
Regulatory Role of Ribonucleotide Reductase Subunit M2 in Hepatocyte Growth and Pathogenesis of Hepatitis C Virus


Abstract
:1. Introduction
2. RRM2, Its Expression throughout the Cell Cycle, Subcellular Localization, and Functions
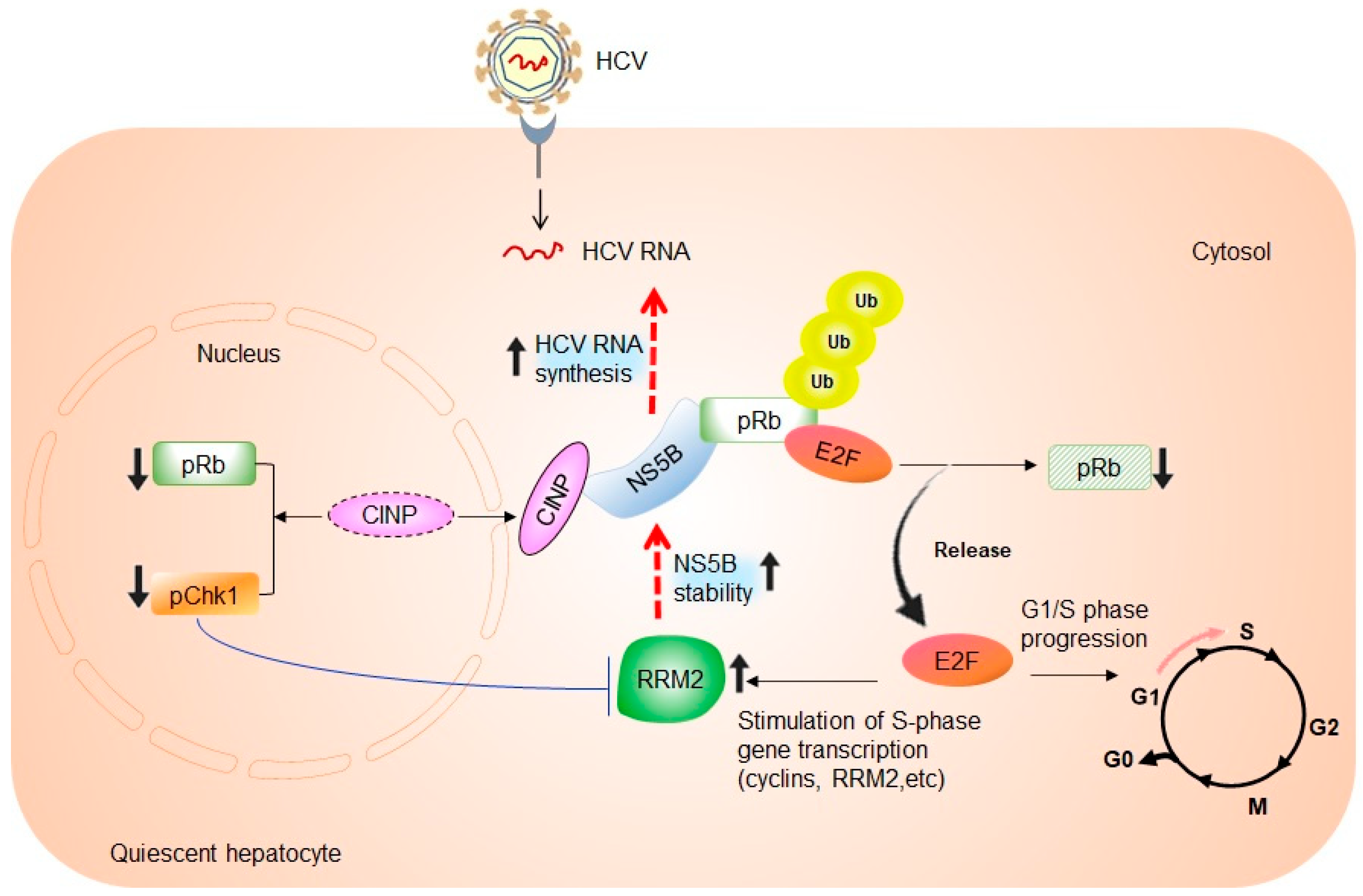
3. Cell Cycle-Dependent Regulation of HCV RNA Replication
4. The Link between RRM2 Expression, S-Phase of Cell Cycle, and HCV RNA Synthesis

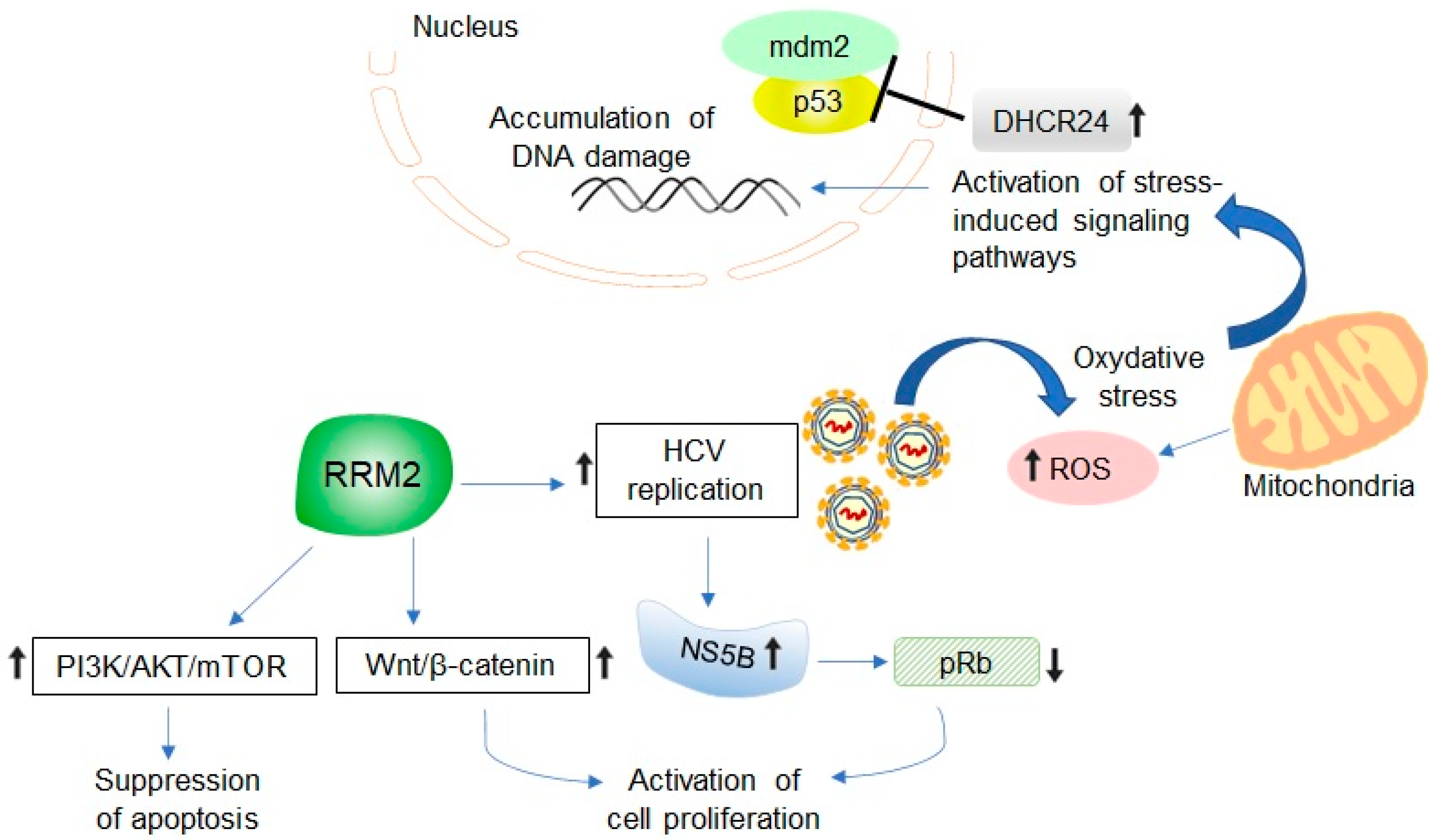
5. Potential Relevance of HCV-Induced RRM2 Upregulation in Promoting Liver Tumorigenesis
6. Developing RRM2 as Molecular Biomarker and Therapeutic Target in HCV-Associated HCC
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Nordlund, P.; Reichard, P. Ribonucleotide reductases. Annu. Rev. Biochem. 2006, 75, 681–706. [Google Scholar] [CrossRef]
- Engström, Y.; Rozell, B.; Hansson, H.A.; Thelander, L. Localization of ribonucleotide reductase in mammalian cells. EMBO J. 1984, 3, 863–867. [Google Scholar] [CrossRef] [PubMed]
- Aye, Y.; Li, M.; Long, M.J.; Weiss, R.S. Ribonucleotide reductase and cancer: Biological mechanisms and targeted therapies. Oncogene 2015, 34, 2011–2021. [Google Scholar] [CrossRef] [PubMed]
- Polaris Observatory HCV Collaborators. Global change in hepatitis C virus prevalence and cascade of care between 2015 and 2020: A modelling study. Lancet Gastroenterol. Hepatol. 2022, 7, 396–415. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Conti, F.; Buonfiglioli, F.; Scuteri, A.; Crespi, C.; Bolondi, L.; Caraceni, P.; Foschi, F.G.; Lenzi, M.; Mazzella, G.; Verucchi, G.; et al. Early occurrence and recurrence of hepatocellular carcinoma in HCV-related cirrhosis treated with direct-acting antivirals. J. Hepatol. 2016, 65, 727–733. [Google Scholar] [CrossRef]
- Hayes, C.N.; Zhang, P.; Zhang, Y.; Chayama, K. Molecular mechanisms of hepatocarcinogenesis following sustained virological response in patients with chronic hepatitis C virus infection. Viruses 2018, 10, 531. [Google Scholar] [CrossRef]
- Van der Meer, A.J.; Berenguer, M. Reversion of disease manifestations after HCV eradication. J. Hepatol. 2016, 65, 95–108. [Google Scholar] [CrossRef] [Green Version]
- Choo, Q.L.; Richman, K.H.; Han, J.H.; Berger, K.; Lee, C.; Dong, C.; Gallegos, C.; Coit, D.; Medina-Selby, R.; Barr, P.J.; et al. Genetic organization and diversity of the hepatitis C virus. Proc. Natl. Acad. Sci. USA 1991, 88, 2451–2455. [Google Scholar] [CrossRef] [Green Version]
- Tsukiyama-Kohara, K.; Iizuka, N.; Kohara, M.; Nomoto, A. Internal ribosome entry site within hepatitis C virus RNA. J. Virol. 1992, 66, 1476–1483. [Google Scholar] [CrossRef]
- Moradpour, D.; Penin, F. Hepatitis C virus proteins: From structure to function. Curr. Top. Microbiol. Immunol. 2013, 369, 113–142. [Google Scholar] [PubMed]
- Otsuka, M.; Kato, N.; Lan, K.; Yoshida, H.; Kato, J.; Goto, T.; Shiratori, Y.; Omata, M. Hepatitis C virus core protein enhances p53 function through augmentation of DNA binding affinity and transcriptional ability. J. Biol. Chem. 2000, 275, 34122–34130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marshall, A.; Rushbrook, S.; Morris, L.S.; Scott, I.S.; Vowler, S.L.; Davies, S.E.; Coleman, N.; Alexander, G. Hepatocyte expression of minichromosome maintenance protein-2 predicts fibrosis progression after transplantation for chronic hepatitis C virus: A pilot study. Liver Transplant. 2005, 11, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Walters, K.A.; Syder, A.J.; Lederer, S.L.; Diamond, D.L.; Paeper, B.; Rice, C.M.; Katze, M.G. Genomic analysis reveals a potential role for cell cycle perturbation in HCV-mediated apoptosis of cultured hepatocytes. PLoS Pathog. 2009, 5, e1000269. [Google Scholar] [CrossRef] [PubMed]
- Kitab, B.; Satoh, M.; Ohmori, Y.; Munakata, T.; Sudoh, M.; Kohara, M.; Tsukiyama-Kohara, K. Ribonucleotide reductase M2 promotes RNA replication of hepatitis C virus by protecting NS5B protein from hPLIC1-dependent proteasomal degradation. J. Biol. Chem. 2019, 294, 5759–5773. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.; Ha, S.Y.; Song, D.H.; Lee, H.W.; Cho, S.Y.; Park, C.K. High expression of ribonucleotide reductase subunit M2 correlates with poor prognosis of hepatocellular carcinoma. Gut Liver 2014, 8, 662–668. [Google Scholar] [CrossRef] [Green Version]
- Satow, R.; Shitashige, M.; Kanai, Y.; Takeshita, F.; Ojima, H.; Jigami, T.; Honda, K.; Kosuge, T.; Ochiya, T.; Hirohashi, S.; et al. Combined functional genome survey of therapeutic targets for hepatocellular carcinoma. Clin. Cancer Res. 2010, 16, 2518–2528. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.M.; Lin, L.S.; Liu, T.P. Sorafenib inhibits Ribonucleotide Reductase Regulatory Subunit M2 (RRM2) in hepatocellular carcinoma cells. Biomolecules 2020, 10, 117. [Google Scholar] [CrossRef] [Green Version]
- Fairman, J.W.; Wijerathna, S.R.; Ahmad, M.F.; Xu, H.; Nakano, R.; Jha, S.; Prendergast, J.; Welin, R.M.; Flodin, S.; Roos, A.; et al. Structural basis for allosteric regulation of human ribonucleotide reductase by nucleotide-induced oligomerization. Nat. Struct. Mol. Biol. 2011, 18, 316–322. [Google Scholar] [CrossRef] [Green Version]
- Nordlund, P.; Sjoberg, B.M.; Eklund, H. Three-dimensional structure of the free radical protein of ribonucleotide reductase. Nature 1990, 345, 593–598. [Google Scholar] [CrossRef]
- Stubbe, J. Di-iron-tyrosyl radical ribonucleotide reductases. Curr. Opin. Chem. Biol. 2003, 7, 183–188. [Google Scholar] [CrossRef]
- Eriksson, S.; Gräslund, A.; Skog, S.; Thelander, L.; Tribukait, B. Cell cycle-dependent regulation of mammalian ribonucleotide reductase. The S phase-correlated increase in subunit M2 is regulated by de novo protein synthesis. J. Biol. Chem. 1984, 259, 11695–11700. [Google Scholar] [CrossRef] [PubMed]
- Björklund, S.; Skog, S.; Tribukait, B.; Thelander, L. S-phase-specific expression of mammalian ribonucleotide reductase R1 and R2 subunit mRNAs. Biochemistry 1990, 29, 5452–5458. [Google Scholar] [CrossRef] [PubMed]
- Filatov, D.; Thelander, L. Role of a proximal NF-Y binding promoter element in S phase-specific expression of mouse ribonucleotide reductase R2 gene. J. Biol. Chem. 1995, 270, 25239–25243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chabes, A.L.; Björklund, S.; Thelander, L. S phase-specific transcription of the mouse ribonucleotide reductase R2 gene requires both a proximal repressive E2F-binding site and an upstream promoter activating region. J. Biol. Chem. 2004, 279, 10796–10807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Angiolella, V.; Donato, V.; Forrester, F.M.; Jeong, Y.T.; Pellacani, C.; Kudo, Y.; Saraf, A.; Florens, L.; Washburn, M.P.; Pagano, M. Cyclin F-mediated degradation of ribonucleotide reductase M2 controls genome integrity and DNA repair. Cell 2012, 149, 1023–1034. [Google Scholar] [CrossRef] [Green Version]
- Engström, Y.; Eriksson, S.; Jildevik, I.; Skog, S.; Thelander, L.; Tribukait, B. Cell cycle-dependent expression of mammalian ribonucleotide reductase. Differential regulation of the two subunits. J. Biol. Chem. 1985, 260, 9114–9116. [Google Scholar] [CrossRef]
- Chabes, A.; Thelander, L. Controlled protein degradation regulates ribonucleotide reductase activity in proliferating mammalian cells during the normal cell cycle and in response to DNA damage and replication blocks. J. Biol. Chem. 2000, 275, 17747–17753. [Google Scholar] [CrossRef] [Green Version]
- Chabes, A.L.; Pfleger, C.M.; Kirschner, M.W.; Thelander, L. Mouse ribonucleotide reductase R2 protein: A new target for anaphase-promoting complex-Cdh1-mediated proteolysis. Proc. Natl. Acad. Sci. USA 2003, 100, 3925–3929. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, H.; Arakawa, H.; Yamaguchi, T.; Shiraishi, K.; Fukuda, S.; Matsui, K.; Takei, Y.; Nakamura, Y. A ribonucleotide reductase gene involved in a p53-dependent cell-cycle checkpoint for DNA damage. Nature 2000, 404, 42–49. [Google Scholar] [CrossRef]
- Guittet, O.; Hakansson, P.; Voevodskaya, N.; Fridd, S.; Graslund, A.; Arakawa, H.; Nakamura, Y.; Thelander, L. Mammalian p53R2 protein forms an active ribonucleotide reductase in vitro with the R1 protein, which is expressed both in resting cells in response to DNA damage and in proliferating cells. J. Biol. Chem. 2001, 276, 40647–40651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pontarin, G.; Ferraro, P.; Bee, L.; Reichard, P.; Bianchi, V. Mammalian ribonucleotide reductase subunit p53R2 is required for mitochondrial DNA replication and DNA repair in quiescent cells. Proc. Natl. Acad. Sci. USA 2012, 109, 13302–13307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engström, Y.; Rozell, B. Immunocytochemical evidence for the cytoplasmic localization and differential expression during the cell cycle of the M1 and M2 subunits of mammalian ribonucleotide reductase. EMBO J. 1988, 7, 1615–1620. [Google Scholar] [CrossRef] [PubMed]
- Pontarin, G.; Fijolek, A.; Pizzo, P.; Ferraro, P.; Rampazzo, C.; Pozzan, T.; Thelander, L.; Reichard, P.A.; Bianchi, V. Ribonucleotide reduction is a cytosolic process in mammalian cells independently of DNA damage. Proc. Natl. Acad. Sci. USA 2008, 105, 17801–17806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niida, H.; Katsuno, Y.; Sengoku, M.; Shimada, M.; Yukawa, M.; Ikura, M.; Ikura, T.; Kohno, K.; Shima, H.; Suzuki, H.; et al. Essential role of Tip60-dependent recruitment of ribonucleotide reductase at DNA damage sites in DNA repair during G1 phase. Genes Dev. 2010, 24, 333–338. [Google Scholar] [CrossRef] [Green Version]
- Michalopoulos, G.K.; DeFrances, M.C. Liver regeneration. Science 1997, 276, 60–66. [Google Scholar] [CrossRef]
- Sainz, B., Jr.; Chisari, F.V. Production of infectious hepatitis C virus by well-differentiated, growth-arrested human hepatoma-derived cells. J. Virol. 2006, 80, 10253–10257. [Google Scholar] [CrossRef] [Green Version]
- Bauhofer, O.; Ruggieri, A.; Schmid, B.; Schirmacher, P.; Bartenschlager, R. Persistence of HCV in quiescent hepatic cells under conditions of an interferon-induced antiviral response. Gastroenterology 2012, 143, 429–438. [Google Scholar] [CrossRef]
- Pietschmann, T.; Lohmann, V.; Rutter, G.; Kurpanek, K.; Bartenschlager, R. Characterization of cell lines carrying self-replicating hepatitis C virus RNAs. J. Virol. 2001, 75, 1252–1264. [Google Scholar] [CrossRef] [Green Version]
- Windisch, M.P.; Frese, M.; Kaul, A.; Trippler, M.; Lohmann, V.; Bartenschlager, R. Dissecting the interferon-induced inhibition of hepatitis C virus replication by using a novel host cell line. J. Virol. 2005, 79, 13778–13793. [Google Scholar] [CrossRef]
- Nelson, H.B.; Tang, H. Effect of cell growth on hepatitis C virus (HCV) replication and a mechanism of cell confluence-based inhibition of HCV RNA and protein expression. J. Virol. 2006, 80, 1181–1190. [Google Scholar] [CrossRef] [Green Version]
- Honda, M.; Kaneko, S.; Matsushita, E.; Kobayashi, K.; Abell, G.A.; Lemon, S.M. Cell cycle regulation of hepatitis C virus internal ribosomal entry site-directed translation. Gastroenterology 2000, 118, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Scholle, F.; Li, K.; Bodola, F.; Ikeda, M.; Luxon, B.A.; Lemon, S.M. Virus-host cell interactions during hepatitis C virus RNA replication: Impact of polyprotein expression on the cellular transcriptome and cell cycle association with viral RNA synthesis. J. Virol. 2004, 78, 1513–1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stuyver, L.J.; McBrayer, T.R.; Tharnish, P.M.; Hassan, A.E.; Chu, C.K.; Pankiewicz, K.W.; Watanabe, K.A.; Schinazi, R.F.; Otto, M.J. Dynamics of subgenomic hepatitis C virus replicon RNA levels in Huh-7 cells after exposure to nucleoside antimetabolites. J. Virol. 2003, 77, 10689–10694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honda, M.; Kaneko, S.; Shimazaki, T.; Matsushita, E.; Kobayashi, K.; Ping, L.H.; Zhang, H.C.; Lemon, S.M. Hepatitis C virus core protein induces apoptosis and impairs cell-cycle regulation in stably transformed Chinese hamster ovary cells. Hepatology 2000, 31, 1351–1359. [Google Scholar] [CrossRef]
- Munakata, T.; Nakamura, M.; Liang, Y.; Li, K.; Lemon, S.M. Down-regulation of the retinoblastoma tumor suppressor by the hepatitis C virus NS5B RNA-dependent RNA polymerase. Proc. Natl. Acad. Sci. USA 2005, 102, 18159–18164. [Google Scholar] [CrossRef] [Green Version]
- Munakata, T.; Liang, Y.; Kim, S.; McGivern, D.R.; Huibregtse, J.; Nomoto, A.; Lemon, S.M. Hepatitis C virus induces E6AP-dependent degradation of the retinoblastoma protein. PLoS Pathog. 2007, 3, 1335–1347. [Google Scholar] [CrossRef] [Green Version]
- Stevaux, O.; Dyson, N.J. A revised picture of the E2F transcriptional network and RB function. Curr. Opin. Cell Biol. 2002, 14, 684–691. [Google Scholar] [CrossRef]
- Tsukiyama-Kohara, K.; Tone, S.; Maruyama, I.; Inoue, K.; Katsume, A.; Nuriya, H.; Ohmori, H.; Ohkawa, J.; Taira, K.; Hoshikawa, Y.; et al. Activation of the CKI-CDK-Rb-E2F pathway in full genome hepatitis C virus-expressing cells. J. Biol. Chem. 2004, 279, 14531–14541. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wang, Y.; Xu, Y.; Tong, W.; Pan, T.; Li, J.; Sun, S.; Shao, J.; Ding, H.; Toyoda, T.; et al. Hepatitis C virus NS5B protein delays S phase progression in human hepatocyte-derived cells by relocalizing cyclin-dependent kinase 2-interacting protein (CINP). J. Biol. Chem. 2011, 286, 26603–26615. [Google Scholar] [CrossRef]
- Zhao, H.; Piwnica-Worms, H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol. Cell. Biol. 2001, 21, 4129–4139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Z.; Chen, Z.; Gunasekera, A.H.; Sowin, T.J.; Rosenberg, S.H.; Fesik, S.; Zhang, H. Chk1 mediates S and G2 arrests through Cdc25A degradation in response to DNA-damaging agents. J. Biol. Chem. 2003, 278, 21767–21773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, L.; Elledge, S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jirmanova, L.; Bulavin, D.V.; Fornace, A.J., Jr. Inhibition of the ATR/Chk1 pathway induces a p38-dependent S-phase delay in mouse embryonic stem cells. Cell Cycle 2005, 10, 1428–1434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, M.; Niida, H.; Zineldeen, D.H.; Tagami, H.; Tanaka, M.; Saito, H.; Nakanishi, M. Chk1 is a histone H3 threonine 11 kinase that regulates DNA damage-induced transcriptional repression. Cell 2008, 132, 221–232. [Google Scholar] [CrossRef] [Green Version]
- Naruyama, H.; Shimada, M.; Niida, H.; Zineldeen, D.H.; Hashimoto, Y.; Kohri, K.; Nakanishi, M. Essential role of Chk1 in S phase progression through regulation of RNR2 expression. Biochem. Biophys. Res. Commun. 2008, 374, 79–83. [Google Scholar] [CrossRef]
- Ruggieri, A.; Murdolo, M.; Harada, T.; Miyamura, T.; Rapicetta, M. Cell cycle perturbation in a human hepatoblastoma cell line constitutively expressing hepatitis C virus core protein. Arch. Virol. 2004, 149, 61–74. [Google Scholar] [CrossRef]
- Yang, X.J.; Liu, J.; Ye, L.; Liao, Q.J.; Wu, J.G.; Gao, J.R.; She, Y.L.; Wu, Z.H.; Ye, L.B. HCV NS2 protein inhibits cell proliferation and induces cell cycle arrest in the S-phase in mammalian cells through down-regulation of cyclin A expression. Virus Res. 2006, 121, 134–143. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Steele, R.; Meyer, K.; Ray, R.; Ray, R.B. Hepatitis C virus NS5A protein modulates cell cycle regulatory genes and promotes cell growth. J. Gen. Virol. 1999, 80, 1179–1183. [Google Scholar] [CrossRef] [Green Version]
- Heidel, J.D.; Liu, J.Y.; Yen, Y.; Zhou, B.; Heale, B.S.; Rossi, J.J.; Bartlett, D.W.; Davis, M.E. Potent siRNA inhibitors of ribonucleotide reductase subunit RRM2 reduce cell proliferation in vitro and in vivo. Clin. Cancer Res. 2007, 13, 2207–2215. [Google Scholar] [CrossRef]
- Ding, Y.; Zhong, T.; Wang, M.; Xiang, X.; Ren, G.; Jia, Z.; Lin, Q.; Liu, Q.; Dong, J.; Li, L.; et al. Integrative analysis reveals across-cancer expression patterns and clinical relevance of ribonucleotide reductase in human cancers. Front. Oncol. 2019, 9, 956. [Google Scholar] [CrossRef]
- Liu, X.; Zhou, B.; Xue, L.; Yen, F.; Chu, P.; Un, F.; Yen, Y. Ribonucleotide reductase subunits M2 and p53R2 are potential biomarkers for metastasis of colon cancer. Clin. Color. Cancer 2007, 6, 374–381. [Google Scholar] [CrossRef]
- Duxbury, M.S.; Whang, E.E. RRM2 induces NF-kappaB-dependent MMP-9 activation and enhances cellular invasiveness. Biochem. Biophys. Res. Commun. 2007, 354, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Souglakos, J.; Boukovinas, I.; Taron, M.; Mendez, P.; Mavroudis, D.; Tripaki, M.; Hatzidaki, D.; Koutsopoulos, A.; Stathopoulos, E.; Georgoulias, V.; et al. Ribonucleotide reductase subunits M1 and M2 mRNA expression levels and clinical outcome of lung adenocarcinoma patients treated with docetaxel/gemcitabine. Br. J. Cancer 2008, 98, 1710–1715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, H.; Villegas, C.; Huang, A.; Wright, J.A. The mammalian ribonucleotide reductase R2 component cooperates with a variety of oncogenes in mechanisms of cellular transformation. Cancer Res. 1998, 58, 1650–1653. [Google Scholar]
- Zhang, M.; Wang, J.; Yao, R.; Wang, L. Small interfering RNA (siRNA)-mediated silencing of the M2 subunit of ribonucleotide reductase: A novel therapeutic strategy in ovarian cancer. Int. J. Gynecol. Cancer 2013, 23, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Li, Y.; Zhou, J. Downregulation of ribonucleotide reductase subunits M2 induces apoptosis and G1 arrest of cervical cancer cells. Oncol. Lett. 2018, 15, 3719–3725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Chen, H.; Yu, Y.; Song, J.; Song, H.; Su, X.; Li, W.; Tong, X.; Qian, W.; Wang, H.; et al. Inhibition of hepatocellular carcinoma growth using immunoliposomes for co-delivery of adriamycin and ribonucleotide reductase M2 siRNA. Biomaterials 2013, 34, 10084–10098. [Google Scholar] [CrossRef]
- Zhou, X.; Zhu, H.Q.; Lu, J. Regulation of gene expression in HBV- and HCV-related hepatocellular carcinoma: Integrated GWRS and GWGS analyses. Int. J. Clin. Exp. Med. 2014, 7, 4038–4050. [Google Scholar]
- He, B.; Yin, J.; Gong, S.; Gu, J.; Xiao, J.; Shi, W.; Ding, W.; He, Y. Bioinformatics analysis of key genes and pathways for hepatocellular carcinoma transformed from cirrhosis. Medicine 2017, 96, e6938. [Google Scholar] [CrossRef]
- Wang, N.; Zhan, T.; Ke, T.; Huang, X.; Ke, D.; Wang, Q.; Li, H. Increased expression of RRM2 by human papillomavirus E7 oncoprotein promotes angiogenesis in cervical cancer. Br. J. Cancer 2014, 110, 1034–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukiyama-Kohara, K. Role of oxidative stress in hepatocarcinogenesis induced by hepatitis C virus. Int. J. Mol. Sci. 2012, 13, 15271–15278. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.M.; Kim, S.J.; Kim, J.H.; Lee, W.; Kim, G.W.; Lee, K.H.; Choi, K.Y.; Oh, J.W. Interaction of hepatitis C virus core protein with Hsp60 triggers the production of reactive oxygen species and enhances TNF-alpha-mediated apoptosis. Cancer Lett. 2009, 279, 230–237. [Google Scholar] [CrossRef]
- Dionisio, N.; Garcia-Mediavilla, M.V.; Sanchez-Campos, S.; Majano, P.L.; Benedicto, I.; Rosado, J.A.; Salido, G.M.; Gonzalez-Gallego, J. Hepatitis C virus NS5A and core proteins induce oxidative stress-mediated calcium signalling alterations in hepatocytes. J. Hepatol. 2009, 50, 872–882. [Google Scholar] [CrossRef] [PubMed]
- Moriya, K.; Fujie, H.; Shintani, Y.; Yotsuyanagi, H.; Tsutsumi, T.; Ishibashi, K.; Matsuura, Y.; Kimura, S.; Miyamura, T.; Koike, K. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat. Med. 1998, 4, 1065–1067. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, T.; Kohara, M.; Izumi, K.; Kasama, Y.; Hirata, Y.; Huang, Y.; Shuda, M.; Mukaidani, C.; Takano, T.; Tokunaga, Y.; et al. Hepatitis C virus impairs p53 via persistent overexpression of 3beta-hydroxysterol delta24-reductase. J. Biol. Chem. 2009, 284, 36442–36452. [Google Scholar] [CrossRef] [Green Version]
- Samarin, J.; Laketa, V.; Malz, M.; Roessler, S.; Stein, I.; Horwitz, E.; Singer, S.; Dimou, E.; Cigliano, A.; Bissinger, M.; et al. PI3K/AKT/mTOR-dependent stabilization of oncogenic far-upstream element binding proteins in hepatocellular carcinoma cells. Hepatology 2016, 63, 813–826. [Google Scholar] [CrossRef] [Green Version]
- He, Z.; Hu, X.; Liu, W.; Dorrance, A.; Garzon, R.; Houghton, P.J.; Shen, C. P53 suppresses ribonucleotide reductase via inhibiting mTORC1. Oncotarget 2017, 8, 41422–41431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, J.; Wang, Z.; Mo, Q.; Long, J.; Fan, Y.; Cheng, L.; Zhang, T.; Liu, X.; Wang, X. Ribonucleotide reductase M2 subunit silencing suppresses tumorigenesis in pancreatic cancer via inactivation of PI3K/AKT/mTOR pathway. Pancreatology 2022, 22, 401–413. [Google Scholar] [CrossRef]
- Vasuri, F.; Visani, M.; Acquaviva, G.; Brand, T.; Fiorentino, M.; Pession, A.; Tallini, G.; D’Errico, A.; de Biase, D. Role of microRNAs in the main molecular pathways of hepatocellular carcinoma. World J. Gastroenterol. 2018, 24, 2647–2660. [Google Scholar] [CrossRef]
- Teufel, M.; Seidel, H.; Köchert, K.; Meinhardt, G.; Finn, R.S.; Llovet, J.M.; Bruix, J. Biomarkers associated with response to regorafenib in patients with hepatocellular carcinoma. Gastroenterology 2019, 156, 1731–1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Li, B. MicroRNA-582-3p targeting ribonucleotide reductase regulatory subunit M2 inhibits the tumorigenesis of hepatocellular carcinoma by regulating the Wnt/β-catenin signaling pathway. Bioengineered 2022, 13, 12876–12887. [Google Scholar] [CrossRef] [PubMed]
- Falade-Nwulia, O.; Suarez-Cuervo, C.; Nelson, D.R.; Fried, M.W.; Segal, J.B.; Sulkowski, M.S. Oral Direct-Acting Agent Therapy for Hepatitis C Virus Infection: A Systematic Review. Ann. Intern. Med. 2017, 166, 637–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, L.; Bias, T.E.; Mathias, C.B.; Trooskin, S.B.; Fong, J.J. Sofosbuvir: A Nucleotide NS5B Inhibitor for the Treatment of Chronic Hepatitis C Infection. Ann. Pharmacother. 2014, 48, 1019–1029. [Google Scholar] [CrossRef]
- Beran, R.K.; Sharma, R.; Corsa, A.C.; Tian, Y.; Golde, J.; Lundgaard, G.; Delaney, W.E.; Zhong, W.; Greenstein, A.E. Cellular growth kinetics distinguish a cyclophilin inhibitor from an HSP90 inhibitor as a selective inhibitor of hepatitis C virus. PLoS ONE 2012, 7, e30286. [Google Scholar] [CrossRef]
- Cerqueira, N.M.; Fernandes, P.A.; Ramos, M.J. Understanding ribonucleotide reductase inactivation by gemcitabine. Chemistry 2007, 13, 8507–8515. [Google Scholar] [CrossRef]
- Konyn, P.; Ahmed, A.; Kim, D. Current epidemiology in hepatocellular carcinoma. Expert Rev. Gastroenterol. Hepatol. 2021, 15, 1295–1307. [Google Scholar] [CrossRef]
- Liu, X.; Xu, Z.; Hou, C.; Wang, M.; Chen, X.; Lin, Q.; Song, R.; Lou, M.; Zhu, L.; Qiu, Y.; et al. Inhibition of hepatitis B virus replication by targeting ribonucleotide reductase M2 protein. Biochem. Pharmacol. 2016, 103, 118–128. [Google Scholar] [CrossRef]
- Wu, X.; Li, J.; Gassa, A.; Buchner, D.; Alakus, H.; Dong, Q.; Ren, N.; Liu, M.; Odenthal, M.; Stippel, D.; et al. Circulating tumor DNA as an emerging liquid biopsy biomarker for early diagnosis and therapeutic monitoring in hepatocellular carcinoma. Int. J. Biol. Sci. 2020, 16, 1551–1562. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [Green Version]
- Galmiche, A.; Chauffert, B.; Barbare, J.C. New biological perspectives for the improvement of the efficacy of sorafenib in hepatocellular carcinoma. Cancer Lett. 2014, 346, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Niu, X.; Chen, R.; He, W.; Chen, D.; Kang, R.; Tang, D. Metallothionein-1G facilitates sorafenib resistance through inhibition of ferroptosis. Hepatology 2016, 64, 488–500. [Google Scholar] [CrossRef] [PubMed]
- Yamane, D.; Hayashi, Y.; Matsumoto, M.; Nakanishi, H.; Imagawa, H.; Kohara, M.; Lemon, S.M.; Ichi, I. FADS2-dependent fatty acid desaturation dictates cellular sensitivity to ferroptosis and permissiveness for hepatitis C virus replication. Cell Chem. Biol. 2022, 29, 799–810. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| First Author [Ref.] | Samples and Methodology | Main Results |
|---|---|---|
| Lee et al. [16] |
|
|
| Zhou et al. [69] |
|
|
| He et al. [70] |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kitab, B.; Tsukiyama-Kohara, K. Regulatory Role of Ribonucleotide Reductase Subunit M2 in Hepatocyte Growth and Pathogenesis of Hepatitis C Virus. Int. J. Mol. Sci. 2023, 24, 2619. https://doi.org/10.3390/ijms24032619
Kitab B, Tsukiyama-Kohara K. Regulatory Role of Ribonucleotide Reductase Subunit M2 in Hepatocyte Growth and Pathogenesis of Hepatitis C Virus. International Journal of Molecular Sciences. 2023; 24(3):2619. https://doi.org/10.3390/ijms24032619
Chicago/Turabian StyleKitab, Bouchra, and Kyoko Tsukiyama-Kohara. 2023. "Regulatory Role of Ribonucleotide Reductase Subunit M2 in Hepatocyte Growth and Pathogenesis of Hepatitis C Virus" International Journal of Molecular Sciences 24, no. 3: 2619. https://doi.org/10.3390/ijms24032619
APA StyleKitab, B., & Tsukiyama-Kohara, K. (2023). Regulatory Role of Ribonucleotide Reductase Subunit M2 in Hepatocyte Growth and Pathogenesis of Hepatitis C Virus. International Journal of Molecular Sciences, 24(3), 2619. https://doi.org/10.3390/ijms24032619