
Myofilament Alterations Associated with Human R14del-Phospholamban Cardiomyopathy




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
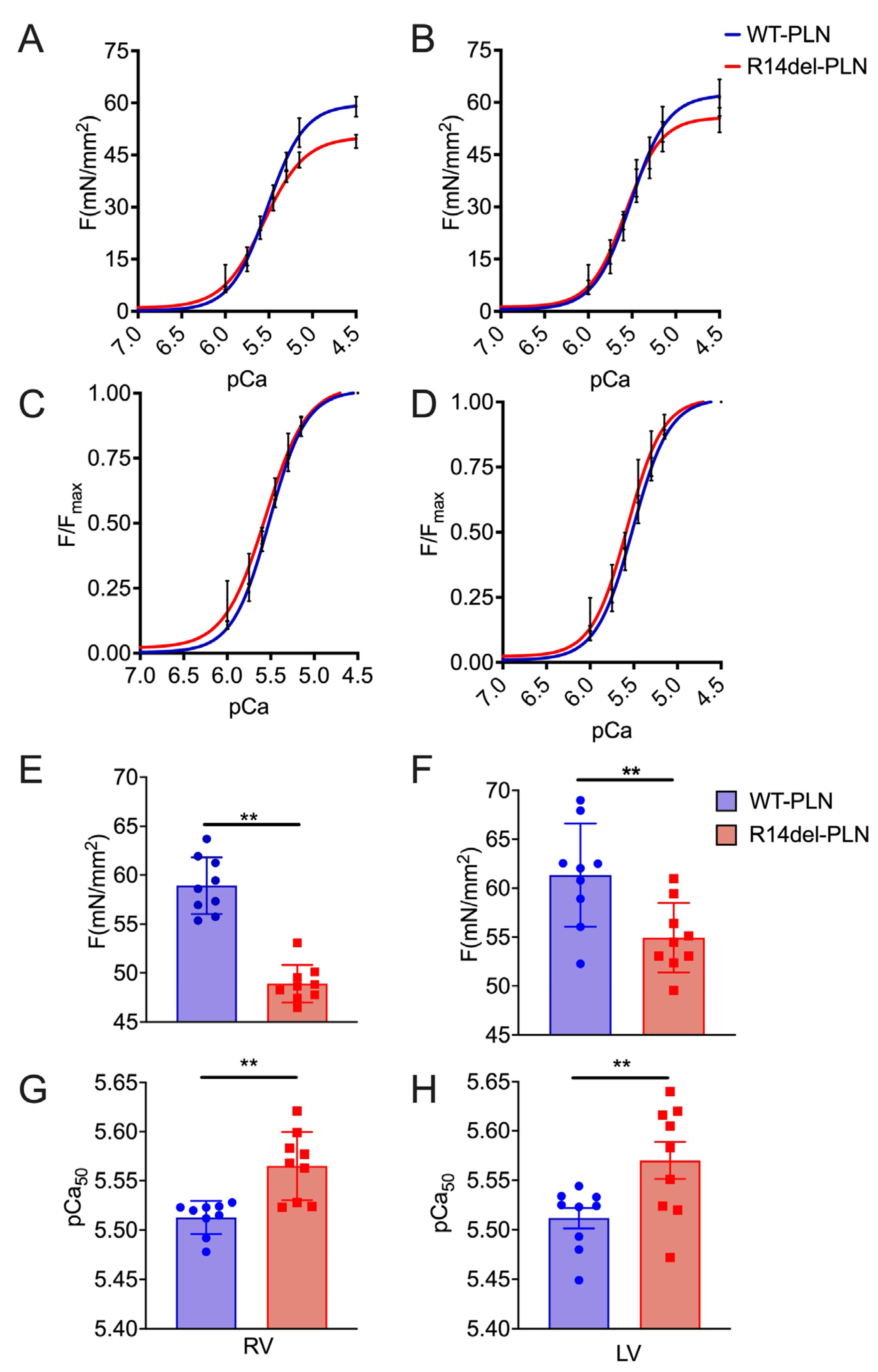
2.1. Decreased Maximal Force in R14del-PLN Cardiomyocytes
2.2. Omecamtiv Mecarbil as a Potential Drug to Treat R14del-PLN-Mediated Contractile Dysfunction
2.3. Decreased Contractility and Calcium Transients in R14del-PLN Cardiomyocytes at 12 Months
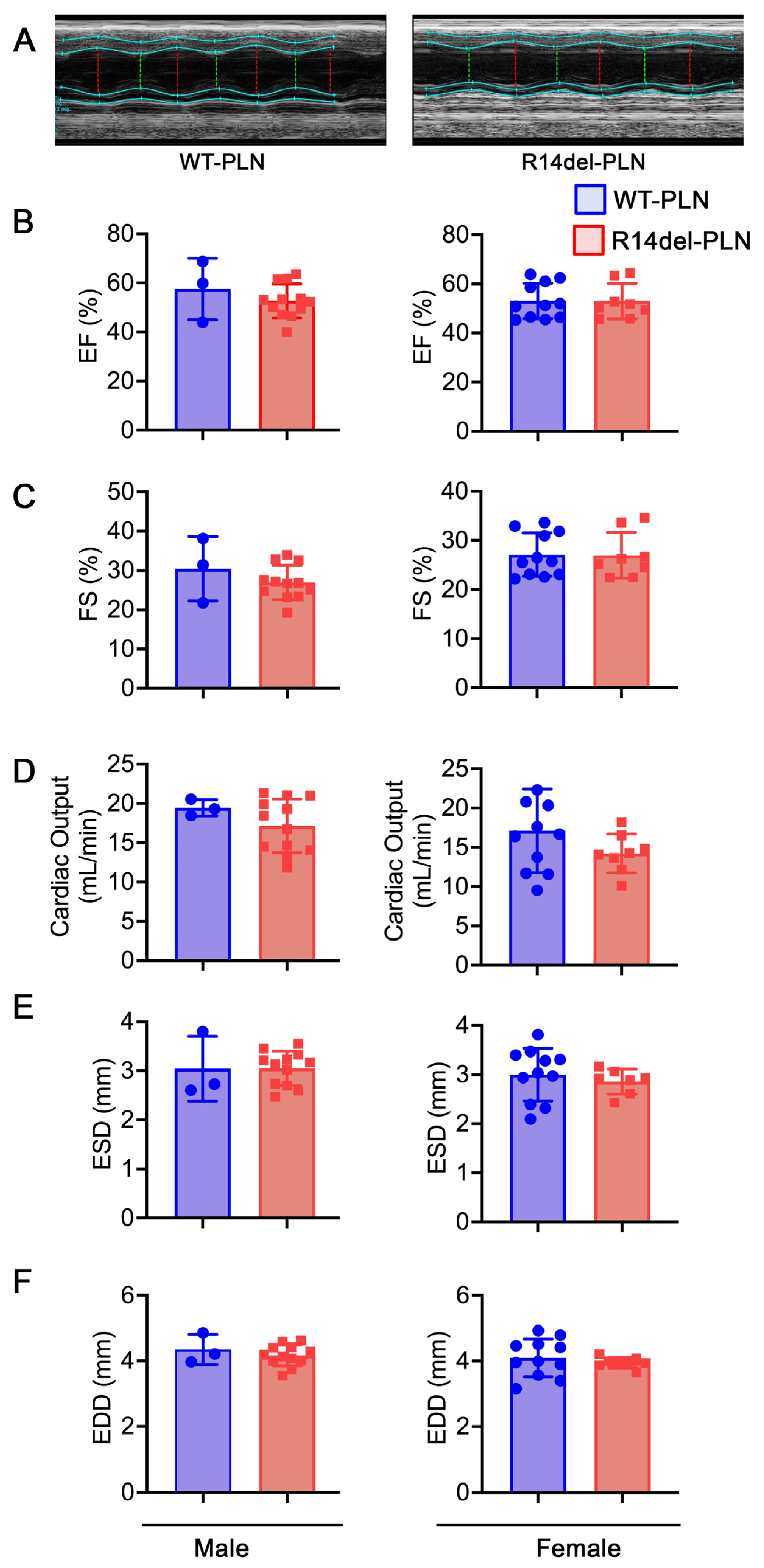
2.4. Normal Cardiac Function and Geometry in R14del-PLN Mice at 12 Months of Age
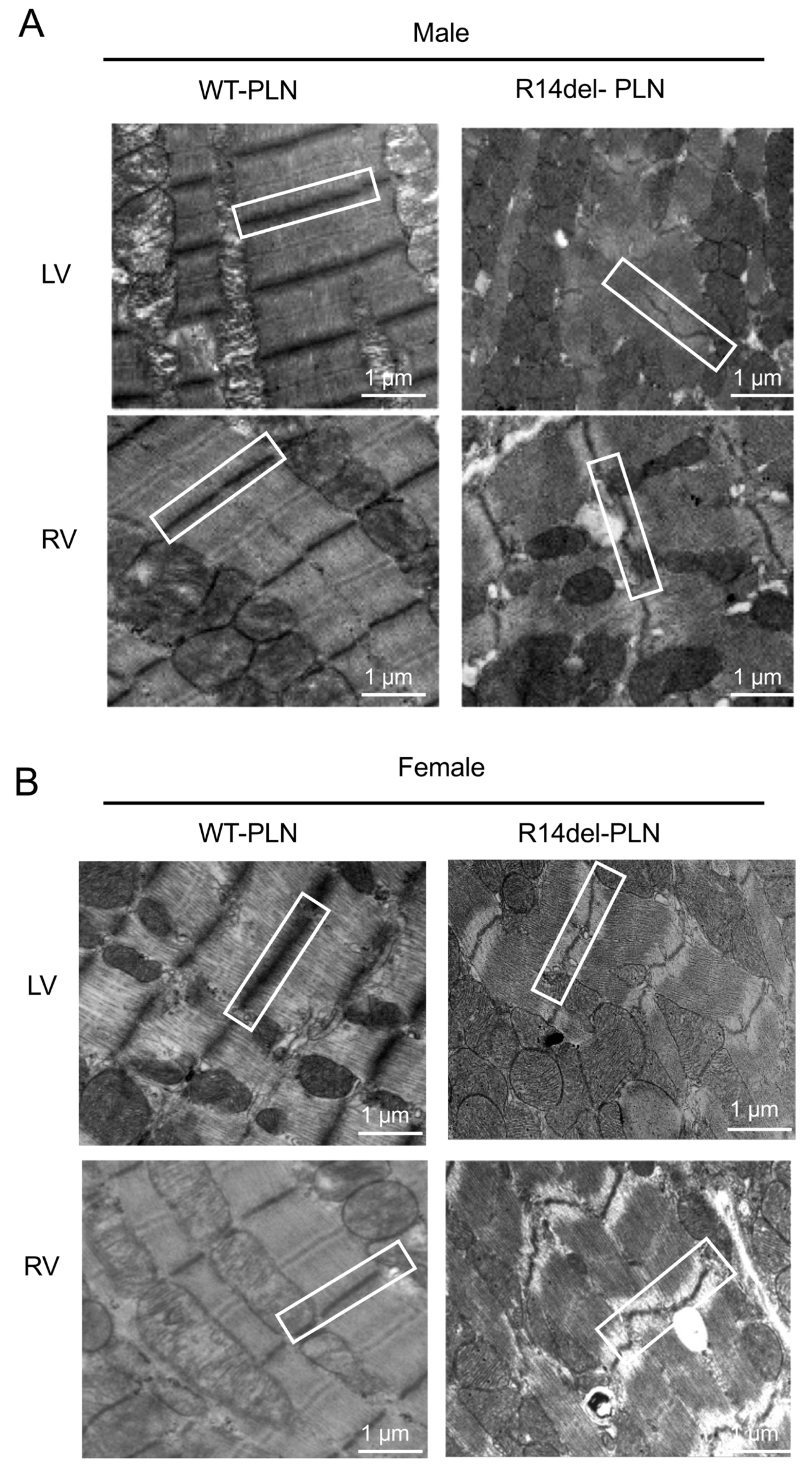
2.5. Increased Fibrosis and Altered Sarcomere Structures in R14del-PLN Hearts at 12 Months of Age
3. Discussion
4. Materials and Methods
4.1. Humanized WT-PLN and R14del-PLN Knock-in Mice
4.2. Measurements of Cardiomyocyte Contraction Mechanics and Ca2+ Kinetics
4.3. Measurements of pCa–Force and Myofilament Ca2+ Muscle Sensitivity Using Skinned Myocytes
4.4. Assessment of Cardiac Function with Echocardiography
4.5. Histopathological Analysis
4.6. Ultrastructure Analysis of Cardiomyocytes with Electron Microscopy
4.7. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kranias, E.G.; Hajjar, R.J. Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ. Res. 2012, 110, 1646–1660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Austin, K.M.; Trembley, M.A.; Chandler, S.F.; Sanders, S.P.; Saffitz, J.E.; Abrams, D.J.; Pu, W.T. Molecular mechanisms of arrhythmogenic cardiomyopathy. Nat. Rev. Cardiol. 2019, 16, 519–537. [Google Scholar] [CrossRef] [PubMed]
- Gerull, B.; Brodehl, A. Insights Into Genetics and Pathophysiology of Arrhythmogenic Cardiomyopathy. Curr. Heart Fail. Rep. 2021, 18, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Haghighi, K.; Kolokathis, F.; Gramolini, A.O.; Waggoner, J.R.; Pater, L.; Lynch, R.A.; Fan, G.C.; Tsiapras, D.; Parekh, R.R.; Dorn, G.W.; et al. A mutation in the human phospholamban gene, deleting arginine 14, results in lethal, hereditary cardiomyopathy. Proc. Natl. Acad. Sci. USA 2006, 103, 1388–1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Zwaag, P.A.; van Rijsingen, I.A.; Asimaki, A.; Jongbloed, J.D.; van Veldhuisen, D.J.; Wiesfeld, A.C.; Cox, M.G.; van Lochem, L.T.; de Boer, R.A.; Hofstra, R.M.; et al. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: Evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur. J. Heart Fail. 2012, 14, 1199–1207. [Google Scholar] [CrossRef] [Green Version]
- de Brouwer, R.; Meems, L.M.G.; Verstraelen, T.E.; Mahmoud, B.; Proost, V.; Wilde, A.A.M.; Bosman, L.P.; van Drie, E.; van der Zwaag, P.A.; van Tintelen, J.P.; et al. Sex-specific aspects of phospholamban cardiomyopathy: The importance and prognostic value of low-voltage electrocardiograms. Heart Rhythm. 2022, 19, 427–434. [Google Scholar] [CrossRef]
- van Rijsingen, I.A.; van der Zwaag, P.A.; Groeneweg, J.A.; Nannenberg, E.A.; Jongbloed, J.D.; Zwinderman, A.H.; Pinto, Y.M.; Dit Deprez, R.H.; Post, J.G.; Tan, H.L.; et al. Outcome in phospholamban R14del carriers: Results of a large multicentre cohort study. Circ. Cardiovasc. Genet. 2014, 7, 455–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kranias, E.G.; Doevendans, P.A.; Glijnis, P.C.; Hajjar, R.J. PLN Foundation. Circ. Res. 2018, 123, 1276–1278. [Google Scholar] [CrossRef]
- Doevendans, P.A.; Glijnis, P.C.; Kranias, E.G. Leducq Transatlantic Network of Excellence to Cure Phospholamban-Induced Cardiomyopathy (CURE-PLaN). Circ. Res. 2019, 125, 720–724. [Google Scholar] [CrossRef]
- Karakikes, I.; Stillitano, F.; Nonnenmacher, M.; Tzimas, C.; Sanoudou, D.; Termglinchan, V.; Kong, C.W.; Rushing, S.; Hansen, J.; Ceholski, D.; et al. Correction of human phospholamban R14del mutation associated with cardiomyopathy using targeted nucleases and combination therapy. Nat. Commun. 2015, 6, 6955. [Google Scholar] [CrossRef]
- Stillitano, F.; Turnbull, I.C.; Karakikes, I.; Nonnenmacher, M.; Backeris, P.; Hulot, J.S.; Kranias, E.G.; Hajjar, R.J.; Costa, K.D. Genomic correction of familial cardiomyopathy in human engineered cardiac tissues. Eur. Heart J. 2016, 37, 3282–3284. [Google Scholar] [CrossRef] [Green Version]
- Badone, B.; Ronchi, C.; Lodola, F.; Knaust, A.E.; Hansen, A.; Eschenhagen, T.; Zaza, A. Characterization of the PLN p.Arg14del Mutation in Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Int. J. Mol. Sci. 2021, 22, 13500. [Google Scholar] [CrossRef]
- Zhao, W.; Yuan, Q.; Qian, J.; Waggoner, J.R.; Pathak, A.; Chu, G.; Mitton, B.; Sun, X.; Jin, J.; Braz, J.C.; et al. The presence of Lys27 instead of Asn27 in human phospholamban promotes sarcoplasmic reticulum Ca2+-ATPase superinhibition and cardiac remodeling. Circulation 2006, 113, 995–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vafiadaki, E.; Haghighi, K.; Arvanitis, D.A.; Kranias, E.G.; Sanoudou, D. Aberrant PLN-R14del Protein Interactions Intensify SERCA2a Inhibition, Driving Impaired Ca2+ Handling and Arrhythmogenesis. Int. J. Mol. Sci. 2022, 23, 6947. [Google Scholar] [CrossRef]
- Haghighi, K.; Gardner, G.; Vafiadaki, E.; Kumar, M.; Green, L.C.; Ma, J.; Crocker, J.S.; Koch, S.; Arvanitis, D.A.; Bidwell, P.; et al. Impaired Right Ventricular Calcium Cycling Is an Early Risk Factor in R14del-Phospholamban Arrhythmias. J. Pers. Med. 2021, 11, 502. [Google Scholar] [CrossRef]
- Dave, J.; Raad, N.; Mittal, N.; Zhang, L.; Fargnoli, A.; Oh, J.G.; Savoia, M.E.; Hansen, J.; Fava, M.; Yin, X.; et al. Gene editing reverses arrhythmia susceptibility in humanized PLN-R14del mice: Modeling a European cardiomyopathy with global impact. Cardiovasc. Res. 2022, 188, 3140–3150. [Google Scholar] [CrossRef]
- Cheng, Y.; Li, W.; McElfresh, T.A.; Chen, X.; Berthiaume, J.M.; Castel, L.; Yu, X.; Van Wagoner, D.R.; Chandler, M.P. Changes in myofilament proteins, but not Ca2+ regulation, are associated with a high-fat diet-induced improvement in contractile function in heart failure. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1438–H1446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huke, S.; Knollmann, B.C. Increased myofilament Ca2+-sensitivity and arrhythmia susceptibility. J. Mol. Cell. Cardiol. 2010, 48, 824–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baudenbacher, F.; Schober, T.; Pinto, J.R.; Sidorov, V.Y.; Hilliard, F.; Solaro, R.J.; Potter, J.D.; Knollmann, B.C. Myofilament Ca2+ sensitization causes susceptibility to cardiac arrhythmia in mice. J. Clin. Investig. 2008, 118, 3893–3903. [Google Scholar] [CrossRef] [Green Version]
- Kumar, M.; Haghighi, K.; Kranias, E.G.; Sadayappan, S. Phosphorylation of cardiac myosin-binding protein-C contributes to calcium homeostasis. J. Biol. Chem. 2020, 295, 11275–11291. [Google Scholar] [CrossRef]
- Pei, J.; Maas, R.G.C.; Nagyova, E.; Gho, J.M.I.H.; Blok, C.S.; van Adrichem, I.; Calis, J.J.A.; van Es, R.; Sepehrkhouy, S.; Feyen, D.; et al. Transcriptional regulation profiling reveals disrupted lipid metabolism in failing hearts with a pathogenic phospholamban mutation. bioRxiv 2020. [Google Scholar] [CrossRef]
- Blair, C.A.; Brundage, E.A.; Thompson, K.L.; Stromberg, A.; Guglin, M.; Biesiadecki, B.J.; Campbell, K.S. Heart Failure in Humans Reduces Contractile Force in Myocardium From Both Ventricles. JACC Basic Transl. Sci. 2020, 5, 786–798. [Google Scholar] [CrossRef] [PubMed]
- Teerlink, J.R.; Diaz, R.; Felker, G.M.; McMurray, J.J.V.; Metra, M.; Solomon, S.D.; Adams, K.F.; Anand, I.; Arias-Mendoza, A.; Biering-Sorensen, T.; et al. Cardiac Myosin Activation with Omecamtiv Mecarbil in Systolic Heart Failure. N. Engl. J. Med. 2021, 384, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Sparrow, A.J.; Watkins, H.; Daniels, M.J.; Redwood, C.; Robinson, P. Mavacamten rescues increased myofilament calcium sensitivity and dysregulation of Ca2+ flux caused by thin filament hypertrophic cardiomyopathy mutations. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H715–H722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, M.; Govindan, S.; Zhang, M.; Khairallah, R.J.; Martin, J.L.; Sadayappan, S.; de Tombe, P.P. Cardiac Myosin-binding Protein C and Troponin-I Phosphorylation Independently Modulate Myofilament Length-dependent Activation. J. Biol. Chem. 2015, 290, 29241–29249. [Google Scholar] [CrossRef] [Green Version]
- Felker, G.M.; Solomon, S.D.; Claggett, B.; Diaz, R.; McMurray, J.J.V.; Metra, M.; Anand, I.; Crespo-Leiro, M.G.; Dahlstrom, U.; Goncalvesova, E.; et al. Assessment of Omecamtiv Mecarbil for the Treatment of Patients With Severe Heart Failure: A Post Hoc Analysis of Data From the GALACTIC-HF Randomized Clinical Trial. JAMA Cardiol. 2022, 7, 26–34. [Google Scholar] [CrossRef]
- Dash, R.; Frank, K.F.; Carr, A.N.; Moravec, C.S.; Kranias, E.G. Gender influences on sarcoplasmic reticulum Ca2+-handling in failing human myocardium. J. Mol. Cell Cardiol. 2001, 33, 1345–1353. [Google Scholar] [CrossRef]
- Hall, C.; Gehmlich, K.; Denning, C.; Pavlovic, D. Complex Relationship Between Cardiac Fibroblasts and Cardiomyocytes in Health and Disease. J. Am. Heart Assoc. 2021, 10, e019338. [Google Scholar] [CrossRef]
- Gho, J.M.; van Es, R.; Stathonikos, N.; Harakalova, M.; te Rijdt, W.P.; Suurmeijer, A.J.; van der Heijden, J.F.; de Jonge, N.; Chamuleau, S.A.; de Weger, R.A.; et al. High resolution systematic digital histological quantification of cardiac fibrosis and adipose tissue in phospholamban p.Arg14del mutation associated cardiomyopathy. PLoS ONE 2014, 9, e94820. [Google Scholar] [CrossRef] [PubMed]
- Te Rijdt, W.P.; Ten Sande, J.N.; Gorter, T.M.; van der Zwaag, P.A.; van Rijsingen, I.A.; Boekholdt, S.M.; van Tintelen, J.P.; van Haelst, P.L.; Planken, R.N.; de Boer, R.A.; et al. Myocardial fibrosis as an early feature in phospholamban p.Arg14del mutation carriers: Phenotypic insights from cardiovascular magnetic resonance imaging. Eur. Heart J. Cardiovasc. Imaging 2019, 20, 92–100. [Google Scholar] [CrossRef]
- Santulli, G.; Xie, W.; Reiken, S.R.; Marks, A.R. Mitochondrial calcium overload is a key determinant in heart failure. Proc. Natl. Acad. Sci. USA 2015, 112, 11389–11394. [Google Scholar] [CrossRef] [Green Version]
- Raad, N.; Bittihn, P.; Cacheux, M.; Jeong, D.; Ilkan, Z.; Ceholski, D.; Kohlbrenner, E.; Zhang, L.; Cai, C.L.; Kranias, E.G.; et al. Arrhythmia Mechanism and Dynamics in a Humanized Mouse Model of Inherited Cardiomyopathy Caused by Phospholamban R14del Mutation. Circulation 2021, 144, 441–454. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Wan, X.; McElfresh, T.A.; Chen, X.; Gresham, K.S.; Rosenbaum, D.S.; Chandler, M.P.; Stelzer, J.E. Impaired contractile function due to decreased cardiac myosin binding protein C content in the sarcomere. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H52–H65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bidwell, P.A.; Haghighi, K.; Kranias, E.G. The antiapoptotic protein HAX-1 mediates half of phospholamban’s inhibitory activity on calcium cycling and contractility in the heart. J. Biol. Chem. 2018, 293, 359–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haghighi, K.; Pritchard, T.J.; Liu, G.S.; Singh, V.P.; Bidwell, P.; Lam, C.K.; Vafiadaki, E.; Das, P.; Ma, J.; Kunduri, S.; et al. Human G109E-inhibitor-1 impairs cardiac function and promotes arrhythmias. J. Mol. Cell. Cardiol. 2015, 89, 349–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.S.; Morales, A.; Vafiadaki, E.; Lam, C.K.; Cai, W.F.; Haghighi, K.; Adly, G.; Hershberger, R.E.; Kranias, E.G. A novel human R25C-phospholamban mutation is associated with super-inhibition of calcium cycling and ventricular arrhythmia. Cardiovasc. Res. 2015, 107, 164–174. [Google Scholar] [CrossRef] [Green Version]
- Barefield, D.Y.; McNamara, J.W.; Lynch, T.L.; Kuster, D.W.D.; Govindan, S.; Haar, L.; Wang, Y.; Taylor, E.N.; Lorenz, J.N.; Nieman, M.L.; et al. Ablation of the calpain-targeted site in cardiac myosin binding protein-C is cardioprotective during ischemia-reperfusion injury. J. Mol. Cell. Cardiol. 2019, 129, 236–246. [Google Scholar] [CrossRef] [Green Version]
- Kuster, D.W.D.; Lynch, T.L.; Barefield, D.Y.; Sivaguru, M.; Kuffel, G.; Zilliox, M.J.; Lee, K.H.; Craig, R.; Namakkal-Soorappan, R.; Sadayappan, S. Altered C10 domain in cardiac myosin binding protein-C results in hypertrophic cardiomyopathy. Cardiovasc. Res. 2019, 115, 1986–1997. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, M.; Haghighi, K.; Koch, S.; Rubinstein, J.; Stillitano, F.; Hajjar, R.J.; Kranias, E.G.; Sadayappan, S. Myofilament Alterations Associated with Human R14del-Phospholamban Cardiomyopathy. Int. J. Mol. Sci. 2023, 24, 2675. https://doi.org/10.3390/ijms24032675
Kumar M, Haghighi K, Koch S, Rubinstein J, Stillitano F, Hajjar RJ, Kranias EG, Sadayappan S. Myofilament Alterations Associated with Human R14del-Phospholamban Cardiomyopathy. International Journal of Molecular Sciences. 2023; 24(3):2675. https://doi.org/10.3390/ijms24032675
Chicago/Turabian StyleKumar, Mohit, Kobra Haghighi, Sheryl Koch, Jack Rubinstein, Francesca Stillitano, Roger J. Hajjar, Evangelia G. Kranias, and Sakthivel Sadayappan. 2023. "Myofilament Alterations Associated with Human R14del-Phospholamban Cardiomyopathy" International Journal of Molecular Sciences 24, no. 3: 2675. https://doi.org/10.3390/ijms24032675
APA StyleKumar, M., Haghighi, K., Koch, S., Rubinstein, J., Stillitano, F., Hajjar, R. J., Kranias, E. G., & Sadayappan, S. (2023). Myofilament Alterations Associated with Human R14del-Phospholamban Cardiomyopathy. International Journal of Molecular Sciences, 24(3), 2675. https://doi.org/10.3390/ijms24032675