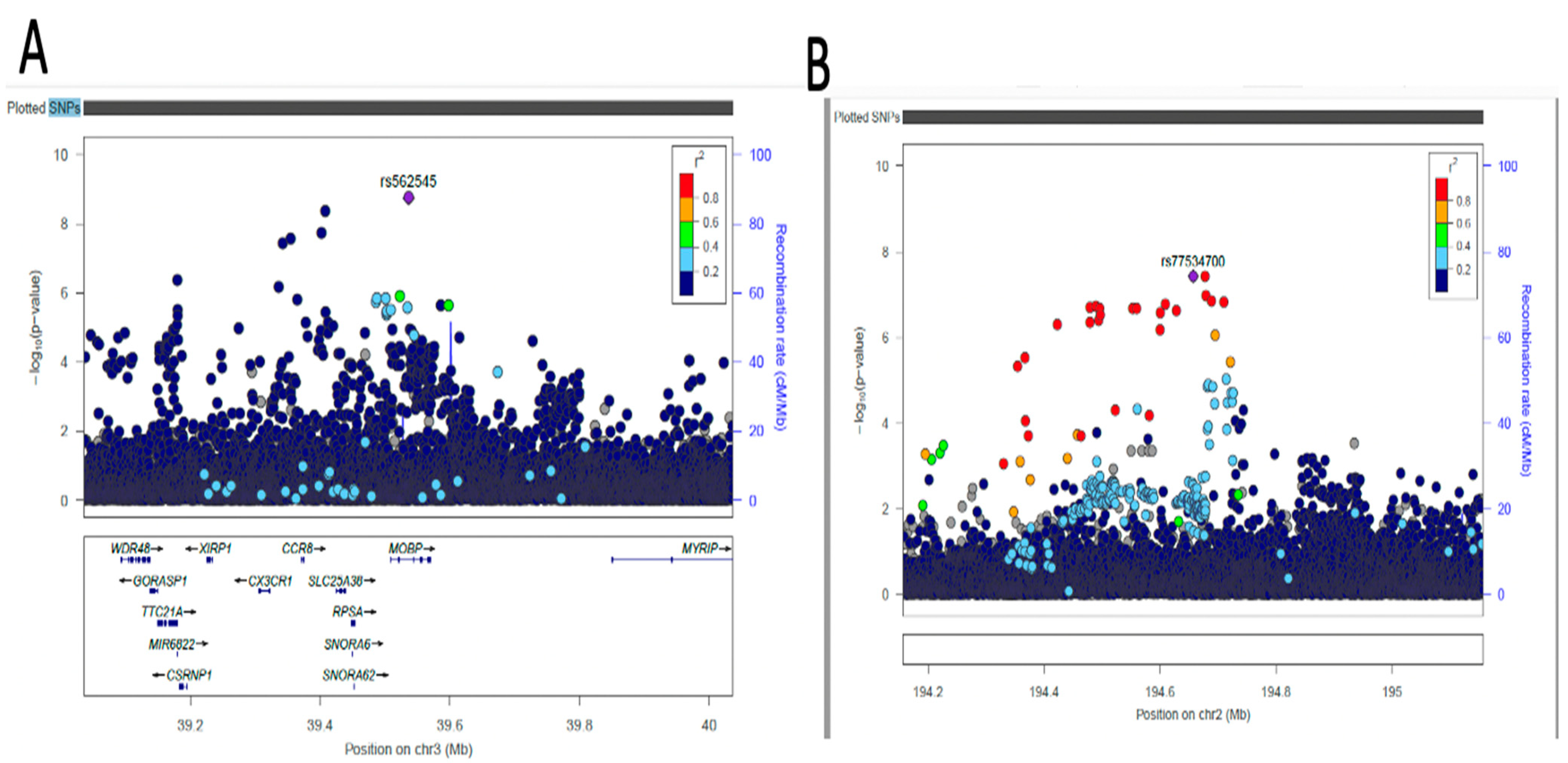
2.4. Fine-Mapping of Putatively Causal Variants
2.5. Multivariate GWAS Analysis of Blood Pressure Traits Identifies Additional Novel Loci
3. Discussion
4. Materials and Methods
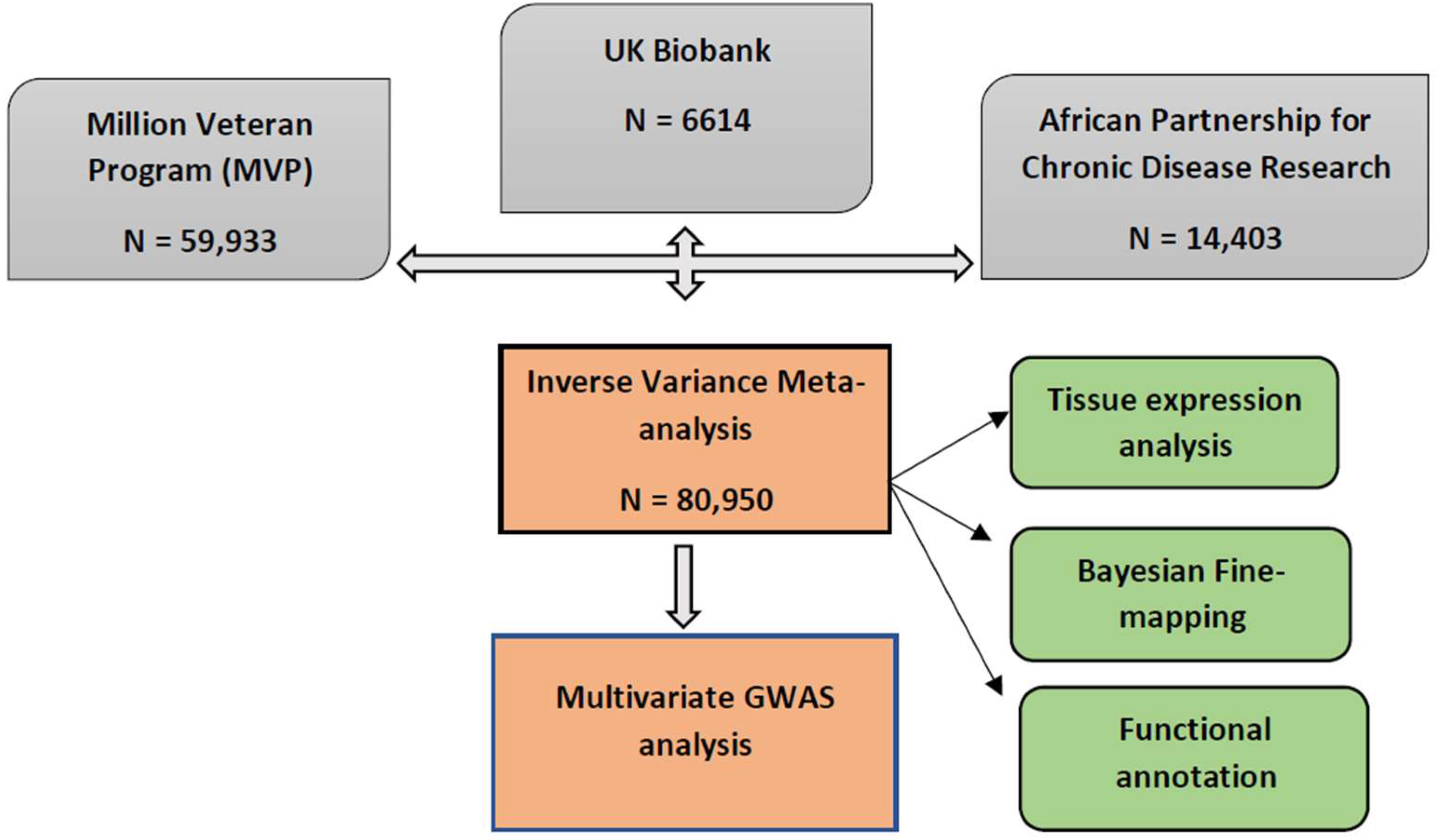
4.1. Study Population
4.2. Meta-Analysis of BP Summary Statistics in African-Ancestry Individuals
4.3. Tissue Expression Enrichment Pathway Analysis
4.4. Functional Mapping and Annotation Analysis
4.5. Locus Definition
4.6. Fine-Mapping Analysis of Sentinel Variants
4.7. Multivariate GWAS Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Muñoz, M.; Pong-Wong, R.; Canela-Xandri, O.; Rawlik, K.; Haley, C.S.; Tenesa, A. Evaluating the contribution of genetics and familial shared environment to common disease using the UK Biobank. Nat. Genet. 2016, 48, 980–983. [Google Scholar] [CrossRef] [PubMed]
- Feinleib, M.; Garrison, R.J.; Fabsitz, R.; Christian, J.C.; Hrubec, Z.; Borhani, N.O.; Kannel, W.B.; Rosenman, R.; Schwartz, J.T.; Wagner, J.O. The Nhlbi Twin Study of Cardiovascular Disease Risk Factors: Methodology and Summary of Results. Am. J. Epidemiol. 1977, 106, 284–295. [Google Scholar] [CrossRef]
- Poulter, N.R.; Prabhakaran, D.; Caulfield, M. Hypertension. Lancet 2015, 386, 801–812. [Google Scholar] [CrossRef]
- Levy, D.; Larson, M.G.; Benjamin, E.J.; Newton-Cheh, C.; Wang, T.J.; Hwang, S.-J.; Vasan, R.S.; Mitchell, G.F. Framingham Heart Study 100K Project: Genome-wide associations for blood pressure and arterial stiffness. BMC Med. Genet. 2007, 8 (Suppl. 1). [Google Scholar] [CrossRef] [PubMed]
- Allen, C.L.; Bayraktutan, U. Risk factors for ischaemic stroke. Int. J. Stroke Off. J. Int. Stroke Soc. 2008, 3, 105–116. [Google Scholar] [CrossRef]
- European Stroke Organisation (ESO). Executive Committee; ESO Writing Committee Guidelines for management of ischaemic stroke and transient ischaemic attack 2008. Cerebrovasc. Dis. Basel Switz. 2008, 25, 457–507. [Google Scholar] [CrossRef] [PubMed]
- NCD Risk Factor Collaboration (NCD-RisC) Worldwide trends in hypertension prevalence and progress in treatment and control from 1990 to 2019: A pooled analysis of 1201 population-representative studies with 104 million participants. Lancet Lond. Engl. 2021, 398, 957–980. [CrossRef] [PubMed]
- Carson, A.P.; Howard, G.; Burke, G.L.; Shea, S.; Levitan, E.B.; Muntner, P. Ethnic differences in hypertension incidence among middle-aged and older adults: The multi-ethnic study of atherosclerosis. Hypertens. Dallas Tex 1979 2011, 57, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Berenson, G.S.; Wattigney, W.A.; Webber, L.S. Epidemiology of hypertension from childhood to young adulthood in black, white, and Hispanic population samples. Public Health Rep. Wash. DC 1974 1996, 111 (Suppl. 2), 3–6. [Google Scholar]
- Writing Group Members; Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; Das, S.R.; de Ferranti, S.; Després, J.-P.; et al. Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation 2016, 133, e38–e360. [Google Scholar] [CrossRef]
- Chor, D.; Pinho Ribeiro, A.L.; Sá Carvalho, M.; Duncan, B.B.; Andrade Lotufo, P.; Araújo Nobre, A.; de Aquino, E.M.L.L.; Schmidt, M.I.; Griep, R.H.; Molina, M.D.C.B.; et al. Prevalence, Awareness, Treatment and Influence of Socioeconomic Variables on Control of High Blood Pressure: Results of the ELSA-Brasil Study. PloS One 2015, 10, e0127382. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.; Ehret, G.B.; Rice, K.; Verwoert, G.C.; Launer, L.J.; Dehghan, A.; Glazer, N.L.; Morrison, A.C.; Johnson, A.D.; Aspelund, T.; et al. Genome-wide association study of blood pressure and hypertension. Nat. Genet. 2009, 41, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Newton-Cheh, C.; Johnson, T.; Gateva, V.; Tobin, M.D.; Bochud, M.; Coin, L.; Najjar, S.S.; Zhao, J.H.; Heath, S.C.; Eyheramendy, S.; et al. Eight blood pressure loci identified by genome-wide association study of 34,433 people of European ancestry. Nat. Genet. 2009, 41, 666–676. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Brandenburg, J.T.; Choudhury, A.; Gómez-Olivé, F.X.; Ramsay, M. Systematic Review of Genomic Associations with Blood Pressure and Hypertension in Populations with African-Ancestry. Front Genet. 2021, 12, 699445. [Google Scholar] [CrossRef]
- Evangelou, E.; Warren, H.R.; Mosen-Ansorena, D.; Mifsud, B.; Pazoki, R.; Gao, H.; Ntritsos, G.; Dimou, N.; Cabrera, C.P.; Karaman, I.; et al. Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat. Genet. 2018, 50, 1412–1425. [Google Scholar] [CrossRef]
- Ehret, G.B.; Munroe, P.B.; Rice, K.M.; Bochud, M.; Johnson, A.D.; Chasman, D.I.; Smith, A.V.; Tobin, M.D.; Verwoert, G.C.; Hwang, S.-J.; et al. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature 2011, 478, 103–109. [Google Scholar] [CrossRef]
- Kato, N.; Takeuchi, F.; Tabara, Y.; Kelly, T.N.; Go, M.J.; Sim, X.; Tay, W.T.; Chen, C.-H.; Zhang, Y.; Yamamoto, K.; et al. Meta-analysis of genome-wide association studies identifies common variants associated with blood pressure variation in east Asians. Nat. Genet. 2011, 43, 531–538. [Google Scholar] [CrossRef]
- Warren, H.R.; Evangelou, E.; Cabrera, C.P.; Gao, H.; Ren, M.; Mifsud, B.; Ntalla, I.; Surendran, P.; Liu, C.; Cook, J.P.; et al. Genome-wide association analysis identifies novel blood pressure loci and offers biological insights into cardiovascular risk. Nat. Genet. 2017, 49, 403–415. [Google Scholar] [CrossRef]
- Adeyemo, A.; Gerry, N.; Chen, G.; Herbert, A.; Doumatey, A.; Huang, H.; Zhou, J.; Lashley, K.; Chen, Y.; Christman, M.; et al. A genome-wide association study of hypertension and blood pressure in African Americans. PLoS Genet. 2009, 5, e1000564. [Google Scholar] [CrossRef]
- Hoffmann, T.J.; Ehret, G.B.; Nandakumar, P.; Ranatunga, D.; Schaefer, C.; Kwok, P.Y.; Iribarren, C.; Chakravarti, A.; Risch, N. Genome-wide association analyses using electronic health records identify new loci influencing blood pressure variation. Nat. Genet. 2017, 49, 54–64. [Google Scholar] [CrossRef]
- Giri, A.; Hellwege, J.N.; Keaton, J.M.; Park, J.; Qiu, C.; Warren, H.R.; Torstenson, E.S.; Kovesdy, C.P.; Sun, Y.V.; Wilson, O.D.; et al. Trans-ethnic association study of blood pressure determinants in over 750,000 individuals. Nat. Genet. 2019, 51, 51–62. [Google Scholar] [CrossRef]
- Liang, J.; Le, T.H.; Edwards, D.R.V.; Tayo, B.O.; Gaulton, K.J.; Smith, J.A.; Lu, Y.; Jensen, R.A.; Chen, G.; Yanek, L.R.; et al. Single-trait and multi-trait genome-wide association analyses identify novel loci for blood pressure in African-ancestry populations. PLOS Genet. 2017, 13, e1006728. [Google Scholar] [CrossRef]
- Fox, E.R.; Young, J.H.; Li, Y.; Dreisbach, A.W.; Keating, B.J.; Musani, S.K.; Liu, K.; Morrison, A.C.; Ganesh, S.; Kutlar, A.; et al. Association of genetic variation with systolic and diastolic blood pressure among African Americans: The Candidate Gene Association Resource study. Hum. Mol. Genet. 2011, 20, 2273–2284. [Google Scholar] [CrossRef] [PubMed]
- Franceschini, N.; Fox, E.; Zhang, Z.; Edwards, T.; Nalls, M.A.; Sung, Y.; Tayo, B.; Sun, Y.; Gottesman, O.; Adeyemo, A.; et al. Genome-wide Association Analysis of Blood-Pressure Traits in African-Ancestry Individuals Reveals Common Associated Genes in African and Non-African Populations. Am. J. Hum. Genet. 2013, 93, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Hendry, L.M.; Sahibdeen, V.; Choudhury, A.; Norris, S.A.; Ramsay, M.; Lombard, Z. Insights into the genetics of blood pressure in black South African individuals: The Birth to Twenty cohort. BMC Med. Genom. 2018, 11, 2. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Kelly, T.N.; Zhao, Q.; Li, H.; Huang, J.; Wang, L.; Jaquish, C.E.; Sung, Y.J.; Shimmin, L.C.; Lu, F.; et al. Genome-wide association study identifies 8 novel loci associated with blood pressure responses to interventions in Han Chinese. Circ. Cardiovasc. Genet. 2013, 6, 598–607. [Google Scholar] [CrossRef]
- Fatumo, S.; Carstensen, T.; Nashiru, O.; Gurdasani, D.; Sandhu, M.; Kaleebu, P. Complimentary Methods for Multivariate Genome-Wide Association Study Identify New Susceptibility Genes for Blood Cell Traits. Front. Genet. 2019, 10. Available online: https://www.frontiersin.org/article/10.3389/fgene.2019.00334 (accessed on 2 June 2022). [CrossRef] [PubMed]
- Nudel, R.; Wang, Y.; Appadurai, V.; Schork, A.J.; Buil, A.; Agerbo, E.; Bybjerg-Grauholm, J.; Børglum, A.D.; Daly, M.J.; Mors, O.; et al. A large-scale genomic investigation of susceptibility to infection and its association with mental disorders in the Danish population. Transl. Psychiatry 2019, 9, 283. [Google Scholar] [CrossRef]
- Okbay, A.; Wu, Y.; Wang, N.; Jayashankar, H.; Bennett, M.; Nehzati, S.M.; Sidorenko, J.; Kweon, H.; Goldman, G.; Gjorgjieva, T.; et al. Polygenic prediction of educational attainment within and between families from genome-wide association analyses in 3 million individuals. Nat. Genet. 2022, 54, 437–449. [Google Scholar] [CrossRef]
- Fagerberg, L.; Hallström, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteom. MCP 2014, 13, 397–406. [Google Scholar] [CrossRef]
- Yang, B.; Wang, T.; Li, N.; Zhang, W.; Hu, Y. The High Expression of RRM2 Can Predict the Malignant Transformation of Endometriosis. Adv. Ther. 2021, 38, 5178–5190. [Google Scholar] [CrossRef]
- Sherva, R.; Tripodis, Y.; Bennett, D.A.; Chibnik, L.B.; Crane, P.K.; de Jager, P.L.; Farrer, L.A.; Saykin, A.J.; Shulman, J.M.; Naj, A.; et al. Genome-wide association study of the rate of cognitive decline in Alzheimer’s disease. Alzheimers Dement. J. Alzheimers Assoc. 2014, 10, 45–52. [Google Scholar] [CrossRef]
- Need, A.C.; Attix, D.K.; McEvoy, J.M.; Cirulli, E.T.; Linney, K.L.; Hunt, P.; Ge, D.; Heinzen, E.L.; Maia, J.M.; Shianna, K.V.; et al. A genome-wide study of common SNPs and CNVs in cognitive performance in the CANTAB. Hum. Mol. Genet. 2009, 18, 4650–4661. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Jose, P.A.; Zeng, C. Gastrointestinal–Renal Axis: Role in the Regulation of Blood Pressure. J. Am. Heart Assoc. 2017, 6, e005536. [Google Scholar] [CrossRef]
- Anand, K.S.; Dhikav, V. Hippocampus in health and disease: An overview. Ann. Indian Acad. Neurol. 2012, 15, 239–246. [Google Scholar] [CrossRef]
- Feng, R.; Rolls, E.T.; Cheng, W.; Feng, J. Hypertension is associated with reduced hippocampal connectivity and impaired memory. EBioMedicine 2020, 61, 103082. [Google Scholar] [CrossRef] [PubMed]
- Mägi, R.; Morris, A. GWAMA: Software for genome-wide association meta-analysis. BMC Bioinform. 2010, 11, 288. [Google Scholar] [CrossRef] [PubMed]
- de Leeuw, C.A.; Mooij, J.M.; Heskes, T.; Posthuma, D. MAGMA: Generalized Gene-Set Analysis of GWAS Data. PLoS Comput. Biol. 2015, 11, e1004219. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Taskesen, E.; Van Bochoven, A.; Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 2017, 8, 1826. [Google Scholar] [CrossRef] [PubMed]
- Westra, H.J.; Peters, M.J.; Esko, T.; Yaghootkar, H.; Schurmann, C.; Kettunen, J.; Christiansen, M.W.; Fairfax, B.P.; Schramm, K.; Powell, J.E.; et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet. 2013, 45, 1238–1243. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Kircher, M.; Witten, D.M.; Jain, P.; O’roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46. [Google Scholar] [CrossRef] [PubMed]
- 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef]
- Hutchinson, A.; Watson, H.; Wallace, C. Improving the coverage of credible sets in Bayesian genetic fine-mapping. PLOS Comput. Biol. 2020, 16, e1007829. [Google Scholar] [CrossRef]
- Li, X.; Zhu, X. Cross-Phenotype Association Analysis Using Summary Statistics from GWAS. Methods Mol. Biol. Clifton NJ 2017, 1666, 455–467. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cohort | Continent | Country | Sample Size (N) | Phenotype | Imputation Panel and Genome Build |
|---|---|---|---|---|---|
| APCDR-UGR (27) | Africa | Uganda | 6407 | DBP SBP | Africa genome panel, hg19 |
| APCDR-DCC (27) | Africa | South Africa | 1600 | DBP SBP | Africa genome panel, hg19 |
| APCDR-DDS (27) | Africa | South Africa | 1165 | DBP SBP | Africa genome panel, hg19 |
| APCDR-AADM (27) | Africa | Nigeria Ghana Kenya | 5231 | DBP SBP | Africa genome panel, hg19 |
| MVP–AFR | America | USA | 59,933 | DBP SBP | 1000 Genome, hg19 |
| UKB–AFR (28) | Europe | UK | 6614 | DBP SBP | 1000 Genome, hg19 |
| Nearest Gene | Lead SNPs | Chr | BP | Effect Allele | Other Allele | Trait | Beta | SE | MAF | p-Value | Functional Consequence |
|---|---|---|---|---|---|---|---|---|---|---|---|
| AC074290.1 | rs77534700 | 2 | 194657479 | A | G | DBP | −0.0967 | 0.0176 | 0.0836 | 3.749x10−8 | Intergenic variant |
| MOBP | rs562545 | 3 | 39536524 | A | G | DBP | 0.0593 | 0.0099 | 0.8973 | 1.823x10−9 | Intron variant |
| Nearest Gene | Lead SNPs | Chr | BP | Effect Allele | Other Allele | HET_p Value | Functional Consequence |
|---|---|---|---|---|---|---|---|
| DNAJC17P1/GLULP6 GLULP6GLULP6 GLULP6 | rs138493856 | 2 | 194678067 | A | G | 6.1322 × 10−9 | Intergenic variant |
| RRM2 | rs139235642 | 2 | 10278626 | T | C | 2.7981 × 10−8 | Intron variant NMD transcript variant |
| LOC105377644 | rs72619992 | 3 | 39407952 | A | C | 1.1339 × 10−8 | Intron variant |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Udosen, B.; Soremekun, O.; Kamiza, A.; Machipisa, T.; Cheickna, C.; Omotuyi, O.; Soliman, M.; Wélé, M.; Nashiru, O.; Chikowore, T.; et al. Meta-Analysis and Multivariate GWAS Analyses in 80,950 Individuals of African Ancestry Identify Novel Variants Associated with Blood Pressure Traits. Int. J. Mol. Sci. 2023, 24, 2164. https://doi.org/10.3390/ijms24032164
Udosen B, Soremekun O, Kamiza A, Machipisa T, Cheickna C, Omotuyi O, Soliman M, Wélé M, Nashiru O, Chikowore T, et al. Meta-Analysis and Multivariate GWAS Analyses in 80,950 Individuals of African Ancestry Identify Novel Variants Associated with Blood Pressure Traits. International Journal of Molecular Sciences. 2023; 24(3):2164. https://doi.org/10.3390/ijms24032164
Chicago/Turabian StyleUdosen, Brenda, Opeyemi Soremekun, Abram Kamiza, Tafadzwa Machipisa, Cisse Cheickna, Olaposi Omotuyi, Mahmoud Soliman, Mamadou Wélé, Oyekanmi Nashiru, Tinashe Chikowore, and et al. 2023. "Meta-Analysis and Multivariate GWAS Analyses in 80,950 Individuals of African Ancestry Identify Novel Variants Associated with Blood Pressure Traits" International Journal of Molecular Sciences 24, no. 3: 2164. https://doi.org/10.3390/ijms24032164
APA StyleUdosen, B., Soremekun, O., Kamiza, A., Machipisa, T., Cheickna, C., Omotuyi, O., Soliman, M., Wélé, M., Nashiru, O., Chikowore, T., & Fatumo, S. (2023). Meta-Analysis and Multivariate GWAS Analyses in 80,950 Individuals of African Ancestry Identify Novel Variants Associated with Blood Pressure Traits. International Journal of Molecular Sciences, 24(3), 2164. https://doi.org/10.3390/ijms24032164