
The Interplay between α-Synuclein and Microglia in α-Synucleinopathies




Abstract
:1. Introduction
2. Microglial Interaction with α-Syn Depends on its Structure
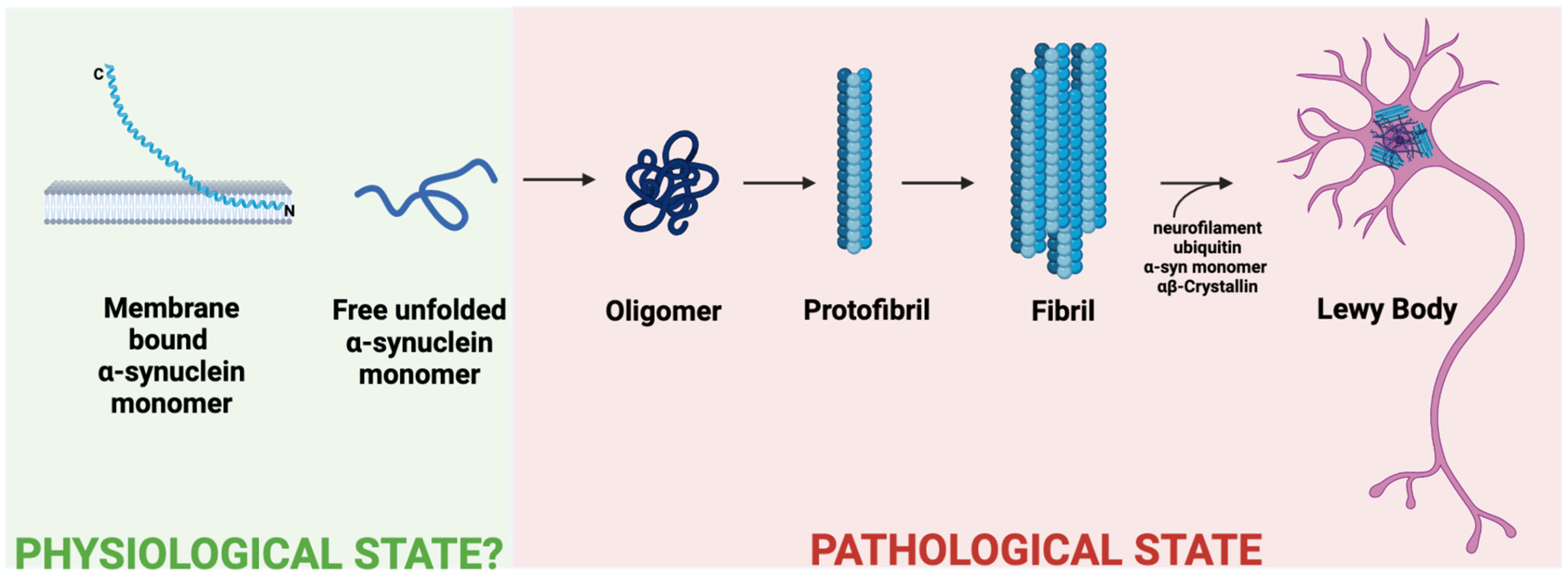
2.1. Monomeric, Oligomeric and Fibrillar Forms of α-Syn
2.2. Strains of α-Syn in Different Synucleinopathies
2.3. Mutant Forms of α-Syn
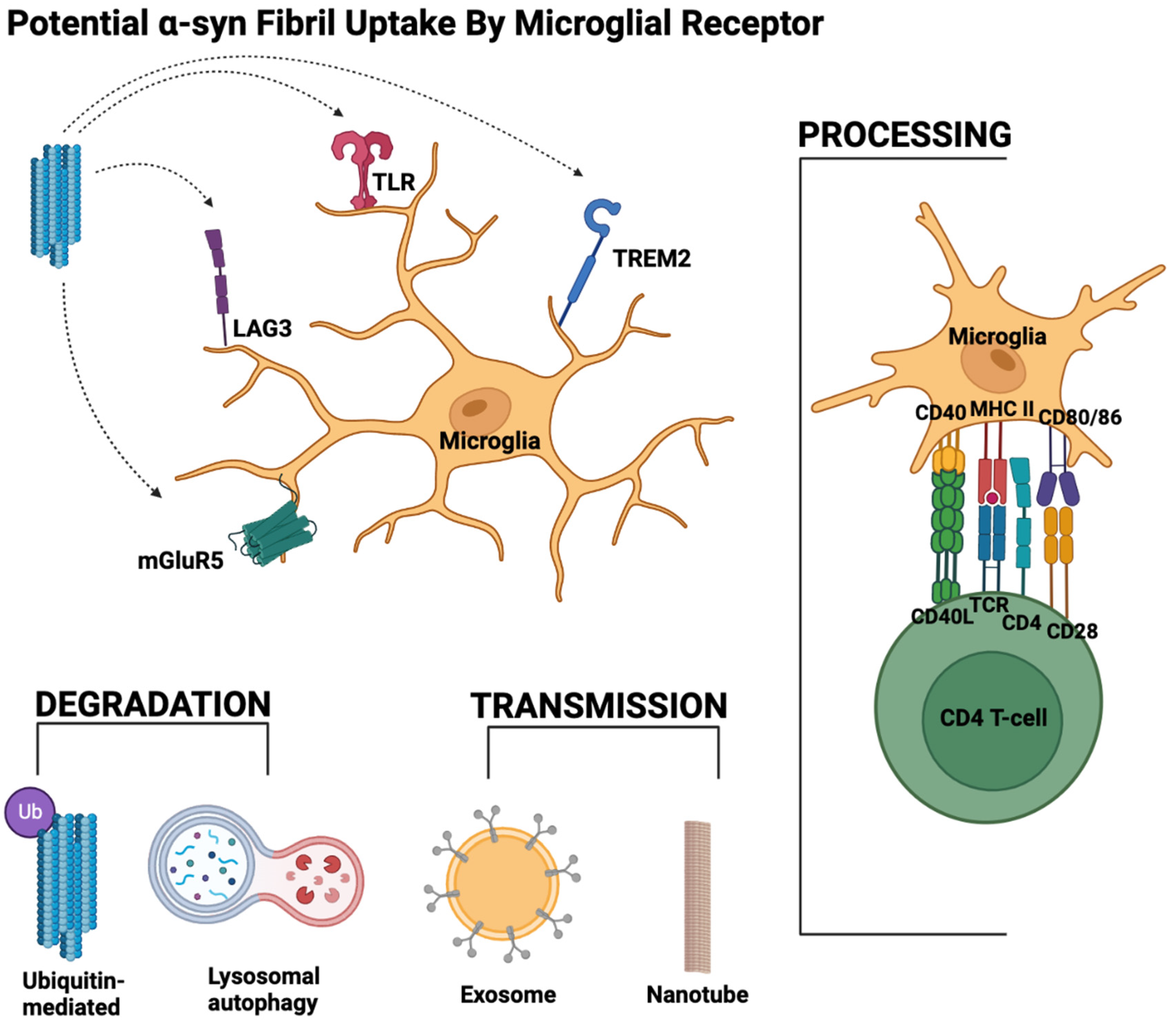
3. Various Microglial Receptors Interact with α-Syn
3.1. Toll-Like Receptors
3.2. Lymphocyte Activation Gene 3
3.3. Triggering Receptor Expressed on Myeloid Cells 2
3.4. Metabotropic Glutamate Receptor 5
3.5. Other α-Syn Receptors
4. α-Syn-Induced Inflammation in Microglia
4.1. Microglia Uptake of α-Syn
4.2. α-Synuclein Induces an Inflammatory Phenotype in Microglia
4.3. Involvement of the Adaptive Immune System-MHC Class II, B Cells and T Cells
5. Processing and Spread of α-Syn
5.1. Degradation of α-Syn Post-Microglial Uptake Linked to Microglial Autophagy
5.2. Secreted Proteins for α-Syn Degradation
5.3. Exosomal Transmission of α-Syn
5.4. α-Syn Transmission via Nanotubes
6. Discussion
7. Conclusions, Limitations, and Future Directions
8. Methods
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Koga, S.; Sekiya, H.; Kondru, N.; Ross, O.A.; Dickson, D.W. Neuropathology and Molecular Diagnosis of Synucleinopathies. Mol. Neurodegener. 2021, 16, 83. [Google Scholar] [CrossRef] [PubMed]
- Galvin, J.E.; Lee, V.M.; Trojanowski, J.Q. Synucleinopathies: Clinical and pathological implications. Arch. Neurol. 2001, 58, 186–190. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.-P.; McKeith, I.G.; Burn, D.J.; Boeve, B.F.; Weintraub, D.; Bamford, C.; Allan, L.M.; Thomas, A.J.; O’Brien, J.T. New evidence on the management of Lewy body dementia. Lancet Neurol. 2020, 19, 157–1699. [Google Scholar] [CrossRef] [PubMed]
- Sezgin, M.; Bilgic, B.; Tinaz, S.; Emre, M. Parkinson’s Disease Dementia and Lewy Body Disease. Semin. Neurol. 2019, 39, 274–282. [Google Scholar] [CrossRef]
- Erkkinen, M.G.; Kim, M.-O.; Geschwind, M.D. Clinical Neurology and Epidemiology of the Major Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2018, 10, a033118. [Google Scholar] [CrossRef] [Green Version]
- Chung, E.J.; Babulal, G.M.; Monsell, S.E.; Cairns, N.J.; Roe, C.M.; Morris, J.C. Clinical Features of Alzheimer Disease With and Without Lewy Bodies. JAMA Neurol. 2015, 72, 789–796. [Google Scholar] [CrossRef]
- Olichney, J.M.; Galasko, D.; Salmon, D.P.; Hofstetter, C.R.; Hansen, L.A.; Katzman, R.; Thal, L.J. Cognitive decline is faster in Lewy body variant than in Alzheimer’s disease. Neurology 1998, 51, 351–357. [Google Scholar] [CrossRef]
- Haider, A.; Spurling, B.C.; Sánchez-Manso, J.C. Lewy Body Dementia. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Ogbodo, J.O.; Agbo, C.P.; Njoku, U.O.; Ogugofor, M.O.; Egba, S.I.; Ihim, S.A.; Echezona, A.C.; Brendan, K.C.; Upaganlawar, A.B.; Upasani, C.D. Alzheimer’s Disease: Pathogenesis and Therapeutic Interventions. Curr. Aging Sci. 2022, 15, 2–25. [Google Scholar] [CrossRef]
- Twohig, D.; Nielsen, H.M. α-synuclein in the pathophysiology of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 23. [Google Scholar] [CrossRef] [Green Version]
- Tysnes, O.-B.; Storstein, A. Epidemiology of Parkinson’s disease. J. Neural Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef]
- Mayeux, R.; Stern, Y. Epidemiology of Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, D.K.; Tanner, C.M.; Brundin, P. Parkinson Disease Epidemiology, Pathology, Genetics, and Pathophysiology. Clin. Geriatr. Med. 2020, 36, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Henderson, M.X.; Trojanowski, J.Q.; Lee, V.M.-Y. α-Synuclein pathology in Parkinson’s disease and related α-synucleinopathies. Neurosci. Lett. 2019, 709, 134316. [Google Scholar] [CrossRef]
- Tarutani, A.; Hasegawa, M. Prion-like Propagation of α-Synuclein in Neurodegenerative Diseases. Prog. Mol. Biol. Transl. Sci. 2019, 168, 323–348. [Google Scholar] [CrossRef]
- Brás, I.C.; Outeiro, T.F. Alpha-Synuclein: Mechanisms of Release and Pathology Progression in Synucleinopathies. Cells 2021, 10, 375. [Google Scholar] [CrossRef]
- Outeiro, T.F.; Koss, D.J.; Erskine, D.; Walker, L.; Kurzawa-Akanbi, M.; Burn, D.; Donaghy, P.; Morris, C.; Taylor, J.-P.; Thomas, A.; et al. Dementia with Lewy bodies: An update and outlook. Mol. Neurodegener. 2019, 14, 5. [Google Scholar] [CrossRef] [PubMed]
- Brettschneider, J.; Del Tredici, K.; Lee, V.M.-Y.; Trojanowski, J.Q. Spreading of pathology in neurodegenerative diseases: A focus on human studies. Nat. Rev. Neurosci. 2015, 16, 109–120. [Google Scholar] [CrossRef] [Green Version]
- Summerfield, C.; Junqué, C.; Tolosa, E.; Salgado-Pineda, P.; Gómez-Ansón, B.; Martí, M.J.; Pastor, P.; Ramírez-Ruíz, B.; Mercader, J. Structural Brain Changes in Parkinson Disease with Dementia: A Voxel-Based Morphometry Study. Arch. Neurol. 2005, 62, 281–285. [Google Scholar] [CrossRef]
- Jobson, D.D.; Hase, Y.; Clarkson, A.N.; Kalaria, R.N. The role of the medial prefrontal cortex in cognition, ageing and dementia. Brain Commun. 2021, 3, fcab125. [Google Scholar] [CrossRef]
- Tanaka, M.; Vécsei, L. Editorial of Special Issue ‘Dissecting Neurological and Neuropsychiatric Diseases: Neurodegeneration and Neuroprotection’. Int. J. Mol. Sci. 2022, 23, 6991. [Google Scholar] [CrossRef]
- Battaglia, S.; Harrison, B.J.; Fullana, M.A. Does the Human Ventromedial Prefrontal Cortex Support Fear Learning, Fear Extinction or Both? A Commentary on Subregional Contributions. Mol. Psychiatry 2022, 27, 784–786. [Google Scholar] [CrossRef] [PubMed]
- Perry, D.C.; Kramer, J.H. Reward Processing in Neurodegenerative Disease. Neurocase 2015, 21, 120–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prinz, M.; Jung, S.; Priller, J. Microglia Biology: One Century of Evolving Concepts. Cell 2019, 179, 292–311. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.Y.; McNeely, T.L.; Baker, D.J. Untangling Senescent and Damage-Associated Microglia in the Aging and Diseased Brain. FEBS J. 2021; Epub ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Cserép, C.; Pósfai, B.; Dénes, Á. Shaping Neuronal Fate: Functional Heterogeneity of Direct Microglia-Neuron Interactions. Neuron 2021, 109, 222–240. [Google Scholar] [CrossRef]
- Pajares, M.; Rojo, A.I.; Manda, G.; Boscá, L.; Cuadrado, A. Inflammation in Parkinson’s Disease: Mechanisms and Therapeutic Implications. Cells 2020, 9, 1687. [Google Scholar] [CrossRef]
- Angelova, D.M.; Brown, D.R. Microglia and the aging brain: Are senescent microglia the key to neurodegeneration? J. Neurochem. 2019, 151, 676–688. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, M.; Szabó, Á.; Spekker, E.; Polyák, H.; Tóth, F.; Vécsei, L. Mitochondrial Impairment: A Common Motif in Neuropsychiatric Presentation? The Link to the Tryptophan–Kynurenine Metabolic System. Cells 2022, 11, 2607. [Google Scholar] [CrossRef]
- Török, N.; Maszlag-Török, R.; Molnár, K.; Szolnoki, Z.; Somogyvári, F.; Boda, K.; Tanaka, M.; Klivényi, P.; Vécsei, L. Single Nucleotide Polymorphisms of Indoleamine 2,3-Dioxygenase 1 Influenced the Age Onset of Parkinson’s Disease. Front. Biosci. 2022, 27, 265. [Google Scholar] [CrossRef]
- Lawrence, G.M.; Holley, C.L.; Schroder, K. Parkinson’s disease: Connecting mitochondria to inflammasomes. Trends Immunol. 2022, 43, 877–885. [Google Scholar] [CrossRef]
- Crapser, J.D.; Spangenberg, E.E.; Barahona, R.A.; Arreola, M.A.; Hohsfield, L.A.; Green, K.N. Microglia Facilitate Loss of Perineuronal Nets in the Alzheimer’s Disease Brain. EBioMedicine 2020, 58, 102919. [Google Scholar] [CrossRef]
- d’Errico, P.; Ziegler-Waldkirch, S.; Aires, V.; Hoffmann, P.; Mezö, C.; Erny, D.; Monasor, L.S.; Liebscher, S.; Ravi, V.M.; Joseph, K.; et al. Microglia Contribute to the Propagation of Aβ into Unaffected Brain Tissue. Nat. Neurosci. 2022, 25, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Spangenberg, E.E.; Lee, R.J.; Najafi, A.R.; Rice, R.A.; Elmore, M.R.P.; Blurton-Jones, M.; West, B.L.; Green, K.N. Eliminating Microglia in Alzheimer’s Mice Prevents Neuronal Loss without Modulating Amyloid-β Pathology. Brain 2016, 139, 1265–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spangenberg, E.; Severson, P.L.; Hohsfield, L.A.; Crapser, J.; Zhang, J.; Burton, E.A.; Zhang, Y.; Spevak, W.; Lin, J.; Phan, N.Y.; et al. Sustained Microglial Depletion with CSF1R Inhibitor Impairs Parenchymal Plaque Development in an Alzheimer’s Disease Model. Nat. Commun. 2019, 10, 3758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.-Y.; Jin, W.-L.; Xu, Y.; Jin, M.-Z. Microglia in neurodegenerative diseases. Neural Regen. Res. 2021, 16, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef]
- Lee, Y.; Lee, S.; Chang, S.-C.; Lee, J. Significant Roles of Neuroinflammation in Parkinson’s Disease: Therapeutic Targets for PD Prevention. Arch. Pharm. Res. 2019, 42, 416–425. [Google Scholar] [CrossRef]
- Weinreb, P.H.; Zhen, W.; Poon, A.W.; Conway, K.A.; Lansbury, P.T., Jr. NACP, A Protein Implicated in Alzheimer’s Disease and Learning, Is Natively Unfolded. Biochemistry 1996, 35, 13709–13715. [Google Scholar] [CrossRef]
- Malfertheiner, K.; Stefanova, N.; Heras-Garvin, A. The Concept of α-Synuclein Strains and How Different Conformations May Explain Distinct Neurodegenerative Disorders. Front. Neurol. 2021, 12, 1776. [Google Scholar] [CrossRef]
- Li, A.; Rastegar, C.; Mao, X. α-Synuclein Conformational Plasticity: Physiologic States, Pathologic Strains, and Biotechnological Applications. Biomolecules 2022, 12, 994. [Google Scholar] [CrossRef]
- Hoffmann, A.; Ettle, B.; Bruno, A.; Kulinich, A.; Hoffmann, A.-C.; von Wittgenstein, J.; Winkler, J.; Xiang, W.; Schlachetzki, J.C. Alpha-synuclein activates BV2 microglia dependent on its aggregation state. Biochem. Biophys. Res. Commun. 2016, 479, 881–886. [Google Scholar] [CrossRef]
- Dettmer, U.; Newman, A.J.; von Saucken, V.; Bartels, T.; Selkoe, D. KTKEGV repeat motifs are key mediators of normal alpha-synuclein tetramerization: Their mutation causes excess monomers and neurotoxicity. Proc. Natl. Acad. Sci. USA 2015, 112, 9596–9601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fakhree, M.A.A.; Nolten, I.S.; Blum, C.; Claessens, M.M.A.E. Different Conformational Subensembles of the Intrinsically Disordered Protein α-Synuclein in Cells. J. Phys. Chem. Lett. 2018, 9, 1249–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fusco, G.; De Simone, A.; Gopinath, T.; Vostrikov, V.; Vendruscolo, M.; Dobson, C.M.; Veglia, G. Direct observation of the three regions in α-synuclein that determine its membrane-bound behaviour. Nat. Commun. 2014, 5, 3827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Zhao, C.; Li, D.; Tian, Z.; Lai, Y.; Diao, J.; Liu, C. Versatile Structures of α-Synuclein. Front. Mol. Neurosci. 2016, 9, 48. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, C.; Luo, F.; Liu, Z.; Gui, X.; Luo, Z.; Zhang, X.; Li, D.; Liu, C.; Li, X. Amyloid fibril structure of α-synuclein determined by cryo-electron microscopy. Cell Res. 2018, 28, 897–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surguchov, A.; Surguchev, A. Synucleins: New Data on Misfolding, Aggregation and Role in Diseases. Biomedicines 2022, 10, 3241. [Google Scholar] [CrossRef]
- Baratono, S.; Press, D. What Are the Key Diagnostic Cognitive Impairment and Dementia Subtypes and How to Integrate all of the Diagnostic Data to Establish a Diagnosis? Clin. Geriatr. Med. 2023, 39, 77–90. [Google Scholar] [CrossRef]
- Jia, L.; Liu, Y.; Wang, W.; Wang, Y.; Liu, H.; Liu, F.; Chen, R.; Dawson, V.L.; Dawson, T.M.; Lu, F.; et al. Molecular Mediation of Prion-like α-Synuclein Fibrillation from Toxic PFFs to Nontoxic Species. ACS Appl. Bio Mater. 2020, 3, 6096–6102. [Google Scholar] [CrossRef]
- Navarro-Paya, C.; Sanz-Hernandez, M.; De Simone, A. Plasticity of Membrane Binding by the Central Region of α-Synuclein. Front. Mol. Biosci. 2022, 9, 857217. [Google Scholar] [CrossRef]
- Gao, V.; Briano, J.A.; Komer, L.E.; Burré, J. Functional and Pathological Effects of α-Synuclein on Synaptic SNARE Complexes. J. Mol. Biol. 2023, 435, 167714. [Google Scholar] [CrossRef] [PubMed]
- Robotta, M.; Braun, P.; van Rooijen, B.; Subramaniam, V.; Huber, M.; Drescher, M. Direct Evidence of Coexisting Horseshoe and Extended Helix Conformations of Membrane-Bound Alpha-Synuclein. ChemPhysChem 2011, 12, 267–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnaoutoglou, N.A.; O’Brien, J.T.; Underwood, B.R. Dementia with Lewy Bodies—From Scientific Knowledge to Clinical Insights. Nat. Rev. Neurol. 2019, 15, 103–112. [Google Scholar] [CrossRef]
- Feng, Y.; Zheng, C.; Zhang, Y.; Xing, C.; Cai, W.; Li, R.; Chen, J.; Duan, Y. Triptolide Inhibits Preformed Fibril-Induced Microglial Activation by Targeting the MicroRNA155-5p/SHIP1 Pathway. Oxid. Med. Cell Longev. 2019, 2019, 6527638. [Google Scholar] [CrossRef] [Green Version]
- La Vitola, P.; Balducci, C.; Cerovic, M.; Santamaria, G.; Brandi, E.; Grandi, F.; Caldinelli, L.; Colombo, L.; Morgese, M.G.; Trabace, L.; et al. Alpha-synuclein oligomers impair memory through glial cell activation and via Toll-like receptor 2. Brain, Behav. Immun. 2018, 69, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Ingelsson, M. Alpha-Synuclein Oligomers—Neurotoxic Molecules in Parkinson’s Disease and Other Lewy Body Disorders. Front. Neurosci. 2016, 10, 408. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Idowu, A.; Rosenthal, L.; Mao, X. Parkinson’s disease fluid biomarkers for differential diagnosis of atypical parkinsonian syndromes. Clin. Transl. Discov. 2023, 3, e150. [Google Scholar] [CrossRef]
- Van der Perren, A.; Gelders, G.; Fenyi, A.; Bousset, L.; Brito, F.; Peelaerts, W.; Haute, C.V.D.; Gentleman, S.; Melki, R.; Baekelandt, V. The structural differences between patient-derived α-synuclein strains dictate characteristics of Parkinson’s disease, multiple system atrophy and dementia with Lewy bodies. Acta Neuropathol. 2020, 139, 977–1000. [Google Scholar] [CrossRef]
- Shahnawaz, M.; Mukherjee, A.; Pritzkow, S.; Mendez, N.; Rabadia, P.; Liu, X.; Hu, B.; Schmeichel, A.; Singer, W.; Wu, G.; et al. Discriminating α-synuclein strains in Parkinson’s disease and multiple system atrophy. Nature 2020, 578, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Sokratian, A.; Zhou, Y.; Xu, E.; Viverette, E.; Dillard, L.; Yuan, Y.; Li, J.Y.; Matarangas, A.; Bouvette, J.; Borgnia, M.; et al. Structural and Functional Landscape of α-Synuclein Fibril Conformations Amplified from Cerebrospinal Fluid. bioRxiv 2022. [Google Scholar] [CrossRef]
- Runfola, M.; De Simone, A.; Vendruscolo, M.; Dobson, C.M.; Fusco, G. The N-Terminal Acetylation of α-Synuclein Changes the Affinity for Lipid Membranes but Not the Structural Properties of the Bound State. Sci. Rep. 2020, 10, 204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, W.; Zibaee, S.; Jakes, R.; Serpell, L.; Davletov, B.; Crowther, R.A.; Goedert, M. Mutation E46K increases phospholipid binding and assembly into filaments of human α-synuclein. FEBS Lett. 2004, 576, 363–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hijaz, B.A.; Volpicelli-Daley, L.A. Initiation and propagation of α-synuclein aggregation in the nervous system. Mol. Neurodegener. 2020, 15, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoenen, C.; Gustin, A.; Birck, C.; Kirchmeyer, M.; Beaume, N.; Felten, P.; Grandbarbe, L.; Heuschling, P.; Heurtaux, T. Alpha-Synuclein Proteins Promote Pro-Inflammatory Cascades in Microglia: Stronger Effects of the A53T Mutant. PLoS ONE 2016, 11, e0162717. [Google Scholar] [CrossRef] [Green Version]
- El-Zayat, S.R.; Sibaii, H.; Mannaa, F.A. Toll-like receptors activation, signaling, and targeting: An overview. Bull. Natl. Res. Cent. 2019, 43, 187. [Google Scholar] [CrossRef] [Green Version]
- Fiebich, B.L.; Batista, C.R.A.; Saliba, S.W.; Yousif, N.M.; De Oliveira, A.C.P. Role of Microglia TLRs in Neurodegeneration. Front. Cell. Neurosci. 2018, 12, 329. [Google Scholar] [CrossRef] [Green Version]
- Campolo, M.; Paterniti, I.; Siracusa, R.; Filippone, A.; Esposito, E.; Cuzzocrea, S. TLR4 absence reduces neuroinflammation and inflammasome activation in Parkinson’s diseases in vivo model. Brain, Behav. Immun. 2019, 76, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.; Jana, M.; Majumder, M.; Mondal, S.; Roy, A.; Pahan, K. Selective targeting of the TLR2/MyD88/NF-κB pathway reduces α-synuclein spreading in vitro and in vivo. Nat. Commun. 2021, 12, 5382. [Google Scholar] [CrossRef] [PubMed]
- Fellner, L.; Irschick, R.B.; Schanda, K.; Reindl, M.; Klimaschewski, L.; Poewe, W.; Wenning, G.K.; Stefanova, N. Toll-like receptor 4 is required for α-synuclein dependent activation of microglia and astroglia. Glia 2013, 61, 349–360. [Google Scholar] [CrossRef] [Green Version]
- Iyer, A.; Roeters, S.J.; Kogan, V.; Woutersen, S.; Claessens, M.M.A.E.; Subramaniam, V. C-Terminal Truncated α-Synuclein Fibrils Contain Strongly Twisted β-Sheets. J. Am. Chem. Soc. 2017, 139, 15392–15400. [Google Scholar] [CrossRef]
- Choi, I.; Zhang, Y.; Seegobin, S.P.; Pruvost, M.; Wang, Q.; Purtell, K.; Zhang, B.; Yue, Z. Microglia clear neuron-released α-synuclein via selective autophagy and prevent neurodegeneration. Nat. Commun. 2020, 11, 1386. [Google Scholar] [CrossRef] [Green Version]
- Sánchez, K.; Maguire-Zeiss, K. MMP13 Expression Is Increased Following Mutant α-Synuclein Exposure and Promotes Inflammatory Responses in Microglia. Front. Neurosci. 2020, 14, 585544. [Google Scholar] [CrossRef]
- Dzamko, N.; Gysbers, A.; Perera, G.; Bahar, A.; Shankar, A.; Gao, J.; Fu, Y.; Halliday, G.M. Toll-like Receptor 2 Is Increased in Neurons in Parkinson’s Disease Brain and May Contribute to Alpha-Synuclein Pathology. Acta Neuropathol. 2017, 133, 303–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, X.; Ou, M.T.; Karuppagounder, S.S.; Kam, T.-I.; Yin, X.; Xiong, Y.; Ge, P.; Umanah, G.E.; Brahmachari, S.; Shin, J.-H.; et al. Pathological α-Synuclein Transmission Initiated by Binding Lymphocyte-Activation Gene 3. Science 2016, 353, aah3374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, X.; Gu, H.; Kim, D.; Kimura, Y.; Wang, N.; Xu, E.; Wang, H.; Chen, C.; Zhang, S.; Jia, C.; et al. Aplp1 and the Aplp1-Lag3 Complex Facilitates Transmission of Pathologic α-Synuclein. bioRxiv 2021. [Google Scholar] [CrossRef]
- Gu, H.; Yang, X.; Mao, X.; Xu, E.; Qi, C.; Wang, H.; Brahmachari, S.; York, B.; Sriparna, M.; Li, A.; et al. Lymphocyte Activation Gene 3 (Lag3) Contributes to α-Synucleinopathy in α-Synuclein Transgenic Mice. Front. Cell. Neurosci. 2021, 15, 656426. [Google Scholar] [CrossRef]
- Rimmerman, N.; Verdiger, H.; Goldenberg, H.; Naggan, L.; Robinson, E.; Kozela, E.; Gelb, S.; Reshef, R.; Ryan, K.M.; Ayoun, L.; et al. Microglia and Their LAG3 Checkpoint Underlie the Antidepressant and Neurogenesis-Enhancing Effects of Electroconvulsive Stimulation. Mol. Psychiatry 2022, 27, 1120–1135. [Google Scholar] [CrossRef] [PubMed]
- Galatro, T.F.; Holtman, I.R.; Lerario, A.M.; Vainchtein, I.D.; Brouwer, N.; Sola, P.R.; Veras, M.M.; Pereira, T.F.; Leite, R.E.P.; Möller, T.; et al. Transcriptomic analysis of purified human cortical microglia reveals age-associated changes. Nat. Neurosci. 2017, 20, 1162–1171. [Google Scholar] [CrossRef] [PubMed]
- Freeze, B.; Acosta, D.; Pandya, S.; Zhao, Y.; Raj, A. Regional expression of genes mediating trans-synaptic alpha-synuclein transfer predicts regional atrophy in Parkinson disease. NeuroImage Clin. 2018, 18, 456–466. [Google Scholar] [CrossRef]
- Rotter, A.; Lenz, B.; Pitsch, R.; Richter-Schmidinger, T.; Kornhuber, J.; Rhein, C. Alpha-Synuclein RNA Expression is Increased in Major Depression. Int. J. Mol. Sci. 2019, 20, 2029. [Google Scholar] [CrossRef] [Green Version]
- Jellinger, K.A. Pathomechanisms of depression in multiple system atrophy. J. Neural Transm. 2022, 130, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Laux, G. Parkinson and depression: Review and outlook. J. Neural Transm. 2022, 129, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Postuma, R.B.; Iranzo, A.; Hu, M.; Högl, B.; Boeve, B.F.; Manni, R.; Oertel, W.H.; Arnulf, I.; Ferini-Strambi, L.; Puligheddu, M.; et al. Risk and predictors of dementia and parkinsonism in idiopathic REM sleep behaviour disorder: A multicentre study. Brain 2019, 142, 744–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Liu, Y.-Q.; Jia, C.; Lim, Y.-J.; Feng, G.; Xu, E.; Long, H.; Kimura, Y.; Tao, Y.; Zhao, C.; et al. Mechanistic basis for receptor-mediated pathological α-synuclein fibril cell-to-cell transmission in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2021, 118, e2011196118. [Google Scholar] [CrossRef] [PubMed]
- Hammond, T.R.; Dufort, C.; Dissing-Olesen, L.; Giera, S.; Young, A.; Wysoker, A.; Walker, A.J.; Gergits, F.; Segel, M.; Nemesh, J.; et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 2019, 50, 253.E6–271E6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Wu, X.; Li, X.; Jiang, L.-L.; Gui, X.; Liu, Y.; Sun, Y.; Zhu, B.; Piña-Crespo, J.C.; Zhang, M.; et al. TREM2 Is a Receptor for β-Amyloid that Mediates Microglial Function. Neuron 2018, 97, 1023–1031. [Google Scholar] [CrossRef] [Green Version]
- Xie, M.; Liu, Y.U.; Zhao, S.; Zhang, L.; Bosco, D.B.; Pang, Y.-P.; Zhong, J.; Sheth, U.; Martens, Y.A.; Zhao, N.; et al. TREM2 Interacts with TDP-43 and Mediates Microglial Neuroprotection against TDP-43-Related Neurodegeneration. Nat. Neurosci. 2022, 25, 26–38. [Google Scholar] [CrossRef]
- Liu, G.; Liu, Y.; Jiang, Q.; Jiang, Y.; Feng, R.; Zhang, L.; Chen, Z.; Li, K.; Liu, J. Convergent Genetic and Expression Datasets Highlight TREM2 in Parkinson’s Disease Susceptibility. Mol. Neurobiol. 2016, 53, 4931–4938. [Google Scholar] [CrossRef]
- Guo, Y.; Wei, X.; Yan, H.; Qin, Y.; Yan, S.; Liu, J.; Zhao, Y.; Jiang, F.; Lou, H. TREM2 deficiency aggravates α-synuclein–induced neurodegeneration and neuroinflammation in Parkinson’s disease models. FASEB J. 2019, 33, 12164–12174. [Google Scholar] [CrossRef] [Green Version]
- Rayaprolu, S.; Mullen, B.; Baker, M.; Lynch, T.; Finger, E.; Seeley, W.W.; Hatanpaa, K.J.; Lomen-Hoerth, C.; Kertesz, A.; Bigio, E.H.; et al. TREM2 in neurodegeneration: Evidence for association of the p.R47H variant with frontotemporal dementia and Parkinson’s disease. Mol. Neurodegener. 2013, 8, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Allen, M.; Sakae, N.; Ertekin-Taner, N.; Graff-Radford, N.R.; Dickson, D.W.; Younkin, S.G.; Sevlever, D. Expression and processing analyses of wild type and p.R47H TREM2 variant in Alzheimer’s disease brains. Mol. Neurodegener. 2016, 11, 72. [Google Scholar] [CrossRef] [Green Version]
- McQuade, A.; Kang, Y.J.; Hasselmann, J.; Jairaman, A.; Sotelo, A.; Coburn, M.; Shabestari, S.K.; Chadarevian, J.P.; Fote, G.; Tu, C.H.; et al. Gene expression and functional deficits underlie TREM2-knockout microglia responses in human models of Alzheimer’s disease. Nat. Commun. 2020, 11, 5370. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Lewis, C.A.; Ulrich, J.D.; Holtzman, D.M. Chronic TREM2 activation exacerbates Aβ-associated tau seeding and spreading. J. Exp. Med. 2022, 220, e20220654. [Google Scholar] [CrossRef]
- Gonzalez-Lozano, M.; Wortel, J.; van der Loo, R.; van Weering, J.; Smit, A.; Li, K. Reduced mGluR5 Activity Modulates Mitochondrial Function. Cells 2021, 10, 1375. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-N.; Fan, J.-K.; Gu, L.; Yang, H.-M.; Zhan, S.-Q.; Zhang, H. Metabotropic glutamate receptor 5 inhibits α-synuclein-induced microglia inflammation to protect from neurotoxicity in Parkinson’s disease. J. Neuroinflamm. 2021, 18, 23. [Google Scholar] [CrossRef]
- Byrnes, K.R.; Stoica, B.; Loane, D.; Riccio, A.; Davis, M.; Faden, A.I. Metabotropic Glutamate Receptor 5 Activation Inhibits Microglial Associated Inflammation and Neurotoxicity. Glia 2009, 57, 550–560. [Google Scholar] [CrossRef] [Green Version]
- Abd-Elrahman, K.S.; Albaker, A.; de Souza, J.M.; Ribeiro, F.M.; Schlossmacher, M.G.; Tiberi, M.; Hamilton, A.; Ferguson, S.S.G. Aβ oligomers induce pathophysiological mGluR5 signaling in Alzheimer’s disease model mice in a sex-selective manner. Sci. Signal. 2020, 13, eabd2494. [Google Scholar] [CrossRef]
- Franco, R.; Aguinaga, D.; Reyes, I.; Canela, E.I.; Lillo, J.; Tarutani, A.; Hasegawa, M.; del Ser-Badia, A.; del Rio, J.A.; Kreutz, M.R.; et al. N-Methyl-D-Aspartate Receptor Link to the MAP Kinase Pathway in Cortical and Hippocampal Neurons and Microglia Is Dependent on Calcium Sensors and Is Blocked by α-Synuclein, Tau, and Phospho-Tau in Non-Transgenic and Transgenic APPSw, Ind Mice. Front. Mol. Neurosci. 2018, 11, 273. [Google Scholar] [CrossRef]
- Hou, L.; Bao, X.; Zang, C.; Yang, H.; Sun, F.; Che, Y.; Wu, X.; Li, S.; Zhang, D.; Wang, Q. Integrin CD11b mediates α-synuclein-induced activation of NADPH oxidase through a Rho-dependent pathway. Redox Biol. 2018, 14, 600–608. [Google Scholar] [CrossRef]
- Jiang, T.; Hoekstra, J.; Heng, X.; Kang, W.; Ding, J.; Liu, J.; Chen, S.; Zhang, J. P2X7 receptor is critical in α-synuclein-mediated microglial NADPH oxidase activation. Neurobiol. Aging 2015, 36, 2304–2318. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.R.; Kang, S.-J.; Kim, J.-M.; Lee, S.-J.; Jou, I.; Joe, E.-H.; Park, S.M. FcγRIIB mediates the inhibitory effect of aggregated α-synuclein on microglial phagocytosis. Neurobiol. Dis. 2015, 83, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Long, H.; Zhang, S.; Zeng, S.; Tong, Y.; Liu, J.; Liu, C.; Li, D. Interaction of RAGE with α-synuclein fibrils mediates inflammatory response of microglia. Cell Rep. 2022, 40, 111401. [Google Scholar] [CrossRef] [PubMed]
- Ihse, E.; Yamakado, H.; van Wijk, X.M.; Lawrence, R.; Esko, J.D.; Masliah, E. Cellular internalization of alpha-synuclein aggregates by cell surface heparan sulfate depends on aggregate conformation and cell type. Sci. Rep. 2017, 7, 9008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, M.S. Microglia in Parkinson’s Disease. In Neuroglia in Neurodegenerative Diseases; Verkhratsky, A., Ho, M.S., Zorec, R., Parpura, V., Eds.; Springer: Singapore, 2019; pp. 335–353. [Google Scholar]
- Valdinocci, D.; Radford, R.A.W.; Siow, S.M.; Chung, R.S.; Pountney, D.L. Potential Modes of Intercellular α-Synuclein Transmission. Int. J. Mol. Sci. 2017, 18, 469. [Google Scholar] [CrossRef] [Green Version]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566.E9–581E9. [Google Scholar] [CrossRef] [Green Version]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276.e17–1290.e17. [Google Scholar] [CrossRef] [Green Version]
- Sobue, A.; Komine, O.; Hara, Y.; Endo, F.; Mizoguchi, H.; Watanabe, S.; Murayama, S.; Saito, T.; Saido, T.C.; Sahara, N.; et al. Microglial gene signature reveals loss of homeostatic microglia associated with neurodegeneration of Alzheimer’s disease. Acta Neuropathol. Commun. 2021, 9, 1. [Google Scholar] [CrossRef]
- Bido, S.; Muggeo, S.; Massimino, L.; Marzi, M.J.; Giannelli, S.G.; Melacini, E.; Nannoni, M.; Gambarè, D.; Bellini, E.; Ordazzo, G.; et al. Microglia-specific overexpression of α-synuclein leads to severe dopaminergic neurodegeneration by phagocytic exhaustion and oxidative toxicity. Nat. Commun. 2021, 12, 6237. [Google Scholar] [CrossRef]
- Bliederhaeuser, C.; Grozdanov, V.; Speidel, A.; Zondler, L.; Ruf, W.P.; Bayer, H.; Kiechle, M.; Feiler, M.S.; Freischmidt, A.; Brenner, D.; et al. Age-dependent defects of alpha-synuclein oligomer uptake in microglia and monocytes. Acta Neuropathol. 2016, 131, 379–391. [Google Scholar] [CrossRef]
- Chen, C.-M.; Yen, C.-Y.; Chen, W.-L.; Lin, C.-H.; Wu, Y.-R.; Chang, K.-H.; Lee-Chen, G.-J. Pathomechanism Characterization and Potential Therapeutics Identification for Parkinson’s Disease Targeting Neuroinflammation. Int. J. Mol. Sci. 2021, 22, 1062. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xia, Y.; Yin, S.; Wan, F.; Hu, J.; Kou, L.; Sun, Y.; Wu, J.; Zhou, Q.; Huang, J.; et al. Targeting Microglial α-Synuclein/TLRs/NF-kappaB/NLRP3 Inflammasome Axis in Parkinson’s Disease. Front. Immunol. 2021, 12, 719807. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Hwang, I.; Park, S.; Hong, S.; Hwang, B.; Cho, Y.; Son, J.; Yu, J.-W. MPTP-driven NLRP3 inflammasome activation in microglia plays a central role in dopaminergic neurodegeneration. Cell Death Differ. 2019, 26, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.W. The MPTP Story. J. Park. Dis. 2017, 7, S11–S19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, K.; Guo, J.; Wang, G.; Ni, Q.; Liu, X. Toll-Like Receptor 2–Mediated Autophagy Promotes Microglial Cell Death by Modulating the Microglial M1/M2 Phenotype. Inflammation 2020, 43, 701–711. [Google Scholar] [CrossRef]
- DeRidder, L.; Sharma, A.; Liaw, K.; Sharma, R.; John, J.; Kannan, S.; Kannan, R.M. Dendrimer–Tesaglitazar Conjugate Induces a Phenotype Shift of Microglia and Enhances β-Amyloid Phagocytosis. Nanoscale 2021, 13, 939–952. [Google Scholar] [CrossRef]
- Thomsen, M.B.; Ferreira, S.A.; Schacht, A.C.; Jacobsen, J.; Simonsen, M.; Betzer, C.; Jensen, P.H.; Brooks, D.J.; Landau, A.M.; Romero-Ramos, M. PET Imaging Reveals Early and Progressive Dopaminergic Deficits after Intra-Striatal Injection of Preformed Alpha-Synuclein Fibrils in Rats. Neurobiol. Dis. 2021, 149, 105229. [Google Scholar] [CrossRef]
- Harms, A.S.; Thome, A.D.; Yan, Z.; Schonhoff, A.M.; Williams, G.P.; Li, X.; Liu, Y.; Qin, H.; Benveniste, E.N.; Standaert, D.G. Peripheral Monocyte Entry Is Required for Alpha-Synuclein Induced Inflammation and Neurodegeneration in a Model of Parkinson Disease. Exp. Neurol. 2018, 300, 179–187. [Google Scholar] [CrossRef]
- Pierce, S.; Coetzee, G.A. Parkinson’s Disease-Associated Genetic Variation Is Linked to Quantitative Expression of Inflammatory Genes. PLoS ONE 2017, 12, e0175882. [Google Scholar] [CrossRef] [Green Version]
- Olesen, M.N.; Christiansen, J.R.; Petersen, S.V.; Jensen, P.H.; Paslawski, W.; Romero-Ramos, M.; Sanchez-Guajardo, V. CD4 T Cells React to Local Increase of α-Synuclein in a Pathology-Associated Variant-Dependent Manner and Modify Brain Microglia in Absence of Brain Pathology. Heliyon 2018, 4, e00513. [Google Scholar] [CrossRef] [Green Version]
- Gate, D.; Tapp, E.; Leventhal, O.; Shahid, M.; Nonninger, T.J.; Yang, A.C.; Strempfl, K.; Unger, M.S.; Fehlmann, T.; Oh, H.; et al. CD4+ T Cells Contribute to Neurodegeneration in Lewy Body Dementia. Science 2021, 374, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Scott, K.M. B Lymphocytes in Parkinson’s Disease. J. Park. Dis. 2022, 12, S75–S81. [Google Scholar] [CrossRef] [PubMed]
- Galiano-Landeira, J.; Torra, A.; Vila, M.; Bové, J. CD8 T Cell Nigral Infiltration Precedes Synucleinopathy in Early Stages of Parkinson’s Disease. Brain 2020, 143, 3717–3733. [Google Scholar] [CrossRef] [PubMed]
- George, S.; Tyson, T.; Rey, N.L.; Sheridan, R.; Peelaerts, W.; Becker, K.; Schulz, E.; Meyerdirk, L.; Burmeister, A.R.; von Linstow, C.U.; et al. T Cells Limit Accumulation of Aggregate Pathology Following Intrastriatal Injection of α-Synuclein Fibrils. J. Park. Dis. 2021, 11, 585–603. [Google Scholar] [CrossRef]
- Sulzer, D.; Alcalay, R.N.; Garretti, F.; Cote, L.; Kanter, E.; Agin-Liebes, J.; Liong, C.; McMurtrey, C.; Hildebrand, W.H.; Mao, X.; et al. T cells from patients with Parkinson’s disease recognize α-synuclein peptides. Nature 2017, 546, 656–661. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. α-Synuclein Is Phosphorylated in Synucleinopathy Lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [CrossRef]
- Tanriöver, G.; Bacioglu, M.; Schweighauser, M.; Mahler, J.; Wegenast-Braun, B.M.; Skodras, A.; Obermüller, U.; Barth, M.; Kronenberg-Versteeg, D.; Nilsson, K.P.R.; et al. Prominent microglial inclusions in transgenic mouse models of α-synucleinopathy that are distinct from neuronal lesions. Acta Neuropathol. Commun. 2020, 8, 133. [Google Scholar] [CrossRef]
- Kamboj, S.; Harms, C.; Wright, D.; Nash, A.; Kumar, L.; Klein-Seetharaman, J.; Sarkar, S.K. Identification of allosteric fingerprints of alpha-synuclein aggregates in matrix metalloprotease-1 and substrate-specific virtual screening with single molecule insights. Sci. Rep. 2022, 12, 5764. [Google Scholar] [CrossRef]
- Sousa, L.; Guarda, M.; Meneses, M.J.; Macedo, M.P.; Miranda, H.V. Insulin-degrading enzyme: An ally against metabolic and neurodegenerative diseases. J. Pathol. 2021, 255, 346–361. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Seow, Y.; Schapira, A.H.; Gardiner, C.; Sargent, I.L.; Wood, M.J.A.; Cooper, J.M. Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol. Dis. 2011, 42, 360–367. [Google Scholar] [CrossRef] [Green Version]
- Danzer, K.M.; Kranich, L.R.; Ruf, W.P.; Cagsal-Getkin, O.; Winslow, A.R.; Zhu, L.; Vanderburg, C.R.; McLean, P.J. Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol. Neurodegener. 2012, 7, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, Y.; Kim, S.; Varkey, J.; Lou, X.; Song, J.-K.; Diao, J.; Langen, R.; Shin, Y.-K. Nonaggregated α-Synuclein Influences SNARE-Dependent Vesicle Docking via Membrane Binding. Biochemistry 2014, 53, 3889–3896. [Google Scholar] [CrossRef] [PubMed]
- Yoo, G.; Yeou, S.; Son, J.B.; Shin, Y.-K.; Lee, N.K. Cooperative inhibition of SNARE-mediated vesicle fusion by α-synuclein monomers and oligomers. Sci. Rep. 2021, 11, 10955. [Google Scholar] [CrossRef] [PubMed]
- Yoo, G.; Shin, Y.-K.; Lee, N.K. The Role of α-Synuclein in SNARE-mediated Synaptic Vesicle Fusion. J. Mol. Biol. 2023, 435, 167775. [Google Scholar] [CrossRef] [PubMed]
- Emmanouilidou, E.; Melachroinou, K.; Roumeliotis, T.; Garbis, S.D.; Ntzouni, M.; Margaritis, L.H.; Stefanis, L.; Vekrellis, K. Cell-Produced alpha-Synuclein Is Secreted in a Calcium-Dependent Manner by Exosomes and Impacts Neuronal Survival. J. Neurosci. 2010, 30, 6838–6851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, J.R. Q&A: What Are Exosomes, Exactly? BMC Biol. 2016, 14, 46. [Google Scholar] [CrossRef] [Green Version]
- Agliardi, C.; Meloni, M.; Guerini, F.R.; Zanzottera, M.; Bolognesi, E.; Baglio, F.; Clerici, M. Oligomeric α-Syn and SNARE Complex Proteins in Peripheral Extracellular Vesicles of Neural Origin Are Biomarkers for Parkinson’s Disease. Neurobiol. Dis. 2021, 148, 105185. [Google Scholar] [CrossRef]
- Magalhães, P.; Lashuel, H.A. Opportunities and Challenges of Alpha-Synuclein as a Potential Biomarker for Parkinson’s Disease and Other Synucleinopathies. NPJ Park. Dis. 2022, 8, 93. [Google Scholar] [CrossRef]
- Guo, M.; Wang, J.; Zhao, Y.; Feng, Y.; Han, S.; Dong, Q.; Cui, M.; Tieu, K. Microglial Exosomes Facilitate α-Synuclein Transmission in Parkinson’s Disease. Brain 2020, 143, 1476–1497. [Google Scholar] [CrossRef]
- Guo, M.; Hao, Y.; Feng, Y.; Li, H.; Mao, Y.; Dong, Q.; Cui, M. Microglial Exosomes in Neurodegenerative Disease. Front. Mol. Neurosci. 2021, 14, 630808. [Google Scholar] [CrossRef]
- Si, X.-L.; Fang, Y.-J.; Li, L.-F.; Gu, L.-Y.; Yin, X.-Z.; Tian, J.; Yan, Y.-P.; Pu, J.-L.; Zhang, B.-R. From inflammasome to Parkinson’s disease: Does the NLRP3 inflammasome facilitate exosome secretion and exosomal alpha-synuclein transmission in Parkinson’s disease? Exp. Neurol. 2021, 336, 113525. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Qin, M.; Bao, P.; Xu, W.; Xu, J. Secretory Carrier Membrane Protein 5 Is an Autophagy Inhibitor That Promotes the Secretion of α-Synuclein via Exosome. PLoS ONE 2017, 12, e0180892. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Zhang, G.; Han, C.; Ma, K.; Guo, X.; Wan, F.; Kou, L.; Yin, S.; Liu, L.; Huang, J.; et al. Microglia as Modulators of Exosomal Alpha-Synuclein Transmission. Cell Death Dis. 2019, 10, 174. [Google Scholar] [CrossRef] [Green Version]
- Scheiblich, H.; Dansokho, C.; Mercan, D.; Schmidt, S.V.; Bousset, L.; Wischhof, L.; Eikens, F.; Odainic, A.; Spitzer, J.; Griep, A.; et al. Microglia jointly degrade fibrillar alpha-synuclein cargo by distribution through tunneling nanotubes. Cell 2021, 184, 5089–5106. [Google Scholar] [CrossRef] [PubMed]
- Schetters, S.T.T.; Gomez-Nicola, D.; Garcia-Vallejo, J.J.; Van Kooyk, Y. Neuroinflammation: Microglia and T Cells Get Ready to Tango. Front. Immunol. 2018, 8, 1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, S.P.; Kam, T.-I.; Panicker, N.; Kim, S.; Oh, Y.; Park, J.-S.; Kwon, S.-H.; Park, Y.J.; Karuppagounder, S.S.; Park, H.; et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat. Med. 2018, 24, 931–938. [Google Scholar] [CrossRef]
- Hinkle, J.T.; Patel, J.; Panicker, N.; Karuppagounder, S.S.; Biswas, D.; Belingon, B.; Chen, R.; Brahmachari, S.; Pletnikova, O.; Troncoso, J.C.; et al. STING mediates neurodegeneration and neuroinflammation in nigrostriatal α-synucleinopathy. Proc. Natl. Acad. Sci. USA 2022, 119. [Google Scholar] [CrossRef]
- Kam, T.-I.; Mao, X.; Park, H.; Chou, S.-C.; Karuppagounder, S.S.; Umanah, G.E.; Yun, S.P.; Brahmachari, S.; Panicker, N.; Chen, R.; et al. Poly(ADP-Ribose) Drives Pathologic α-Synuclein Neurodegeneration in Parkinson’s Disease. Science 2018, 362, eaat8407. [Google Scholar] [CrossRef] [Green Version]
- Li, A.; Tyson, J.; Patel, S.; Patel, M.; Katakam, S.; Mao, X.; He, W. Emerging Nanotechnology for Treatment of Alzheimer’s and Parkinson’s Disease. Front. Bioeng. Biotechnol. 2021, 9, 672594. [Google Scholar] [CrossRef]
- Liu, Y.-Q.; Mao, Y.; Xu, E.; Jia, H.; Zhang, S.; Dawson, V.L.; Dawson, T.M.; Li, Y.-M.; Zheng, Z.; He, W.; et al. Nanozyme Scavenging ROS for Prevention of Pathologic α-Synuclein Transmission in Parkinson’s Disease. Nano Today 2021, 36, 101027. [Google Scholar] [CrossRef]
- Butler, Y.R.; Liu, Y.; Kumbhar, R.; Zhao, P.; Gadhave, K.; Wang, N.; Li, Y.; Mao, X.; Wang, W. α-Synuclein Fibril-Specific Nanobody Reduces Prion-like α-Synuclein Spreading in Mice. Nat. Commun. 2022, 13, 4060. [Google Scholar] [CrossRef] [PubMed]
- Tansey, M.G.; Wallings, R.L.; Houser, M.C.; Herrick, M.K.; Keating, C.E.; Joers, V. Inflammation and immune dysfunction in Parkinson disease. Nat. Rev. Immunol. 2022, 22, 657–673. [Google Scholar] [CrossRef] [PubMed]
- Arena, G.; Sharma, K.; Agyeah, G.; Krüger, R.; Grünewald, A.; Fitzgerald, J.C. Neurodegeneration and Neuroinflammation in Parkinson’s Disease: A Self-Sustained Loop. Curr. Neurol. Neurosci. Rep. 2022, 22, 427–440. [Google Scholar] [CrossRef] [PubMed]
- Troncoso-Escudero, P.; Parra, A.; Nassif, M.; Vidal, R.L. Outside in: Unraveling the Role of Neuroinflammation in the Progression of Parkinson’s Disease. Front. Neurol. 2018, 9, 860. [Google Scholar] [CrossRef]
- Russo, C.; Valle, M.S.; Russo, A.; Malaguarnera, L. The Interplay between Ghrelin and Microglia in Neuroinflammation: Implications for Obesity and Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 13432. [Google Scholar] [CrossRef]
- Kim, T.-K.; Bae, E.-J.; Jung, B.C.; Choi, M.; Shin, S.J.; Park, S.J.; Kim, J.T.; Jung, M.K.; Ulusoy, A.; Song, M.-Y.; et al. Inflammation promotes synucleinopathy propagation. Exp. Mol. Med. 2022, 54, 2148–2161. [Google Scholar] [CrossRef]
- Gerhard, A.; Pavese, N.; Hotton, G.; Turkheimer, F.; Es, M.; Hammers, A.; Eggert, K.; Oertel, W.; Banati, R.B.; Brooks, D.J. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol. Dis. 2006, 21, 404–412. [Google Scholar] [CrossRef]
- Abud, E.M.; Ramirez, R.N.; Martinez, E.S.; Healy, L.M.; Nguyen, C.H.H.; Newman, S.A.; Yeromin, A.V.; Scarfone, V.M.; Marsh, S.E.; Fimbres, C.; et al. iPSC-Derived Human Microglia-like Cells to Study Neurological Diseases. Neuron 2017, 94, 278–293.e9. [Google Scholar] [CrossRef] [Green Version]
- Dräger, N.M.; Sattler, S.M.; Huang, C.T.-L.; Teter, O.M.; Leng, K.; Hashemi, S.H.; Hong, J.; Aviles, G.; Clelland, C.D.; Zhan, L.; et al. A CRISPRi/A platform in human iPSC-derived microglia uncovers regulators of disease states. Nat. Neurosci. 2022, 25, 1149–1162. [Google Scholar] [CrossRef]
- Brownjohn, P.W.; Smith, J.; Solanki, R.; Lohmann, E.; Houlden, H.; Hardy, J.; Dietmann, S.; Livesey, F.J. Functional Studies of Missense TREM2 Mutations in Human Stem Cell-Derived Microglia. Stem Cell Rep. 2018, 10, 1294–1307. [Google Scholar] [CrossRef] [Green Version]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic Reactive Astrocytes Are Induced by Activated Microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef] [PubMed]
- Surguchev, A.A.; Emamzadeh, F.N.; Surguchov, A. Cell Responses to Extracellular α-Synuclein. Molecules 2019, 24, 305. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| BV2 Cells | Primary Microglia | Mouse (in vivo) | |
|---|---|---|---|
| Method of Detection | qPCR (from BV2 RNA) and ELISA (from cell culture supernatant) for inflammatory mediator production (TNFα and IL-1β) | ELISA (from cell culture supernatant) for inflammatory cytokines (TNFα and IL-1β) | Various methods, including analysis of microglial morphology with Iba1 staining and IHC for IL-1β |
| Monomer | + | + | ? |
| Oligomer | - | - | X |
| Fibrillar | ++ | ++ | X |
| Receptor | Known Interactor? | Aggregation State of α-Syn Known to Interact with Receptor | Downstream Effect |
|---|---|---|---|
| TLR | Yes | Monomeric and aggregated forms | Phagocytosis of α-syn Secretion of ROS and pro-inflammatory cytokines Synucleinphagy |
| Lag3 | Yes, but not yet involving microglia | Aggregated forms | Unknown |
| TREM2 | Unknown | Not yet known | Unknown, but likely survival, phagocytosis and proliferation |
| mGluR5 | Yes | Monomeric and aggregated forms | Neuroprotection Dampens immune response |
| NMDAR | Yes | Aggregated forms | Decreased homeostatic microglial activity |
| Cd11b | Yes | Aggregated forms | Increased microglial oxidative stress |
| P2X7 | Yes | Aggregated forms | Increased microglial oxidative stress |
| FcγRIIB | Yes | Aggregated forms | Inhibition of phagocytosis |
| RAGE | Yes | Monomeric and preferential binding of aggregated forms | Neuroinflammation evidenced by secretion of TNF-α, IL-1β, and IL-6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deyell, J.S.; Sriparna, M.; Ying, M.; Mao, X. The Interplay between α-Synuclein and Microglia in α-Synucleinopathies. Int. J. Mol. Sci. 2023, 24, 2477. https://doi.org/10.3390/ijms24032477
Deyell JS, Sriparna M, Ying M, Mao X. The Interplay between α-Synuclein and Microglia in α-Synucleinopathies. International Journal of Molecular Sciences. 2023; 24(3):2477. https://doi.org/10.3390/ijms24032477
Chicago/Turabian StyleDeyell, Jacob S., Manjari Sriparna, Mingyao Ying, and Xiaobo Mao. 2023. "The Interplay between α-Synuclein and Microglia in α-Synucleinopathies" International Journal of Molecular Sciences 24, no. 3: 2477. https://doi.org/10.3390/ijms24032477
APA StyleDeyell, J. S., Sriparna, M., Ying, M., & Mao, X. (2023). The Interplay between α-Synuclein and Microglia in α-Synucleinopathies. International Journal of Molecular Sciences, 24(3), 2477. https://doi.org/10.3390/ijms24032477