
Investigating Therapeutic Effects of Indole Derivatives Targeting Inflammation and Oxidative Stress in Neurotoxin-Induced Cell and Mouse Models of Parkinson’s Disease

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Indole Compounds and Cytotoxicity
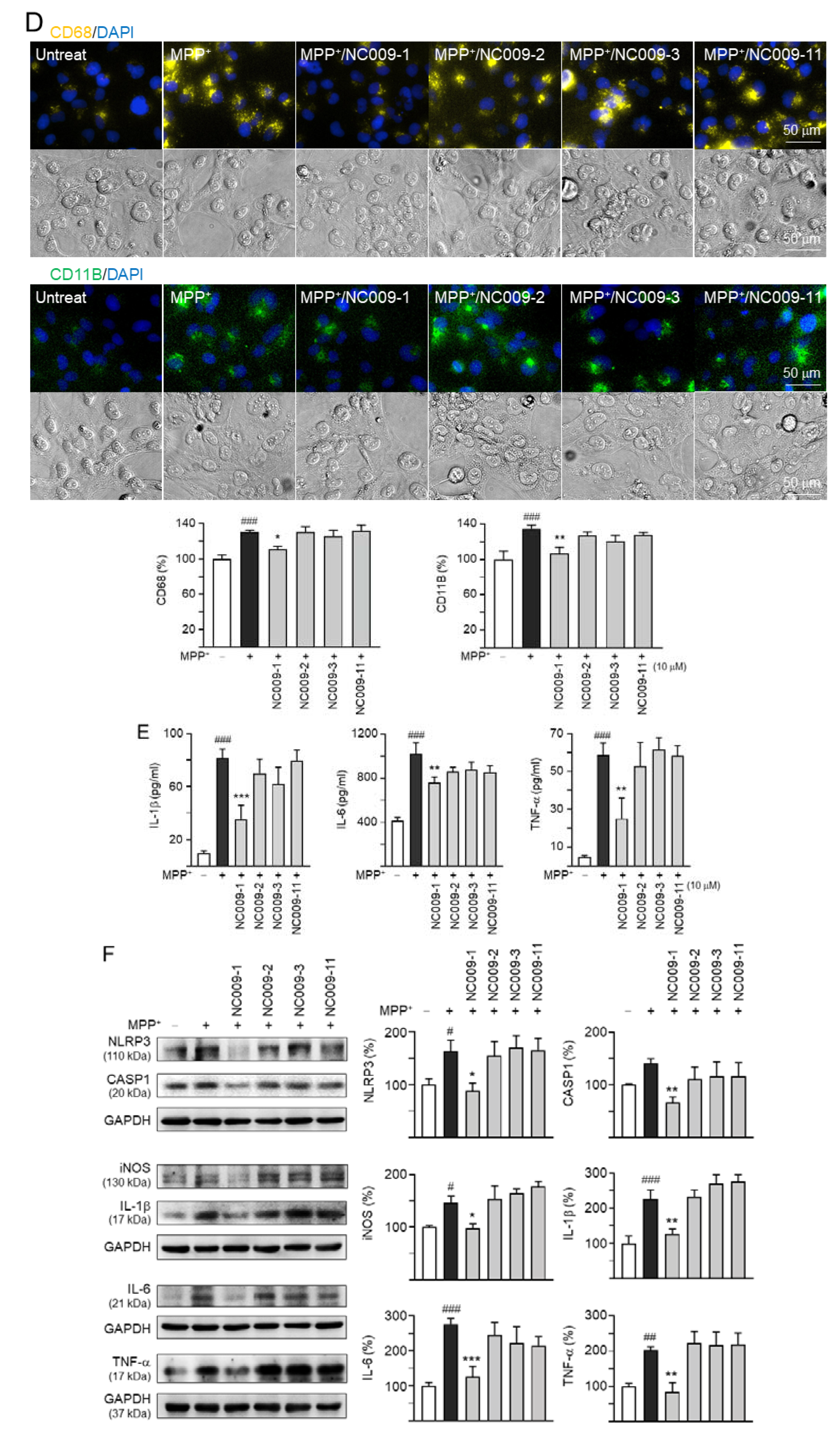
2.2. Activation of HMC3 Microglia and Anti-Inflammatory Potentials of NC009 Compounds
2.3. Effect of NC009-1 on MPTP-Induced Motor Behavior in Mice
2.4. Effect of NC009-1 on Dopamine, DAT, 4-HNE/TH, IBA1, and GFAP Levels in MPTP-Treated Mice
2.5. Down-Regulation of Neuroinflammation and Up-Regulation of Cellular Redox Signaling by NC009-1 in MPTP-Treated Mice
3. Discussion
4. Materials and Methods
4.1. Compounds and Cell Culture
4.2. Bioavailability and BBB Permeation Prediction
4.3. Cytotoxicity Assay
4.4. Antioxidant Assay
4.5. HMC3 Microglia Activation and Inflammatory Mediators Detection
4.6. Sub-Chronic MPTP Mouse Model
4.7. Behavioral Tests
4.8. HPLC Analysis of Dopamine
4.9. Immunohistochemistry Analysis
4.10. Neuroinflammation and Oxidative Stress Analyses in Mice
4.11. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jankovic, J. Parkinson’s disease: Clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surmeier, D.J.; Obeso, J.A.; Halliday, G.M. Selective neuronal vulnerability in Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 101–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef] [PubMed]
- Jenner, P. Oxidative stress in Parkinson’s disease. Ann. Neurol. 2003, 53 (Suppl. 3), S26–S36. [Google Scholar] [CrossRef] [PubMed]
- Tuite, P.J.; Krawczewski, K. Parkinsonism: A review-of-systems approach to diagnosis. Semin. Neurol. 2007, 27, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Nicklas, W.J.; Youngster, S.K.; Kindt, M.V.; Heikkila, R.E., IV. MPTP, MPP+ and mitochondrial function. Life Sci. 1987, 40, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Lofrumento, D.D.; Saponaro, C.; Cianciulli, A.; De Nuccio, F.; Mitolo, V.; Nicolardi, G.; Panaro, M.A. MPTP-induced neuroinflammation increases the expression of pro-inflammatory cytokines and their receptors in mouse brain. Neuroimmunomodulation 2011, 18, 79–88. [Google Scholar] [CrossRef]
- Phani, S.; Loike, J.D.; Przedborski, S. Neurodegeneration and inflammation in Parkinson’s disease. Parkinsonism Relat. Disord. 2012, 18 (Suppl. 1), S207–S209. [Google Scholar] [CrossRef]
- Sanchez-Guajardo, V.; Barnum, C.J.; Tansey, M.G.; Romero-Ramos, M. Neuroimmunological processes in Parkinson’s disease and their relation to α-synuclein: Microglia as the referee between neuronal processes and peripheral immunity. ASN Neuro 2013, 5, 113–139. [Google Scholar] [CrossRef] [Green Version]
- Bassani, T.B.; Vital, M.A.; Rauh, L.K. Neuroinflammation in the pathophysiology of Parkinson’s disease and therapeutic evidence of anti-inflammatory drugs. Arq. Neuropsiquiatr. 2015, 7, 616–623. [Google Scholar] [CrossRef]
- Kumar, D.; Sharma, S.; Kalra, S.; Singh, G.; Monga, V.; Kumar, B. Medicinal perspective of indole derivatives: Recent developments and structure-activity relationship studies. Curr. Drug Targets. 2020, 21, 864–891. [Google Scholar] [PubMed]
- Chang, K.H.; Lin, C.H.; Chen, H.C.; Huang, H.Y.; Chen, S.L.; Lin, T.H.; Ramesh, C.; Huang, C.C.; Fung, H.C.; Wu, Y.R.; et al. The potential of indole/indolylquinoline compounds in tau misfolding reduction by enhancement of HSPB1. CNS Neurosci. Ther. 2017, 23, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.M.; Chen, W.L.; Hung, C.T.; Lin, T.H.; Chao, C.Y.; Lin, C.H.; Wu, Y.R.; Chang, K.H.; Yao, C.F.; Lee-Chen, G.J.; et al. The indole compound NC009-1 inhibits aggregation and promotes neurite outgrowth through enhancement of HSPB1 in SCA17 cells and ameliorates the behavioral deficits in SCA17 mice. Neurotoxicology 2018, 67, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Chiu, Y.J.; Lin, C.H.; Hsu, W.C.; Wu, J.L.; Huang, C.H.; Lin, C.W.; Yao, C.F.; Huang, H.J.; Lo, Y.S.; et al. Indole compound NC009-1 augments APOE and TRKA in Alzheimer’s disease cell and mouse models for neuroprotection and cognitive improvement. J. Alzheimers Dis. 2019, 67, 737–756. [Google Scholar] [CrossRef]
- Chiu, Y.J.; Lin, S.A.; Chen, W.L.; Lin, T.H.; Lin, C.H.; Yao, C.F.; Lin, W.; Wu, Y.R.; Chang, K.H.; Lee-Chen, G.J.; et al. Pathomechanism characterization and potential therapeutics identification for SCA3 targeting neuroinflammation. Aging 2020, 12, 23619–23646. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Hitchcock, S.A.; Pennington, L.D. Structure—Brain exposure relationships. J. Med. Chem. 2006, 49, 7559–7583. [Google Scholar] [CrossRef]
- Liu, H.; Wang, L.; Lv, M.; Pei, R.; Li, P.; Pei, Z.; Wang, Y.; Su, W.; Xie, X.Q. AlzPlatform: An Alzheimer’s disease domain-specific chemogenomics knowledgebase for polypharmacology and target identification research. J. Chem. Inf. Model. 2014, 54, 1050–1060. [Google Scholar] [CrossRef]
- Boots, A.W.; Haenen, G.R.; Bast, A. Health effects of quercetin: From antioxidant to nutraceutical. Eur. J. Pharmacol. 2008, 585, 325–337. [Google Scholar] [CrossRef]
- Kotake, Y.; Ohta, S. MPP+ analogs acting on mitochondria and inducing neuro-degeneration. Curr. Med. Chem. 2003, 10, 2507–2516. [Google Scholar] [CrossRef]
- Bournival, J.; Plouffe, M.; Renaud, J.; Provencher, C.; Martinoli, M.G. Quercetin and sesamin protect dopaminergic cells from MPP+-induced neuroinflammation in a microglial (N9)-neuronal (PC12) coculture system. Oxid. Med. Cell. Longev. 2012, 2012, 921941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoogland, I.C.; Houbolt, C.; van Westerloo, D.J.; van Gool, W.A.; van de Beek, D. Systemic inflammation and microglial activation: Systematic review of animal experiments. J. Neuroinflamm. 2015, 12, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, S.; Li, L.; Sun, X.; Hua, J.; Zhang, K.; Hao, L.; Liu, L.; Shi, D.; Zhou, H. FTY720 inhibits MPP+-induced microglial activation by affecting NLRP3 inflammasome activation. J. Neuroimmune Pharmacol. 2019, 14, 478–492. [Google Scholar] [CrossRef] [PubMed]
- Blandini, F.; Armentero, M.T. Animal models of Parkinson’s disease. FEBS J. 2012, 279, 1156–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steru, L.; Chermat, R.; Thierry, B.; Simon, P. The tail suspension test: A new method for screening antidepressants in mice. Psychopharmacology 1985, 85, 367–370. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Shao, X.Y.; Qi, G.J.; Chen, Q.; Bu, L.L.; Chen, L.J.; Shi, J.; Ming, J.; Tian, B. Cdk5-dependent activation of neuronal inflammasomes in Parkinson’s disease. Mov. Disord. 2016, 31, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Hwang, I.; Park, S.; Hong, S.; Hwang, B.; Cho, Y.; Son, J.; Yu, J.W. MPTP-driven NLRP3 inflammasome activation in microglia plays a central role in dopaminergic neurodegeneration. Cell Death Differ. 2019, 26, 213–228. [Google Scholar] [CrossRef]
- Block, M.L.; Zecca, L.; Hong, J.S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef]
- Gerhard, A.; Pavese, N.; Hotton, G.; Turkheimer, F.; Es, M.; Hammers, A.; Eggert, K.; Oertel, W.; Banati, R.B.; Brooks, D.J. In vivo imaging of microglial ctivation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol. Dis. 2006, 21, 404–412. [Google Scholar] [CrossRef]
- Subhramanyam, C.S.; Wang, C.; Hu, Q.; Dheen, S.T. Microglia-mediated neuroinflammation in neurodegenerative diseases. Semin. Cell Dev. Biol. 2019, 94, 112–120. [Google Scholar] [CrossRef]
- McGeer, P.L.; Itagaki, S.; Boyes, B.E.; McGeer, E.G. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 1988, 38, 1285–1291. [Google Scholar] [CrossRef] [PubMed]
- Walker, M.D.; Dinelle, K.; Kornelsen, R.; Lee, N.V.; Miao, Q.; Adam, M.; Takhar, C.; Mak, E.; Schulzer, M.; Farrer, M.J.; et al. [11C]PBR28 PET imaging is sensitive to neuroinflammation in the aged rat. J. Cereb. Blood Flow Metab. 2015, 35, 1331–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Wang, J.; Zhao, C.; Bi, W.; Yue, Z.; Ma, Z.A. Genetic ablation of PLA2G6 in mice leads to cerebellar atrophy characterized by Purkinje cell loss and glial cell activation. PLoS ONE 2011, 6, e26991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Xia, Y.; Yin, S.; Wan, F.; Hu, J.; Kou, L.; Sun, Y.; Wu, J.; Zhou, Q.; Huang, J.; et al. Targeting microglial α-synuclein/TLRs/NF-kappaB/NLRP3 inflammasome axis in Parkinson’s disease. Front. Immunol. 2021, 12, 719807. [Google Scholar] [CrossRef] [PubMed]
- Gordon, R.; Albornoz, E.A.; Christie, D.C.; Langley, M.R.; Kumar, V.; Mantovani, S.; Robertson, A.A.B.; Butler, M.S.; Rowe, D.B.; O’Neill, L.A.; et al. Inflammasome inhibition prevents α-synuclein pathology and dopaminergic neurodegeneration in mice. Sci. Transl. Med. 2018, 10, eaah4066. [Google Scholar] [CrossRef] [Green Version]
- Mogi, M.; Harada, M.; Narabayashi, H.; Inagaki, H.; Minami, M.; Nagatsu, T. Interleukin (IL)-lβ.; IL-2.; IL-4.; IL-6 and transforming growth factor-α levels are elevated in ventricular cerebrospinal fluid in juvenile parkinsonism and Parkinson’s disease. Neurosci. Lett. 1996, 211, 13–16. [Google Scholar] [CrossRef]
- Nagatsu, T.; Mogi, M.; Ichinose, H.; Togari, A. Cytokines in Parkinson’s disease. J. Neural. Transm. Suppl. 2000, 58, 143–151. [Google Scholar]
- Williams-Gray, C.H.; Wijeyekoon, R.; Yarnall, A.J.; Lawson, R.A.; Breen, D.P.; Evans, J.R.; Cummins, G.A.; Duncan, G.W.; Khoo, T.K.; Burn, D.J.; et al. Serum immune markers and disease progression in an incident Parkinson’s disease cohort (ICICLE-PD). Mov. Disord. 2016, 31, 995–1003. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, C.C.; Pott Godoy, M.C.; Tarelli, R.; Chertoff, M.; Depino, A.M.; Pitossi, F.J. Progressive neurodegeneration and motor disabilities induced by chronic expression of IL-1β in the substantia nigra. Neurobiol. Dis. 2006, 24, 183–193. [Google Scholar] [CrossRef]
- Béraud, D.; Maguire-Zeiss, K.A. Misfolded α-synuclein and Toll-like receptors: Therapeutic targets for Parkinson’s disease. Parkinsonism Relat. Disord. 2012, 18 (Suppl. 1), S17–S20. [Google Scholar] [CrossRef] [Green Version]
- Lowenstein, C.J.; Padalko, E. iNOS (NOS2) at a glance. J. Cell Sci. 2004, 117, 2865–2867. [Google Scholar] [CrossRef] [PubMed]
- Hunot, S.; Boissiere, F.; Faucheux, B.; Brugg, B.; Mouatt-Prigent, A.; Agid, Y.; Hirsch, E.C. Nitric oxide synthase and neuronal vulnerability in Parkinson’s disease. Neuroscience 1996, 72, 355–363. [Google Scholar] [CrossRef]
- Zhu, J.; Gao, W.; Shan, X.; Wang, C.; Wang, H.; Shao, Z.; Dou, S.; Jiang, Y.; Wang, C.; Cheng, B. Apelin-36 mediates neuroprotective effects by regulating oxidative stress.; autophagy and apoptosis in MPTP-induced Parkinson’s disease model mice. Brain Res. 2020, 1726, 146493. [Google Scholar] [CrossRef]
- Broom, L.; Marinova-Mutafchieva, L.; Sadeghian, M.; Davis, J.B.; Medhurst, A.D.; Dexter, D.T. Neuroprotection by the selective iNOS inhibitor GW274150 in a model of Parkinson disease. Free Radic. Biol. Med. 2011, 50, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Tapias, V.; Hu, X.; Luk, K.C.; Sanders, L.H.; Lee, V.M.; Greenamyre, J.T. Synthetic alpha-synuclein fibrils cause mitochondrial impairment and selective dopamine neurodegeneration in part via iNOS-mediated nitric oxide production. Cell. Mol. Life Sci. 2017, 74, 2851–2874. [Google Scholar] [CrossRef] [PubMed]
- Stykel, M.G.; Ryan, S.D. Nitrosative stress in Parkinson’s disease. NPJ Parkinsonism Dis. 2022, 8, 104. [Google Scholar] [CrossRef] [PubMed]
- Dehmer, T.; Lindenau, J.; Haid, S.; Dichgans, J.; Schulz, J.B. Deficiency of inducible nitric oxide synthase protects against MPTP toxicity in vivo. J. Neurochem. 2000, 74, 2213–2216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruol, D.L.; Nelson, T.E. Physiological and pathological roles of interleukin-6 in the central nervous system. Mol. Neurobiol. 1997, 15, 307–339. [Google Scholar] [CrossRef]
- Blum-Degen, D.; Müller, T.; Kuhn, W.; Gerlach, M.; Przuntek, H.; Riederer, P. Interleukin-1β and interleukin-6 are elevated in the cerebrospinal fluid of Alzheimer’s and de novo Parkinson’s disease patients. Neurosci. Lett. 1995, 202, 17–20. [Google Scholar] [CrossRef]
- Brodacki, B.; Staszewski, J.; Toczylowska, B.; Kozlowska, E.; Drela, N.; Chalimoniuk, M.; Stepien, A. Serum interleukin (IL-2.; IL-10.; IL-6.; IL-4).; TNFα.; and INFγ concentrations are elevated in patients with atypical and idiopathic parkinsonism. Neurosci. Lett. 2008, 441, 158–162. [Google Scholar] [CrossRef]
- Dobbs, R.J.; Charlett, A.; Purkiss, A.G.; Dobbs, S.M.; Weller, C.; Peterson, D.W. Association of circulating TNF-α and IL-6 with ageing and parkinsonism. Acta Neurol. Scand. 1999, 100, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Dursun, E.; Gezen-Ak, D.; Hanagasi, H.; Bilgic, B.; Lohmann, E.; Ertan, S.; Atasoy, İ.L.; Alaylıoğlu, M.; Araz, Ö.S.; Önal, B.; et al. The interleukin 1 alpha.; interleukin 1 beta.; interleukin 6 and alpha-2-macroglobulin serum levels in patients with early or late onset Alzheimer’s disease.; mild cognitive impairment or Parkinson’s disease. J. Neuroimmunol. 2015, 283, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Gruden, M.A.; Yanamandra, K.; Kucheryanu, V.G.; Bocharova, O.R.; Sherstnev, V.V.; Morozova-Roche, L.A.; Sewell, R.D. Correlation between protective immunity to α-synuclein aggregates.; oxidative stress and inflammation. Neuroimmunomodulation 2012, 19, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, K.W.; Schuh, A.F.; Saute, J.; Townsend, R.; Fricke, D.; Leke, R.; Souza, D.O.; Portela, L.V.; Chaves, M.L.F.; Rieder, C.R.M. Interleukin-6 serum levels in patients with Parkinson’s disease. Neurochem. Res. 2009, 34, 1401–1404. [Google Scholar] [CrossRef] [Green Version]
- Lindqvist, D.; Kaufman, E.; Brundin, L.; Hall, S.; Surova, Y.; Hansson, O. Non-motor symptoms in patients with Parkinson’s disease—Correlations with inflammatory cytokines in serum. PLoS ONE 2012, 7, e47387. [Google Scholar] [CrossRef] [Green Version]
- Miiller, T.; Blum-Degen, D.; Przuntek, H.; Kuhn, W. Interleukin-6 levels in cerebrospinal fluid inversely correlate to severity of Parkinson’s disease. Acta Neurol. Scand. 1998, 98, 142–144. [Google Scholar] [CrossRef]
- Mogi, M.; Harada, M.; Kondo, T.; Riederer, P.; Inagaki, H.; Minami, M.; Nagatsu, T. Interleukin-1β.; interleukin-6.; epidermal growth factor and transforming growth factor-α are elevated in the brain from parkinsonian patients. Neurosci. Lett. 1994, 180, 147–150. [Google Scholar] [CrossRef]
- Tang, P.; Chong, L.; Li, X.; Liu, Y.; Liu, P.; Hou, C.; Li, R. Correlation between serum RANTES levels and the severity of Parkinson’s disease. Oxid. Med. Cell. Longev. 2014, 2014, 208408. [Google Scholar] [CrossRef] [Green Version]
- Mogi, M.; Harada, M.; Riederer, P.; Narabayashi, H.; Fujita, K.; Nagatsu, T. Tumor necrosis factor-α (TNF-α) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci. Lett. 1994, 165, 208–210. [Google Scholar] [CrossRef]
- Qin, L.; Wu, X.; Block, M.L.; Liu, Y.; Breese, G.R.; Hong, J.S.; Knapp, D.J.; Crews, F.T. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 2007, 55, 453–462. [Google Scholar] [CrossRef] [Green Version]
- Teismann, P.; Tieu, K.; Cohen, O.; Choi, D.K.; Wu, D.C.; Marks, D.; Vila, M.; Jackson-Lewis, V.; Przedborski, S. Pathogenic role of glial cells in Parkinson’s disease. Mov. Disord. 2003, 18, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Boka, G.; Anglade, P.; Wallach, D.; Javoy-Agid, F.; Agid, Y.; Hirsch, E.C. Immunocytochemical analysis of tumor necrosis factor and its receptors in Parkinson’s disease. Neurosci. Lett. 1994, 172, 151–154. [Google Scholar] [CrossRef] [PubMed]
- Ferger, B.; Leng, A.; Mura, A.; Hengerer, B.; Feldon, J. Genetic ablation of tumor necrosis factor-alpha (TNF-α) and pharmacological inhibition of TNF-synthesis attenuates MPTP toxicity in mouse striatum. J. Neurochem. 2004, 89, 822–833. [Google Scholar] [CrossRef] [PubMed]
- Sriram, K.; Matheson, J.M.; Benkovic, S.A.; Miller, D.B.; Luster, M.I.; O’Callaghan, J.P. Mice deficient in TNF receptors are protected against dopaminergic neurotoxicity: Implications for Parkinson’s disease. FASEB J. 2002, 16, 1474–1476. [Google Scholar] [CrossRef] [Green Version]
- Butterfield, D.A.; Reed, T.; Sultana, R. Roles of 3-nitrotyrosine- and 4-hydroxynonenal-modified brain proteins in the progression and pathogenesis of Alzheimer’s disease. Free Radic. Res. 2011, 45, 59–72. [Google Scholar] [CrossRef]
- Chang, K.H.; Cheng, M.L.; Chiang, M.C.; Chen, C.M. Lipophilic antioxidants in neurodegenerative diseases. Clin. Chim. Acta 2018, 485, 79–87. [Google Scholar] [CrossRef]
- Chang, K.H.; Chen, C.M. The role of oxidative stress in Parkinson’s disease. Antioxidants 2020, 9, 597. [Google Scholar] [CrossRef]
- Ferrer, I.; Perez, E.; Dalfo, E.; Barrachina, M. Abnormal levels of prohibitin and ATP synthase in the substantia nigra and frontal cortex in Parkinson’s disease. Neurosci. Lett. 2007, 415, 205–209. [Google Scholar] [CrossRef]
- Radunovic, A.; Porto, W.G.; Zeman, S.; Leigh, P.N. Increased mitochondrial superoxide dismutase activity in Parkinson’s disease but not amyotrophic lateral sclerosis motor cortex. Neurosci. Lett. 1997, 239, 105–108. [Google Scholar] [CrossRef]
- Jakel, R.J.; Townsend, J.A.; Kraft, A.D.; Johnson, J.A. Nrf2-mediated protection against 6-hydroxydopamine. Brain Res. 2007, 1144, 192–201. [Google Scholar] [CrossRef] [Green Version]
- Wei, P.C.; Lee-Chen, G.J.; Chen, C.M.; Wu, Y.R.; Chen, Y.J.; Lin, J.L.; Lo, Y.S.; Yao, C.F.; Chang, K.H. Neuroprotection of indole-derivative compound NC001-8 by the regulation of the NRF2 pathway in Parkinson’s disease cell models. Oxid. Med. Cell. Longev. 2019, 2019, 5074367. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.H.; Lee-Chen, G.J.; Wu, Y.R.; Chen, Y.J.; Lin, J.L.; Li, M.; Chen, I.C.; Lo, Y.S.; Wu, H.C.; Chen, C.M. Impairment of proteasome and anti-oxidative pathways in the induced pluripotent stem cell model for sporadic Parkinson’s disease. Parkinsonism Relat. Disord. 2016, 24, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Petrillo, S.; Schirinzi, T.; Di Lazzaro, G.; D’Amico, J.; Colona, V.L.; Bertini, E.; Pierantozzi, M.; Mari, L.; Mercuri, N.B.; Piemonte, F.; et al. Systemic activation of Nrf2 pathway in Parkinson’s disease. Mov. Disord. 2020, 35, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Niu, L.; Gao, L.; Li, W.X.; Jia, D.; Wang, X.L.; Gao, G.D. Neuroprotective effect of gypenosides against oxidative injury in the substantia nigra of a mouse model of Parkinson’s disease. J. Int. Med. Res. 2010, 38, 1084–1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campolo, M.; Casili, G.; Biundo, F.; Crupi, R.; Cordaro, M.; Cuzzocrea, S.; Esposito, E. The Neuroprotective effect of dimethyl fumarate in an MPTP-mouse model of Parkinson’s disease: Involvement of reactive oxygen species/nuclear factor-κB/nuclear transcription factor related to NF-E2. Antioxid. Redox Signal. 2017, 27, 453–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lastres-Becker, I.; Garcia-Yague, A.J.; Scannevin, R.H.; Casarejos, M.J.; Kugler, S.; Rabano, A.; Cuadrado, A. Repurposing the NRF2 activator dimethyl fumarate as therapy against synucleinopathy in Parkinson’s disease. Antioxid. Redox Signal. 2016, 25, 61–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.M.; Lin, C.H.; Wu, Y.R.; Yen, C.Y.; Huang, Y.T.; Lin, J.L.; Lin, C.Y.; Chen, W.L.; Chao, C.Y.; Lee-Chen, G.J.; et al. Lactulose and melibiose inhibit alpha-synuclein aggregation and up-regulate autophagy to reduce neuronal vulnerability. Cells 2020, 9, 1230. [Google Scholar] [CrossRef]
- Ramesh, C.; Kavala, V.; Raju, B.R.; Kuo, C.W.; Yao, C.F. Novel synthesis of indolylquinoline derivatives via the C-alkylation of Baylis-Hillman adducts. Tetrahedron Lett. 2009, 50, 4037–4041. [Google Scholar] [CrossRef]
- Ou, B.; Hampsch-Woodill, M.; Prior, R.L. Development and validation of an improved oxygen radical absorbance capacity assay using fluorescein as the fluorescent probe. J. Agric. Food Chem. 2001, 49, 4619–4626. [Google Scholar] [CrossRef]
- Lau, Y.S.; Trobough, K.L.; Crampton, J.M.; Wilson, J.A. Effects of probenecid on striatal dopamine depletion in acute and long-term 1-methyl-4-phenyl-1.;2.;3.;6-tetrahydropyridine (MPTP)-treated mice. Gen. Pharmacol. 1990, 21, 181–187. [Google Scholar] [CrossRef]
- Chaudhuri, K.R.; Schapira, A.H. Non-motor symptoms of Parkinson’s disease: Dopaminergic pathophysiology and treatment. Lancet Neurol. 2009, 8, 464–474. [Google Scholar] [CrossRef] [PubMed]
- Dhingra, D.; Sharma, A. Antidepressant-like activity of Glycyrrhiza glabra L. in mouse models of immobility tests. Prog. Neuropsychopharmacol. Biol. Psychiatry 2006, 30, 449–454. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiu, Y.-J.; Lin, C.-H.; Lin, C.-Y.; Yang, P.-N.; Lo, Y.-S.; Chen, Y.-C.; Chen, C.-M.; Wu, Y.-R.; Yao, C.-F.; Chang, K.-H.; et al. Investigating Therapeutic Effects of Indole Derivatives Targeting Inflammation and Oxidative Stress in Neurotoxin-Induced Cell and Mouse Models of Parkinson’s Disease. Int. J. Mol. Sci. 2023, 24, 2642. https://doi.org/10.3390/ijms24032642
Chiu Y-J, Lin C-H, Lin C-Y, Yang P-N, Lo Y-S, Chen Y-C, Chen C-M, Wu Y-R, Yao C-F, Chang K-H, et al. Investigating Therapeutic Effects of Indole Derivatives Targeting Inflammation and Oxidative Stress in Neurotoxin-Induced Cell and Mouse Models of Parkinson’s Disease. International Journal of Molecular Sciences. 2023; 24(3):2642. https://doi.org/10.3390/ijms24032642
Chicago/Turabian StyleChiu, Ya-Jen, Chih-Hsin Lin, Chung-Yin Lin, Pei-Ning Yang, Yen-Shi Lo, Yu-Chieh Chen, Chiung-Mei Chen, Yih-Ru Wu, Ching-Fa Yao, Kuo-Hsuan Chang, and et al. 2023. "Investigating Therapeutic Effects of Indole Derivatives Targeting Inflammation and Oxidative Stress in Neurotoxin-Induced Cell and Mouse Models of Parkinson’s Disease" International Journal of Molecular Sciences 24, no. 3: 2642. https://doi.org/10.3390/ijms24032642
APA StyleChiu, Y. -J., Lin, C. -H., Lin, C. -Y., Yang, P. -N., Lo, Y. -S., Chen, Y. -C., Chen, C. -M., Wu, Y. -R., Yao, C. -F., Chang, K. -H., & Lee-Chen, G. -J. (2023). Investigating Therapeutic Effects of Indole Derivatives Targeting Inflammation and Oxidative Stress in Neurotoxin-Induced Cell and Mouse Models of Parkinson’s Disease. International Journal of Molecular Sciences, 24(3), 2642. https://doi.org/10.3390/ijms24032642