

Leptin Increases: Physiological Roles in the Control of Sympathetic Nerve Activity, Energy Balance, and the Hypothalamic–Pituitary–Thyroid Axis

Abstract
:1. Introduction
2. Dissimilar Impact of Decreases versus Increases in Leptin
3. Physiological Significance of Increases in Leptin
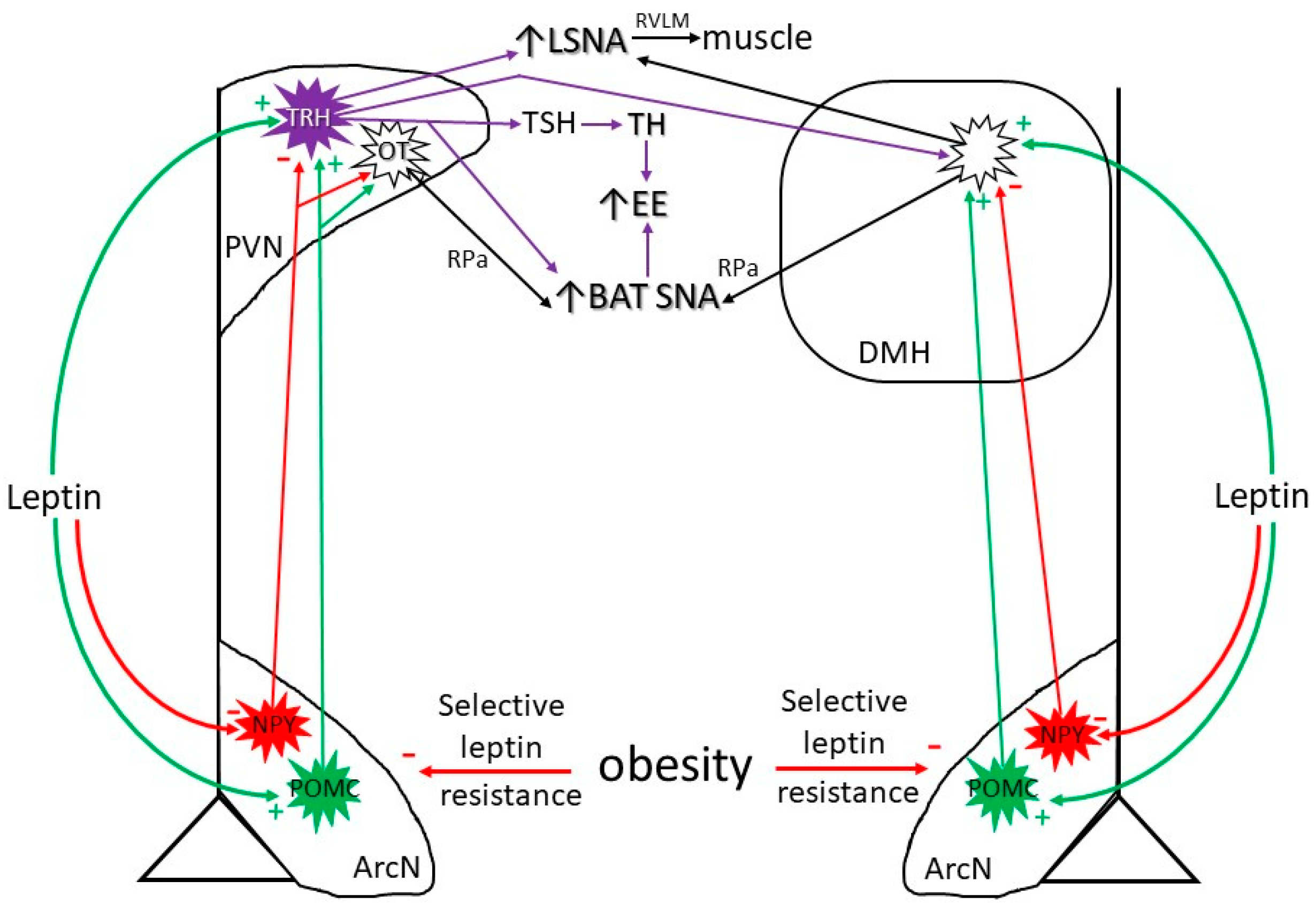
3.1. Neurocircuitry by Which Leptin Increases SNA and Energy Expenditure (Figure 1)

3.2. Diet-Induced Increases in Leptin
4. Obesity-Induced Increases in Leptin
Obesity Suppresses Leptin’s ArcN Anorexic Actions
5. Obesity: A State of Inflammation
5.1. Obesity-Induced Systemic Inflammation
5.2. Obesity-Induced Neuroinflammation
5.3. Potential Actions of Leptin to Facilitate Inflammation with Obesity
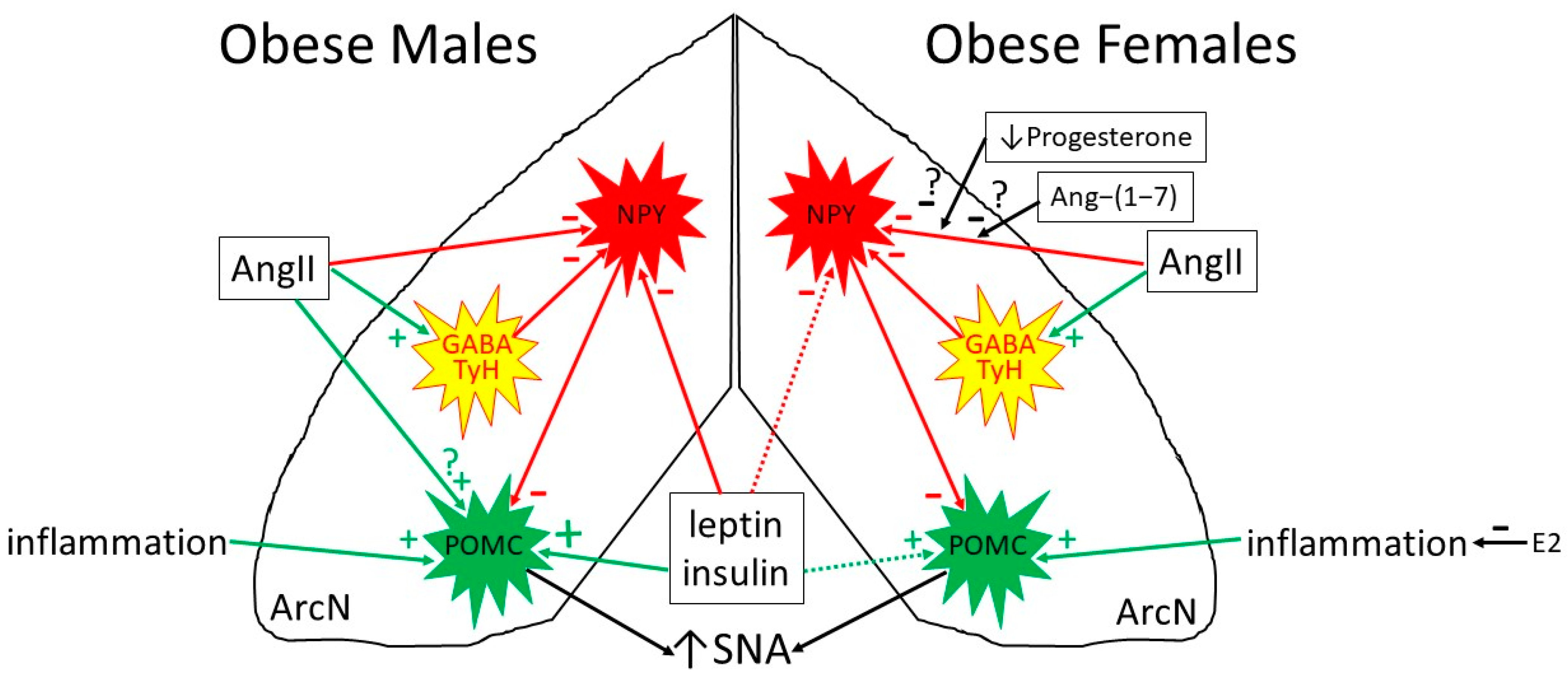
6. Obesity Induces Selective Leptin Resistance
6.1. Selective Leptin Resistance in the ArcN: Preserved Leptin-Induced Increases in SNA (in Obese Males)
6.2. Preserved or Enhanced Leptin Responsiveness in the PVN and DMH: Leptin Support of the HPT Axis and of BAT SNA with Obesity

{kind=link}
{kind=link}
| Males | Females | |||
|---|---|---|---|---|
| Lean | Obese | Lean | Obese | |
| Plasma leptin levels | _ | ↑ | ↑ | ↑↑ |
| Food intake with increased leptin | ↓ | _ | ↓↓ (dependent on estrogen) | _ |
| LSNA with increased leptin | ↑ | ↑↑ | ↑ (dependent on estrogen) | 0 |
| SSNA with increased leptin | ↑ | ? | ↑ | ? |
| Tonic PVN NPY sympathoinhibition | _ | ↓(0) | _ | _ |
| PVN POMC (α-MSH) sympathoexcitation | _ | ↑ | _ | ↓ |
| HPT axis | _ | _ | _ | _ |
| HPT axis: support mediated by ArcN leptin | _ | ↓ | _ | ↓ (?) |
| HPT axis: support mediated by PVN leptin | 0 | ↑ | 0 (?) | ↑ (?) |
6.3. The Role of Leptin in Weight Regain
7. Summary and Future Research Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Taggart, N. Diet, activity and body-weight. A study of variations in a woman. Br. J. Nutr. 1962, 16, 223–235. [Google Scholar] [CrossRef]
- Norberg, M.; Lindvall, K.; Jenkins, P.L.; Emmelin, M.; Lönnberg, G.; Nafziger, A.N. Self-rated health does not predict 10-year weight change among middle-aged adults in a longitudinal population study. BMC Public Health 2011, 11, 748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, C.C.; Hall, K.D. Short and long-term energy intake patterns and their implications for human body weight regulation. Physiol. Behav. 2014, 134, 60–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, J.M. Leptin and the endocrine control of energy balance. Nat. Metab. 2019, 1, 754–764. [Google Scholar] [CrossRef]
- Schwartz, M.W.; Seeley, R.J.; Zeltser, L.M.; Drewnowski, A.; Ravussin, E.; Redman, L.M.; Leibel, R.L. Obesity Pathogenesis: An Endocrine Society Scientific Statement. Endocr. Rev. 2017, 38, 267–296. [Google Scholar] [CrossRef] [Green Version]
- Leibel, R.L. The role of leptin in the control of body weight. Nutr. Rev. 2002, 60, S15–S19. [Google Scholar] [CrossRef]
- Aronne, L.J.; Hall, K.D.; Jakicic, J.M.; Leibel, R.L.; Lowe, M.R.; Rosenbaum, M.; Klein, S. Describing the Weight-Reduced State: Physiology, Behavior, and Interventions. Obesity 2021, 29 (Suppl. S1), S9–S24. [Google Scholar] [CrossRef] [PubMed]
- Berthoud, H.R.; Seeley, R.J.; Roberts, S.B. Physiology of Energy Intake in the Weight-Reduced State. Obesity 2021, 29 (Suppl. S1), S25–S30. [Google Scholar] [CrossRef] [PubMed]
- Flier, J.S. Starvation in the Midst of Plenty: Reflections on the History and Biology of Insulin and Leptin. Endocr. Rev. 2019, 40, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Kusminski, C.M.; Elmquist, J.K.; Scherer, P.E. Leptin: Less Is More. Diabetes 2020, 69, 823–829. [Google Scholar] [CrossRef]
- Halaas, J.L.; Boozer, C.; Blair-West, J.; Fidahusein, N.; Denton, D.A.; Friedman, J.M. Physiological response to long-term peripheral and central leptin infusion in lean and obese mice. Proc. Natl. Acad. Sci. USA 1997, 94, 8878–8883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Z.; Pelletier, N.E.; Wong, J.; Li, B.; Sdrulla, A.D.; Madden, C.J.; Marks, D.L.; Brooks, V.L. Leptin increases sympathetic nerve activity via induction of its own receptor in the paraventricular nucleus. Elife 2020, 9, e55357. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.E.; Nogueiras, R.; Morris, A.; Tovar, S.; Grant, C.; Cruickshank, M.; Rayner, D.V.; Dieguez, C.; Williams, L.M. Leptin receptor gene expression and number in the brain are regulated by leptin level and nutritional status. J. Physiol. 2009, 587, 3573–3585. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.H.; Lu, D.Y.; Yang, R.S.; Tsai, H.Y.; Kao, M.C.; Fu, W.M.; Chen, Y.F. Leptin-induced IL-6 production is mediated by leptin receptor, insulin receptor substrate-1, phosphatidylinositol 3-kinase, Akt, NF-kappaB, and p300 pathway in microglia. J. Immunol. 2007, 179, 1292–1302. [Google Scholar] [CrossRef] [Green Version]
- Shi, Z.; Brooks, V.L. Leptin differentially increases sympathetic nerve activity and its baroreflex regulation in female rats: Role of oestrogen. J. Physiol. 2015, 593, 1633–1647. [Google Scholar] [CrossRef] [Green Version]
- Shi, Z.; Li, B.; Brooks, V.L. Role of the Paraventricular Nucleus of the Hypothalamus in the Sympathoexcitatory Effects of Leptin. Hypertension 2015, 66, 1034–1041. [Google Scholar] [CrossRef] [Green Version]
- Han, C.; Wu, W.; Ale, A.; Kim, M.S.; Cai, D. Central Leptin and Tumor Necrosis Factor-α (TNFα) in Diurnal Control of Blood Pressure and Hypertension. J. Biol. Chem. 2016, 291, 15131–15142. [Google Scholar] [CrossRef] [Green Version]
- Moult, P.R.; Cross, A.; Santos, S.D.; Carvalho, A.L.; Lindsay, Y.; Connolly, C.N.; Irving, A.J.; Leslie, N.R.; Harvey, J. Leptin regulates AMPA receptor trafficking via PTEN inhibition. J. Neurosci. 2010, 30, 4088–4101. [Google Scholar] [CrossRef] [Green Version]
- Münzberg, H.; Singh, P.; Heymsfield, S.B.; Yu, S.; Morrison, C.D. Recent advances in understanding the role of leptin in energy homeostasis. F1000Research 2020, 9, 451. [Google Scholar] [CrossRef]
- Shi, Z.; Madden, C.J.; Brooks, V.L. Arcuate neuropeptide Y inhibits sympathetic nerve activity via multiple neuropathways. J. Clin. Investig. 2017, 127, 2868–2880. [Google Scholar] [CrossRef]
- Harlan, S.M.; Morgan, D.A.; Agassandian, K.; Guo, D.F.; Cassell, M.D.; Sigmund, C.D.; Mark, A.L.; Rahmouni, K. Ablation of the leptin receptor in the hypothalamic arcuate nucleus abrogates leptin-induced sympathetic activation. Circ. Res. 2011, 108, 808–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prior, L.J.; Eikelis, N.; Armitage, J.A.; Davern, P.J.; Burke, S.L.; Montani, J.P.; Barzel, B.; Head, G.A. Exposure to a high-fat diet alters leptin sensitivity and elevates renal sympathetic nerve activity and arterial pressure in rabbits. Hypertension 2010, 55, 862–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassaglia, P.A.; Shi, Z.; Li, B.; Reis, W.L.; Clute-Reinig, N.M.; Stern, J.E.; Brooks, V.L. Neuropeptide Y acts in the paraventricular nucleus to suppress sympathetic nerve activity and its baroreflex regulation. J. Physiol. 2014, 592, 1655–1675. [Google Scholar] [CrossRef]
- Shi, Z.; Wong, J.; Brooks, V.L. Obesity: Sex and sympathetics. Biol. Sex Differ. 2020, 11, 10. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Bonillas, A.C.; Wong, J.; Padilla, S.L.; Brooks, V.L. Neuropeptide Y suppresses thermogenic and cardiovascular sympathetic nerve activity via Y1 receptors in the paraventricular nucleus and dorsomedial hypothalamus. J. Neuroendocrinol. 2021, 33, e13006. [Google Scholar] [CrossRef] [PubMed]
- Chitravanshi, V.C.; Kawabe, K.; Sapru, H.N. Stimulation of the hypothalamic arcuate nucleus increases brown adipose tissue nerve activity via hypothalamic paraventricular and dorsomedial nuclei. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H433–H444. [Google Scholar] [CrossRef] [Green Version]
- Coote, J.H. A role for the paraventricular nucleus of the hypothalamus in the autonomic control of heart and kidney. Exp. Physiol. 2005, 90, 169–173. [Google Scholar] [CrossRef]
- Elsaafien, K.; Kirchner, M.K.; Mohammed, M.; Eikenberry, S.A.; West, C.; Scott, K.A.; de Kloet, A.D.; Stern, J.E.; Krause, E.G. Identification of Novel Cross-Talk between the Neuroendocrine and Autonomic Stress Axes Controlling Blood Pressure. J. Neurosci. 2021, 41, 4641–4657. [Google Scholar] [CrossRef]
- Pandit, R.; Beerens, S.; Adan, R.A.H. Role of leptin in energy expenditure: The hypothalamic perspective. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2017, 312, R938–R947. [Google Scholar] [CrossRef] [Green Version]
- Doslikova, B.; Tchir, D.; McKinty, A.; Zhu, X.; Marks, D.L.; Baracos, V.E.; Colmers, W.F. Convergent neuronal projections from paraventricular nucleus, parabrachial nucleus, and brainstem onto gastrocnemius muscle, white and brown adipose tissue in male rats. J. Comp. Neurol. 2019, 527, 2826–2842. [Google Scholar] [CrossRef]
- Oldfield, B.J.; Giles, M.E.; Watson, A.; Anderson, C.; Colvill, L.M.; McKinley, M.J. The neurochemical characterisation of hypothalamic pathways projecting polysynaptically to brown adipose tissue in the rat. Neuroscience 2002, 110, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Kerem, L.; Lawson, E.A. The Effects of Oxytocin on Appetite Regulation, Food Intake and Metabolism in Humans. Int. J. Mol. Sci. 2021, 22, 7737. [Google Scholar] [CrossRef] [PubMed]
- Hollenberg, A.N. The role of the thyrotropin-releasing hormone (TRH) neuron as a metabolic sensor. Thyroid 2008, 18, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Nillni, E.A. Regulation of the hypothalamic thyrotropin releasing hormone (TRH) neuron by neuronal and peripheral inputs. Front. Neuroendocrinol. 2010, 31, 134–156. [Google Scholar] [CrossRef] [Green Version]
- Enriori, P.J.; Sinnayah, P.; Simonds, S.E.; Garcia Rudaz, C.; Cowley, M.A. Leptin action in the dorsomedial hypothalamus increases sympathetic tone to brown adipose tissue in spite of systemic leptin resistance. J. Neurosci. 2011, 31, 12189–12197. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Kerman, I.A.; Laque, A.; Nguyen, P.; Faouzi, M.; Louis, G.W.; Jones, J.C.; Rhodes, C.; Münzberg, H. Leptin-receptor-expressing neurons in the dorsomedial hypothalamus and median preoptic area regulate sympathetic brown adipose tissue circuits. J. Neurosci. 2011, 31, 1873–1884. [Google Scholar] [CrossRef] [Green Version]
- Marsh, A.J.; Fontes, M.A.; Killinger, S.; Pawlak, D.B.; Polson, J.W.; Dampney, R.A. Cardiovascular responses evoked by leptin acting on neurons in the ventromedial and dorsomedial hypothalamus. Hypertension 2003, 42, 488–493. [Google Scholar] [CrossRef] [Green Version]
- Lim, K.; Barzel, B.; Burke, S.L.; Armitage, J.A.; Head, G.A. Origin of Aberrant Blood Pressure and Sympathetic Regulation in Diet-Induced Obesity. Hypertension 2016, 68, 491–500. [Google Scholar] [CrossRef]
- Seoane-Collazo, P.; Fernø, J.; Gonzalez, F.; Diéguez, C.; Leis, R.; Nogueiras, R.; López, M. Hypothalamic-autonomic control of energy homeostasis. Endocrine 2015, 50, 276–291. [Google Scholar] [CrossRef]
- Shiuchi, T.; Toda, C.; Okamoto, S.; Coutinho, E.A.; Saito, K.; Miura, S.; Ezaki, O.; Minokoshi, Y. Induction of glucose uptake in skeletal muscle by central leptin is mediated by muscle β(2)-adrenergic receptor but not by AMPK. Sci. Rep. 2017, 7, 15141. [Google Scholar] [CrossRef]
- Saito, M.; Matsushita, M.; Yoneshiro, T.; Okamatsu-Ogura, Y. Brown Adipose Tissue, Diet-Induced Thermogenesis, and Thermogenic Food Ingredients: From Mice to Men. Front. Endocrinol. 2020, 11, 222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Din, M.U.; Saari, T.; Raiko, J.; Kudomi, N.; Maurer, S.F.; Lahesmaa, M.; Fromme, T.; Amri, E.Z.; Klingenspor, M.; Solin, O.; et al. Postprandial Oxidative Metabolism of Human Brown Fat Indicates Thermogenesis. Cell Metab. 2018, 28, 207–216.e3. [Google Scholar] [CrossRef]
- Ho, K.K.Y. Diet-induced thermogenesis: Fake friend or foe? J. Endocrinol. 2018, 238, R185–R191. [Google Scholar] [CrossRef]
- Chan, P.C.; Hsieh, P.S. The Role and Regulatory Mechanism of Brown Adipose Tissue Activation in Diet-Induced Thermogenesis in Health and Diseases. Int. J. Mol. Sci. 2022, 23, 9448. [Google Scholar] [CrossRef] [PubMed]
- Seoane-Collazo, P.; Martínez-Sánchez, N.; Milbank, E.; Contreras, C. Incendiary Leptin. Nutrients 2020, 12, 472. [Google Scholar] [CrossRef] [Green Version]
- Romon, M.; Lebel, P.; Velly, C.; Marecaux, N.; Fruchart, J.C.; Dallongeville, J. Leptin response to carbohydrate or fat meal and association with subsequent satiety and energy intake. Am. J. Physiol. 1999, 277, E855–E861. [Google Scholar] [CrossRef]
- Elimam, A.; Marcus, C. Meal timing, fasting and glucocorticoids interplay in serum leptin concentrations and diurnal profile. Eur. J. Endocrinol. 2002, 147, 181–188. [Google Scholar] [CrossRef] [Green Version]
- Perry, R.J.; Lyu, K.; Rabin-Court, A.; Dong, J.; Li, X.; Yang, Y.; Qing, H.; Wang, A.; Yang, X.; Shulman, G.I. Leptin mediates postprandial increases in body temperature through hypothalamus-adrenal medulla-adipose tissue crosstalk. J. Clin. Investig. 2020, 130, 2001–2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothwell, N.J.; Stock, M.J. A role for brown adipose tissue in diet-induced thermogenesis. Nature 1979, 281, 31–35. [Google Scholar] [CrossRef]
- Stock, M.J. Gluttony and thermogenesis revisited. Int. J. Obes. Relat. Metab. Disord. 1999, 23, 1105–1117. [Google Scholar] [CrossRef]
- Trayhurn, P.; Arch, J.R.S. Is energy expenditure reduced in obese mice with mutations in the leptin/leptin receptor genes? J. Nutr. Sci. 2020, 9, e23. [Google Scholar] [CrossRef] [PubMed]
- Trayhurn, P.; Jones, P.M.; McGuckin, M.M.; Goodbody, A.E. Effects of overfeeding on energy balance and brown fat thermogenesis in obese (ob/ob) mice. Nature 1982, 295, 323–325. [Google Scholar] [CrossRef] [PubMed]
- Leibel, R.L.; Rosenbaum, M.; Hirsch, J. Changes in energy expenditure resulting from altered body weight. N. Engl. J. Med. 1995, 332, 621–628. [Google Scholar] [CrossRef]
- Ravussin, Y.; Edwin, E.; Gallop, M.; Xu, L.; Bartolomé, A.; Kraakman, M.J.; LeDuc, C.A.; Ferrante, A.W., Jr. Evidence for a Non-leptin System that Defends against Weight Gain in Overfeeding. Cell Metab. 2018, 28, 289–299.e5. [Google Scholar] [CrossRef] [Green Version]
- Van Baak, M.A.; Mariman, E.C.M. Mechanisms of weight regain after weight loss—The role of adipose tissue. Nat. Rev. Endocrinol. 2019, 15, 274–287. [Google Scholar] [CrossRef]
- Ludwig, D.S.; Aronne, L.J.; Astrup, A.; de Cabo, R.; Cantley, L.C.; Friedman, M.I.; Heymsfield, S.B.; Johnson, J.D.; King, J.C.; Krauss, R.M.; et al. The carbohydrate-insulin model: A physiological perspective on the obesity pandemic. Am. J. Clin. Nutr. 2021, 114, 1873–1885. [Google Scholar] [CrossRef]
- Hall, K.D.; Farooqi, I.S.; Friedman, J.M.; Klein, S.; Loos, R.J.F.; Mangelsdorf, D.J.; O’Rahilly, S.; Ravussin, E.; Redman, L.M.; Ryan, D.H.; et al. The energy balance model of obesity: Beyond calories in, calories out. Am. J. Clin. Nutr. 2022, 115, 1243–1254. [Google Scholar] [CrossRef] [PubMed]
- Greenway, F.L. Physiological adaptations to weight loss and factors favouring weight regain. Int. J. Obes. 2015, 39, 1188–1196. [Google Scholar] [CrossRef] [Green Version]
- Zorbas, C.; Reeve, E.; Naughton, S.; Batis, C.; Whelan, J.; Waqa, G.; Bell, C. The Relationship between Feasting Periods and Weight Gain: A Systematic Scoping Review. Curr. Obes. Rep. 2020, 9, 39–62. [Google Scholar] [CrossRef]
- Yanovski, J.A.; Yanovski, S.Z.; Sovik, K.N.; Nguyen, T.T.; O’Neil, P.M.; Sebring, N.G. A prospective study of holiday weight gain. N. Engl. J. Med. 2000, 342, 861–867. [Google Scholar] [CrossRef]
- Myers, M.G.; Cowley, M.A.; Münzberg, H. Mechanisms of leptin action and leptin resistance. Annu. Rev. Physiol. 2008, 70, 537–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Münzberg, H.; Björnholm, M.; Bates, S.H.; Myers, M.G., Jr. Leptin receptor action and mechanisms of leptin resistance. Cell Mol. Life Sci. 2005, 62, 642–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balland, E.; Chen, W.; Tiganis, T.; Cowley, M.A. Persistent Leptin Signaling in the Arcuate Nucleus Impairs Hypothalamic Insulin Signaling and Glucose Homeostasis in Obese Mice. Neuroendocrinology 2019, 109, 374–390. [Google Scholar] [CrossRef] [PubMed]
- Liao, T.; Zhang, S.L.; Yuan, X.; Mo, W.Q.; Wei, F.; Zhao, S.N.; Yang, W.; Liu, H.; Rong, X. Liraglutide Lowers Body Weight Set Point in DIO Rats and its Relationship with Hypothalamic Microglia Activation. Obesity 2020, 28, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Zhu, Y.; Schultz, R.D.; Li, N.; He, Z.; Zhang, Z.; Caron, A.; Zhu, Q.; Sun, K.; Xiong, W.; et al. Partial Leptin Reduction as an Insulin Sensitization and Weight Loss Strategy. Cell Metab. 2019, 30, 706–719.e706. [Google Scholar] [CrossRef]
- Dorfman, M.D.; Thaler, J.P. Hypothalamic inflammation and gliosis in obesity. Curr. Opin. Endocrinol. Diabetes Obes. 2015, 22, 325–330. [Google Scholar] [CrossRef] [Green Version]
- Bastard, J.P.; Maachi, M.; Lagathu, C.; Kim, M.J.; Caron, M.; Vidal, H.; Capeau, J.; Feve, B. Recent advances in the relationship between obesity, inflammation, and insulin resistance. Eur. Cytokine Netw. 2006, 17, 4–12. [Google Scholar]
- Gregor, M.F.; Hotamisligil, G.S. Inflammatory mechanisms in obesity. Annu. Rev. Immunol. 2011, 29, 415–445. [Google Scholar] [CrossRef] [Green Version]
- Charrière, G.; Cousin, B.; Arnaud, E.; André, M.; Bacou, F.; Penicaud, L.; Casteilla, L. Preadipocyte conversion to macrophage. Evidence of plasticity. J. Biol. Chem. 2003, 278, 9850–9855. [Google Scholar] [CrossRef] [Green Version]
- Phillips, C.L.; Grayson, B.E. The immune remodel: Weight loss-mediated inflammatory changes to obesity. Exp. Biol. Med. 2020, 245, 109–121. [Google Scholar] [CrossRef] [Green Version]
- Aguilar-Valles, A.; Inoue, W.; Rummel, C.; Luheshi, G.N. Obesity, adipokines and neuroinflammation. Neuropharmacology 2015, 96, 124–134. [Google Scholar] [CrossRef]
- Trayhurn, P.; Wood, I.S. Adipokines: Inflammation and the pleiotropic role of white adipose tissue. Br. J. Nutr. 2004, 92, 347–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Cava, A.; Matarese, G. The weight of leptin in immunity. Nat. Rev. Immunol. 2004, 4, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Howard, J.K.; Lord, G.M.; Matarese, G.; Vendetti, S.; Ghatei, M.A.; Ritter, M.A.; Lechler, R.I.; Bloom, S.R. Leptin protects mice from starvation-induced lymphoid atrophy and increases thymic cellularity in ob/ob mice. J. Clin. Investig. 1999, 104, 1051–1059. [Google Scholar] [CrossRef] [Green Version]
- Siegl, D.; Annecke, T.; Johnson, B.L., 3rd; Schlag, C.; Martignoni, A.; Huber, N.; Conzen, P.; Caldwell, C.C.; Tschöp, J. Obesity-induced hyperleptinemia improves survival and immune response in a murine model of sepsis. Anesthesiology 2014, 121, 98–114. [Google Scholar] [CrossRef] [Green Version]
- Dib, L.H.; Ortega, M.T.; Fleming, S.D.; Chapes, S.K.; Melgarejo, T. Bone marrow leptin signaling mediates obesity-associated adipose tissue inflammation in male mice. Endocrinology 2014, 155, 40–46. [Google Scholar] [CrossRef] [Green Version]
- Frühbeck, G.; Catalán, V.; Rodríguez, A.; Ramírez, B.; Becerril, S.; Salvador, J.; Colina, I.; Gómez-Ambrosi, J. Adiponectin-leptin Ratio is a Functional Biomarker of Adipose Tissue Inflammation. Nutrients 2019, 11, 454. [Google Scholar] [CrossRef] [Green Version]
- Fantuzzi, G. Adiponectin and inflammation: Consensus and controversy. J. Allergy Clin. Immunol. 2008, 121, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Pang, S.S.; Le, Y.Y. Role of resistin in inflammation and inflammation-related diseases. Cell Mol. Immunol. 2006, 3, 29–34. [Google Scholar]
- Vendrell, J.; Broch, M.; Vilarrasa, N.; Molina, A.; Gómez, J.M.; Gutiérrez, C.; Simón, I.; Soler, J.; Richart, C. Resistin, adiponectin, ghrelin, leptin, and proinflammatory cytokines: Relationships in obesity. Obes. Res. 2004, 12, 962–971. [Google Scholar] [CrossRef]
- Bradley, J.R. TNF-mediated inflammatory disease. J. Pathol. 2008, 214, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Pereira, S.S.; Alvarez-Leite, J.I. Low-Grade Inflammation, Obesity, and Diabetes. Curr. Obes. Rep. 2014, 3, 422–431. [Google Scholar] [CrossRef] [PubMed]
- Tsigos, C.; Kyrou, I.; Chala, E.; Tsapogas, P.; Stavridis, J.C.; Raptis, S.A.; Katsilambros, N. Circulating tumor necrosis factor alpha concentrations are higher in abdominal versus peripheral obesity. Metabolism 1999, 48, 1332–1335. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.R. Inflammatory markers and bariatric surgery: A meta-analysis. Inflamm. Res. 2012, 61, 789–807. [Google Scholar] [CrossRef]
- Younis, S.; Rosner, I.; Rimar, D.; Boulman, N.; Rozenbaum, M.; Odeh, M.; Slobodin, G. Interleukin 6 blockade-associated weight gain with abdominal enlargement in a patient with rheumatoid arthritis. J. Clin. Rheumatol. 2013, 19, 48–49. [Google Scholar] [CrossRef]
- Park, H.S.; Park, J.Y.; Yu, R. Relationship of obesity and visceral adiposity with serum concentrations of CRP, TNF-alpha and IL-6. Diabetes Res. Clin. Pract. 2005, 69, 29–35. [Google Scholar] [CrossRef]
- Askarpour, M.; Khani, D.; Sheikhi, A.; Ghaedi, E.; Alizadeh, S. Effect of Bariatric Surgery on Serum Inflammatory Factors of Obese Patients: A Systematic Review and Meta-Analysis. Obes. Surg. 2019, 29, 2631–2647. [Google Scholar] [CrossRef]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte chemoattractant protein-1 (MCP-1): An overview. J. Interferon Cytokine Res. 2009, 29, 313–326. [Google Scholar] [CrossRef]
- Dahlman, I.; Kaaman, M.; Olsson, T.; Tan, G.D.; Bickerton, A.S.; Wåhlén, K.; Andersson, J.; Nordström, E.A.; Blomqvist, L.; Sjögren, A.; et al. A unique role of monocyte chemoattractant protein 1 among chemokines in adipose tissue of obese subjects. J. Clin. Endocrinol. Metab. 2005, 90, 5834–5840. [Google Scholar] [CrossRef] [Green Version]
- Tourniaire, F.; Romier-Crouzet, B.; Lee, J.H.; Marcotorchino, J.; Gouranton, E.; Salles, J.; Malezet, C.; Astier, J.; Darmon, P.; Blouin, E.; et al. Chemokine Expression in Inflamed Adipose Tissue Is Mainly Mediated by NF-κB. PLoS ONE 2013, 8, e66515. [Google Scholar] [CrossRef]
- Dalmas, E.; Rouault, C.; Abdennour, M.; Rovere, C.; Rizkalla, S.; Bar-Hen, A.; Nahon, J.L.; Bouillot, J.L.; Guerre-Millo, M.; Clément, K.; et al. Variations in circulating inflammatory factors are related to changes in calorie and carbohydrate intakes early in the course of surgery-induced weight reduction. Am. J. Clin. Nutr. 2011, 94, 450–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baggiolini, M.; Clark-Lewis, I. Interleukin-8, a chemotactic and inflammatory cytokine. FEBS Lett. 1992, 307, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.S.; Park, H.S.; Kawada, T.; Kim, J.H.; Lim, D.; Hubbard, N.E.; Kwon, B.S.; Erickson, K.L.; Yu, R. Circulating levels of MCP-1 and IL-8 are elevated in human obese subjects and associated with obesity-related parameters. Int. J. Obes. 2006, 30, 1347–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruun, J.M.; Verdich, C.; Toubro, S.; Astrup, A.; Richelsen, B. Association between measures of insulin sensitivity and circulating levels of interleukin-8, interleukin-6 and tumor necrosis factor-alpha. Effect of weight loss in obese men. Eur. J. Endocrinol. 2003, 148, 535–542. [Google Scholar] [CrossRef] [Green Version]
- Alvehus, M.; Simonyte, K.; Andersson, T.; Söderström, I.; Burén, J.; Rask, E.; Mattsson, C.; Olsson, T. Adipose tissue IL-8 is increased in normal weight women after menopause and reduced after gastric bypass surgery in obese women. Clin. Endocrinol. 2012, 77, 684–690. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.M.; Zhang, X. Interleukin-10: New perspectives on an old cytokine. Immunol. Rev. 2008, 226, 205–218. [Google Scholar] [CrossRef]
- Gotoh, K.; Fujiwara, K.; Anai, M.; Okamoto, M.; Masaki, T.; Kakuma, T.; Shibata, H. Role of spleen-derived IL-10 in prevention of systemic low-grade inflammation by obesity [Review]. Endocr. J. 2017, 64, 375–378. [Google Scholar] [CrossRef] [Green Version]
- Esposito, K.; Pontillo, A.; Giugliano, F.; Giugliano, G.; Marfella, R.; Nicoletti, G.; Giugliano, D. Association of low interleukin-10 levels with the metabolic syndrome in obese women. J. Clin. Endocrinol. Metab. 2003, 88, 1055–1058. [Google Scholar] [CrossRef]
- Mold, C.; Gewurz, H.; Du Clos, T.W. Regulation of complement activation by C-reactive protein. Immunopharmacology 1999, 42, 23–30. [Google Scholar] [CrossRef]
- Holloszy, J.O.; Fontana, L. Caloric restriction in humans. Exp. Gerontol. 2007, 42, 709–712. [Google Scholar] [CrossRef]
- Ress, C.; Tschoner, A.; Engl, J.; Klaus, A.; Tilg, H.; Ebenbichler, C.F.; Patsch, J.R.; Kaser, S. Effect of bariatric surgery on circulating chemerin levels. Eur. J. Clin. Investig. 2010, 40, 277–280. [Google Scholar] [CrossRef]
- Letterio, J.J.; Roberts, A.B. Regulation of immune responses by TGF-beta. Annu. Rev. Immunol. 1998, 16, 137–161. [Google Scholar] [CrossRef] [PubMed]
- Alessi, M.C.; Bastelica, D.; Morange, P.; Berthet, B.; Leduc, I.; Verdier, M.; Geel, O.; Juhan-Vague, I. Plasminogen activator inhibitor 1, transforming growth factor-beta1, and BMI are closely associated in human adipose tissue during morbid obesity. Diabetes 2000, 49, 1374–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cottam, D.R.; Mattar, S.G.; Barinas-Mitchell, E.; Eid, G.; Kuller, L.; Kelley, D.E.; Schauer, P.R. The chronic inflammatory hypothesis for the morbidity associated with morbid obesity: Implications and effects of weight loss. Obes. Surg. 2004, 14, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Festa, A.; D’Agostino, R., Jr.; Williams, K.; Karter, A.J.; Mayer-Davis, E.J.; Tracy, R.P.; Haffner, S.M. The relation of body fat mass and distribution to markers of chronic inflammation. Int. J. Obes. Relat. Metab. Disord. 2001, 25, 1407–1415. [Google Scholar] [CrossRef] [Green Version]
- Dixon, J.B.; O’Brien, P.E. Obesity and the white blood cell count: Changes with sustained weight loss. Obes. Surg. 2006, 16, 251–257. [Google Scholar] [CrossRef]
- Lynch, L.A.; O’Connell, J.M.; Kwasnik, A.K.; Cawood, T.J.; O’Farrelly, C.; O’Shea, D.B. Are natural killer cells protecting the metabolically healthy obese patient? Obesity 2009, 17, 601–605. [Google Scholar] [CrossRef]
- Fischer, I.P.; Irmler, M.; Meyer, C.W.; Sachs, S.J.; Neff, F.; Hrabě de Angelis, M.; Beckers, J.; Tschöp, M.H.; Hofmann, S.M.; Ussar, S. A history of obesity leaves an inflammatory fingerprint in liver and adipose tissue. Int. J. Obes. 2018, 42, 507–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacLean, P.S.; Higgins, J.A.; Giles, E.D.; Sherk, V.D.; Jackman, M.R. The role for adipose tissue in weight regain after weight loss. Obes. Rev. 2015, 16 (Suppl. S1), 45–54. [Google Scholar] [CrossRef] [Green Version]
- Wood, I.S.; de Heredia, F.P.; Wang, B.; Trayhurn, P. Cellular hypoxia and adipose tissue dysfunction in obesity. Proc. Nutr. Soc. 2009, 68, 370–377. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Kim, J.; Yoon, J.H.; Ghim, J.; Yea, K.; Song, P.; Park, S.; Lee, A.; Hong, C.P.; Jang, M.S.; et al. CXCL12 secreted from adipose tissue recruits macrophages and induces insulin resistance in mice. Diabetologia 2014, 57, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Schenk, S.; Saberi, M.; Olefsky, J.M. Insulin sensitivity: Modulation by nutrients and inflammation. J. Clin. Investig. 2008, 118, 2992–3002. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Haghiac, M.; Surace, P.; Challier, J.C.; Guerre-Millo, M.; Singh, K.; Waters, T.; Minium, J.; Presley, L.; Catalano, P.M.; et al. Pregravid obesity associates with increased maternal endotoxemia and metabolic inflammation. Obesity 2011, 19, 476–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Yu, Y.; Qin, Y.; Zhou, Y.; Tang, R.; Wang, Q.; Li, X.; Wang, H.; Weston-Green, K.; Huang, X.F.; et al. Alterations to the microbiota-colon-brain axis in high-fat-diet-induced obese mice compared to diet-resistant mice. J. Nutr. Biochem. 2019, 65, 54–65. [Google Scholar] [CrossRef] [Green Version]
- Chompre, G.; Sambolin, L.; Cruz, M.L.; Sanchez, R.; Rodriguez, Y.; Rodríguez-Santiago, R.E.; Yamamura, Y.; Appleyard, C.B. A one month high fat diet disrupts the gut microbiome and integrity of the colon inducing adiposity and behavioral despair in male Sprague Dawley rats. Heliyon 2022, 8, e11194. [Google Scholar] [CrossRef]
- Canale, M.P.; Manca di Villahermosa, S.; Martino, G.; Rovella, V.; Noce, A.; De Lorenzo, A.; Di Daniele, N. Obesity-related metabolic syndrome: Mechanisms of sympathetic overactivity. Int. J. Endocrinol. 2013, 2013, 865965. [Google Scholar] [CrossRef] [Green Version]
- Esler, M.; Straznicky, N.; Eikelis, N.; Masuo, K.; Lambert, G.; Lambert, E. Mechanisms of sympathetic activation in obesity-related hypertension. Hypertension 2006, 48, 787–796. [Google Scholar] [CrossRef] [Green Version]
- Occhinegro, A.; Wong, C.Y.; Chua, B.Y.; Jackson, D.C.; McKinley, M.J.; McAllen, R.M.; Martelli, D. The endogenous inflammatory reflex inhibits the inflammatory response to different immune challenges in mice. Brain Behav. Immun. 2021, 97, 371–375. [Google Scholar] [CrossRef]
- McKinley, M.J.; Martelli, D.; Trevizan-Baú, P.; McAllen, R.M. Divergent splanchnic sympathetic efferent nerve pathways regulate interleukin-10 and tumour necrosis factor-α responses to endotoxaemia. J. Physiol. 2022, 600, 4521–4536. [Google Scholar] [CrossRef]
- van der Heijden, C.; Groh, L.; Keating, S.T.; Kaffa, C.; Noz, M.P.; Kersten, S.; van Herwaarden, A.E.; Hoischen, A.; Joosten, L.A.B.; Timmers, H.; et al. Catecholamines Induce Trained Immunity in Monocytes In Vitro and In Vivo. Circ. Res. 2020, 127, 269–283. [Google Scholar] [CrossRef]
- Straub, R.H.; Dufner, B.; Rauch, L. Proinflammatory α-Adrenergic Neuronal Regulation of Splenic IFN-γ, IL-6, and TGF-β of Mice from Day 15 onwards in Arthritis. Neuroimmunomodulation 2020, 27, 58–68. [Google Scholar] [CrossRef]
- Oquendo, M.B.; Lorza-Gil, E.; Juarez-Lopez, D.; Wagner, R.; Birkenfeld, A.L.; Ullrich, S.; Gerst, F. Effects of adrenergic-stimulated lipolysis and cytokine production on in vitro mouse adipose tissue-islet interactions. Sci. Rep. 2022, 12, 15831. [Google Scholar] [CrossRef] [PubMed]
- Thaler, J.P.; Yi, C.X.; Schur, E.A.; Guyenet, S.J.; Hwang, B.H.; Dietrich, M.O.; Zhao, X.; Sarruf, D.A.; Izgur, V.; Maravilla, K.R.; et al. Obesity is associated with hypothalamic injury in rodents and humans. J. Clin. Investig. 2012, 122, 153–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreutzer, C.; Peters, S.; Schulte, D.M.; Fangmann, D.; Türk, K.; Wolff, S.; van Eimeren, T.; Ahrens, M.; Beckmann, J.; Schafmayer, C.; et al. Hypothalamic Inflammation in Human Obesity Is Mediated by Environmental and Genetic Factors. Diabetes 2017, 66, 2407–2415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Souza, C.T.; Araujo, E.P.; Bordin, S.; Ashimine, R.; Zollner, R.L.; Boschero, A.C.; Saad, M.J.; Velloso, L.A. Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology 2005, 146, 4192–4199. [Google Scholar] [CrossRef] [Green Version]
- Le Thuc, O.; Stobbe, K.; Cansell, C.; Nahon, J.L.; Blondeau, N.; Rovère, C. Hypothalamic Inflammation and Energy Balance Disruptions: Spotlight on Chemokines. Front. Endocrinol. 2017, 8, 197. [Google Scholar] [CrossRef] [Green Version]
- Douglass, J.D.; Dorfman, M.D.; Fasnacht, R.; Shaffer, L.D.; Thaler, J.P. Astrocyte IKKβ/NF-κB signaling is required for diet-induced obesity and hypothalamic inflammation. Mol. Metab. 2017, 6, 366–373. [Google Scholar] [CrossRef]
- Benzler, J.; Ganjam, G.K.; Pretz, D.; Oelkrug, R.; Koch, C.E.; Legler, K.; Stöhr, S.; Culmsee, C.; Williams, L.M.; Tups, A. Central inhibition of IKKβ/NF-κB signaling attenuates high-fat diet-induced obesity and glucose intolerance. Diabetes 2015, 64, 2015–2027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- André, C.; Guzman-Quevedo, O.; Rey, C.; Rémus-Borel, J.; Clark, S.; Castellanos-Jankiewicz, A.; Ladeveze, E.; Leste-Lasserre, T.; Nadjar, A.; Abrous, D.N.; et al. Inhibiting Microglia Expansion Prevents Diet-Induced Hypothalamic and Peripheral Inflammation. Diabetes 2017, 66, 908–919. [Google Scholar] [CrossRef] [Green Version]
- Morari, J.; Anhe, G.F.; Nascimento, L.F.; de Moura, R.F.; Razolli, D.; Solon, C.; Guadagnini, D.; Souza, G.; Mattos, A.H.; Tobar, N.; et al. Fractalkine (CX3CL1) is involved in the early activation of hypothalamic inflammation in experimental obesity. Diabetes 2014, 63, 3770–3784. [Google Scholar] [CrossRef] [Green Version]
- Poon, K.; Barson, J.R.; Ho, H.T.; Leibowitz, S.F. Relationship of the Chemokine, CXCL12, to Effects of Dietary Fat on Feeding-Related Behaviors and Hypothalamic Neuropeptide Systems. Front. Behav. Neurosci. 2016, 10, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pflieger, F.J.; Hernandez, J.; Schweighöfer, H.; Herden, C.; Rosengarten, B.; Rummel, C. The role of neutrophil granulocytes in immune-to-brain communication. Temperature 2018, 5, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Brandi, E.; Torres-Garcia, L.; Svanbergsson, A.; Haikal, C.; Liu, D.; Li, W.; Li, J.Y. Brain region-specific microglial and astrocytic activation in response to systemic lipopolysaccharides exposure. Front. Aging Neurosci. 2022, 14, 910988. [Google Scholar] [CrossRef] [PubMed]
- Brochu, S.; Olivier, M.; Rivest, S. Neuronal activity and transcription of proinflammatory cytokines, IkappaBalpha, and iNOS in the mouse brain during acute endotoxemia and chronic infection with Trypanosoma brucei brucei. J. Neurosci. Res. 1999, 57, 801–816. [Google Scholar] [CrossRef]
- Vargas-Caraveo, A.; Sayd, A.; Maus, S.R.; Caso, J.R.; Madrigal, J.L.M.; García-Bueno, B.; Leza, J.C. Lipopolysaccharide enters the rat brain by a lipoprotein-mediated transport mechanism in physiological conditions. Sci. Rep. 2017, 7, 13113. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.H.; Kim, H.J.; Lee, Y.S.; Kang, G.M.; Lim, H.S.; Lee, S.H.; Song, D.K.; Kwon, O.; Hwang, I.; Son, M.; et al. Hypothalamic Macrophage Inducible Nitric Oxide Synthase Mediates Obesity-Associated Hypothalamic Inflammation. Cell Rep. 2018, 25, 934–946.e935. [Google Scholar] [CrossRef] [Green Version]
- Waise, T.M.Z.; Toshinai, K.; Naznin, F.; NamKoong, C.; Md Moin, A.S.; Sakoda, H.; Nakazato, M. One-day high-fat diet induces inflammation in the nodose ganglion and hypothalamus of mice. Biochem. Biophys. Res. Commun. 2015, 464, 1157–1162. [Google Scholar] [CrossRef] [Green Version]
- de La Serre, C.B.; de Lartigue, G.; Raybould, H.E. Chronic exposure to low dose bacterial lipopolysaccharide inhibits leptin signaling in vagal afferent neurons. Physiol. Behav. 2015, 139, 188–194. [Google Scholar] [CrossRef] [Green Version]
- Hosoi, T.; Okuma, Y.; Nomura, Y. Electrical stimulation of afferent vagus nerve induces IL-1beta expression in the brain and activates HPA axis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000, 279, R141–R147. [Google Scholar] [CrossRef] [Green Version]
- Borges Bde, C.; Rorato, R.C.; Uchoa, E.T.; Marangon, P.B.; Elias, C.F.; Antunes-Rodrigues, J.; Elias, L.L. Protein tyrosine phosphatase-1B contributes to LPS-induced leptin resistance in male rats. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E40–E50. [Google Scholar] [CrossRef] [Green Version]
- Van Dyken, P.; Lacoste, B. Impact of Metabolic Syndrome on Neuroinflammation and the Blood-Brain Barrier. Front. Neurosci. 2018, 12, 930. [Google Scholar] [CrossRef] [Green Version]
- Rummel, C.; Inoue, W.; Poole, S.; Luheshi, G.N. Leptin regulates leukocyte recruitment into the brain following systemic LPS-induced inflammation. Mol. Psychiatry 2010, 15, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Clegg, D.J.; Brown, L.M.; Woods, S.C.; Benoit, S.C. Gonadal hormones determine sensitivity to central leptin and insulin. Diabetes 2006, 55, 978–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Côté, I.; Green, S.M.; Toklu, H.Z.; Morgan, D.; Carter, C.S.; Tümer, N.; Scarpace, P.J. Differential physiological responses to central leptin overexpression in male and female rats. J. Neuroendocrinol. 2017, 29, jne12552. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.B.; Bowen, H.M.; Mitchell, T.D. Leptin resistance in mice is determined by gender and duration of exposure to high-fat diet. Physiol. Behav. 2003, 78, 543–555. [Google Scholar] [CrossRef]
- Mark, A.L. Selective leptin resistance revisited. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 305, R566–R581. [Google Scholar] [CrossRef]
- Bell, B.B.; Rahmouni, K. Leptin as a Mediator of Obesity-Induced Hypertension. Curr. Obes. Rep. 2016, 5, 397–404. [Google Scholar] [CrossRef] [Green Version]
- Lambert, E.; Sari, C.I.; Dawood, T.; Nguyen, J.; McGrane, M.; Eikelis, N.; Chopra, R.; Wong, C.; Chatzivlastou, K.; Head, G.; et al. Sympathetic nervous system activity is associated with obesity-induced subclinical organ damage in young adults. Hypertension 2010, 56, 351–358. [Google Scholar] [CrossRef] [Green Version]
- Fu, Q. Sex differences in sympathetic activity in obesity and its related hypertension. Ann. N. Y. Acad. Sci. 2019, 1454, 31–41. [Google Scholar] [CrossRef]
- Lambert, E.; Straznicky, N.; Eikelis, N.; Esler, M.; Dawood, T.; Masuo, K.; Schlaich, M.; Lambert, G. Gender differences in sympathetic nervous activity: Influence of body mass and blood pressure. J. Hypertens. 2007, 25, 1411–1419. [Google Scholar] [CrossRef]
- Tank, J.; Heusser, K.; Diedrich, A.; Hering, D.; Luft, F.C.; Busjahn, A.; Narkiewicz, K.; Jordan, J. Influences of gender on the interaction between sympathetic nerve traffic and central adiposity. J. Clin. Endocrinol. Metab. 2008, 93, 4974–4978. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Cham, J.L.; Badoer, E. High-fat feeding alters the cardiovascular role of the hypothalamic paraventricular nucleus. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R799–R807. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Zhao, D.; Cassaglia, P.A.; Brooks, V.L. Sites and sources of sympathoexcitation in obese male rats: Role of brain insulin. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2020, 318, R634–R648. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.; Burke, S.L.; Head, G.A. Obesity-related hypertension and the role of insulin and leptin in high-fat-fed rabbits. Hypertension 2013, 61, 628–634. [Google Scholar] [CrossRef] [Green Version]
- Simonds, S.E.; Pryor, J.T.; Ravussin, E.; Greenway, F.L.; Dileone, R.; Allen, A.M.; Bassi, J.; Elmquist, J.K.; Keogh, J.M.; Henning, E.; et al. Leptin mediates the increase in blood pressure associated with obesity. Cell 2014, 159, 1404–1416. [Google Scholar] [CrossRef] [Green Version]
- Shi, Z.; Cassaglia, P.A.; Pelletier, N.E.; Brooks, V.L. Sex differences in the sympathoexcitatory response to insulin in obese rats: Role of neuropeptide Y. J. Physiol. 2019, 597, 1757–1775. [Google Scholar] [CrossRef] [Green Version]
- Evans, M.C.; Lord, R.A.; Anderson, G.M. Multiple Leptin Signalling Pathways in the Control of Metabolism and Fertility: A Means to Different Ends? Int. J. Mol. Sci. 2021, 22, 9210. [Google Scholar] [CrossRef]
- Arakawa, H.; Chitravanshi, V.C.; Sapru, H.N. The hypothalamic arcuate nucleus: A new site of cardiovascular action of angiotensin-(1-12) and angiotensin II. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H951–H960. [Google Scholar] [CrossRef]
- Shi, Z.; Stornetta, D.S.; Stornetta, R.L.; Brooks, V.L. Arcuate Angiotensin II Increases Arterial Pressure via Coordinated Increases in Sympathetic Nerve Activity and Vasopressin Secretion. eNeuro 2022, 9, 1–23. [Google Scholar] [CrossRef]
- Shi, Z.; Stornetta, R.L.; Stornetta, D.S.; Abbott, S.B.G.; Brooks, V.L. The arcuate nucleus: A site of synergism between Angiotensin II and leptin to increase sympathetic nerve activity and blood pressure in rats. Neurosci. Lett. 2022, 785, 136773. [Google Scholar] [CrossRef]
- Jöhren, O.; Sanvitto, G.L.; Egidy, G.; Saavedra, J.M. Angiotensin II AT1A receptor mRNA expression is induced by estrogen-progesterone in dopaminergic neurons of the female rat arcuate nucleus. J. Neurosci. 1997, 17, 8283–8292. [Google Scholar] [CrossRef] [Green Version]
- Claflin, K.E.; Sandgren, J.A.; Lambertz, A.M.; Weidemann, B.J.; Littlejohn, N.K.; Burnett, C.M.; Pearson, N.A.; Morgan, D.A.; Gibson-Corley, K.N.; Rahmouni, K.; et al. Angiotensin AT1A receptors on leptin receptor-expressing cells control resting metabolism. J. Clin. Investig. 2017, 127, 1414–1424. [Google Scholar] [CrossRef]
- Mehay, D.; Silberman, Y.; Arnold, A.C. The Arcuate Nucleus of the Hypothalamus and Metabolic Regulation: An Emerging Role for Renin-Angiotensin Pathways. Int. J. Mol. Sci. 2021, 22, 7050. [Google Scholar] [CrossRef]
- Cassis, L.A.; Police, S.B.; Yiannikouris, F.; Thatcher, S.E. Local adipose tissue renin-angiotensin system. Curr. Hypertens. Rep. 2008, 10, 93–98. [Google Scholar] [CrossRef] [Green Version]
- Brooks, V.L.; Shi, Z.; Holwerda, S.W.; Fadel, P.J. Obesity-induced increases in sympathetic nerve activity: Sex matters. Auton. Neurosci. 2015, 187, 18–26. [Google Scholar] [CrossRef] [Green Version]
- Morselli, E.; Frank, A.P.; Palmer, B.F.; Rodriguez-Navas, C.; Criollo, A.; Clegg, D.J. A sexually dimorphic hypothalamic response to chronic high-fat diet consumption. Int. J. Obes. 2016, 40, 206–209. [Google Scholar] [CrossRef]
- Joseph-Bravo, P.; Jaimes-Hoy, L.; Uribe, R.M.; Charli, J.L. 60 YEARS OF NEUROENDOCRINOLOGY: TRH, the first hypophysiotropic releasing hormone isolated: Control of the pituitary-thyroid axis. J. Endocrinol. 2015, 226, T85–T100. [Google Scholar] [CrossRef] [Green Version]
- Perello, M.; Cakir, I.; Cyr, N.E.; Romero, A.; Stuart, R.C.; Chiappini, F.; Hollenberg, A.N.; Nillni, E.A. Maintenance of the thyroid axis during diet-induced obesity in rodents is controlled at the central level. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E976–E989. [Google Scholar] [CrossRef] [Green Version]
- Araujo, R.L.; Andrade, B.M.; Padrón, A.S.; Gaidhu, M.P.; Perry, R.L.; Carvalho, D.P.; Ceddia, R.B. High-fat diet increases thyrotropin and oxygen consumption without altering circulating 3,5,3’-triiodothyronine (T3) and thyroxine in rats: The role of iodothyronine deiodinases, reverse T3 production, and whole-body fat oxidation. Endocrinology 2010, 151, 3460–3469. [Google Scholar] [CrossRef]
- Reinehr, T. Obesity and thyroid function. Mol. Cell Endocrinol. 2010, 316, 165–171. [Google Scholar] [CrossRef]
- Iacobellis, G.; Ribaudo, M.C.; Zappaterreno, A.; Iannucci, C.V.; Leonetti, F. Relationship of thyroid function with body mass index, leptin, insulin sensitivity and adiponectin in euthyroid obese women. Clin. Endocrinol. 2005, 62, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Sari, R.; Balci, M.K.; Altunbas, H.; Karayalcin, U. The effect of body weight and weight loss on thyroid volume and function in obese women. Clin. Endocrinol. 2003, 59, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Landa, M.S.; García, S.I.; Schuman, M.L.; Burgueño, A.; Alvarez, A.L.; Saravia, F.E.; Gemma, C.; Pirola, C.J. Knocking down the diencephalic thyrotropin-releasing hormone precursor gene normalizes obesity-induced hypertension in the rat. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E1388–E1394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravussin, E.; Smith, S.R.; Ferrante, A.W., Jr. Physiology of Energy Expenditure in the Weight-Reduced State. Obesity 2021, 29 (Suppl. S1), S31–S38. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, A.; Doucet, E. Relative changes in resting energy expenditure during weight loss: A systematic review. Obes. Rev. 2010, 11, 531–547. [Google Scholar] [CrossRef] [PubMed]
- Busetto, L.; Bettini, S.; Makaronidis, J.; Roberts, C.A.; Halford, J.C.G.; Batterham, R.L. Mechanisms of weight regain. Eur. J. Intern. Med. 2021, 93, 3–7. [Google Scholar] [CrossRef]
- Fantin, F.; Giani, A.; Zoico, E.; Rossi, A.P.; Mazzali, G.; Zamboni, M. Weight Loss and Hypertension in Obese Subjects. Nutrients 2019, 11, 1667. [Google Scholar] [CrossRef] [Green Version]
- Cohen, J.B. Hypertension in Obesity and the Impact of Weight Loss. Curr. Cardiol. Rep. 2017, 19, 98. [Google Scholar] [CrossRef]
- Knuth, N.D.; Johannsen, D.L.; Tamboli, R.A.; Marks-Shulman, P.A.; Huizenga, R.; Chen, K.Y.; Abumrad, N.N.; Ravussin, E.; Hall, K.D. Metabolic adaptation following massive weight loss is related to the degree of energy imbalance and changes in circulating leptin. Obesity 2014, 22, 2563–2569. [Google Scholar] [CrossRef] [Green Version]
- Rosenbaum, M.; Hirsch, J.; Murphy, E.; Leibel, R.L. Effects of changes in body weight on carbohydrate metabolism, catecholamine excretion, and thyroid function. Am. J. Clin. Nutr. 2000, 71, 1421–1432. [Google Scholar] [CrossRef] [Green Version]
- Kissileff, H.R.; Thornton, J.C.; Torres, M.I.; Pavlovich, K.; Mayer, L.S.; Kalari, V.; Leibel, R.L.; Rosenbaum, M. Leptin reverses declines in satiation in weight-reduced obese humans. Am. J. Clin. Nutr. 2012, 95, 309–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenbaum, M.; Goldsmith, R.; Bloomfield, D.; Magnano, A.; Weimer, L.; Heymsfield, S.; Gallagher, D.; Mayer, L.; Murphy, E.; Leibel, R.L. Low-dose leptin reverses skeletal muscle, autonomic, and neuroendocrine adaptations to maintenance of reduced weight. J. Clin. Investig. 2005, 115, 3579–3586. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, M.; Goldsmith, R.L.; Haddad, F.; Baldwin, K.M.; Smiley, R.; Gallagher, D.; Leibel, R.L. Triiodothyronine and leptin repletion in humans similarly reverse weight-loss-induced changes in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E771–E779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cottam, M.A.; Caslin, H.L.; Winn, N.C.; Hasty, A.H. Multiomics reveals persistence of obesity-associated immune cell phenotypes in adipose tissue during weight loss and weight regain in mice. Nat. Commun. 2022, 13, 2950. [Google Scholar] [CrossRef] [PubMed]
- Zamarron, B.F.; Mergian, T.A.; Cho, K.W.; Martinez-Santibanez, G.; Luan, D.; Singer, K.; DelProposto, J.L.; Geletka, L.M.; Muir, L.A.; Lumeng, C.N. Macrophage Proliferation Sustains Adipose Tissue Inflammation in Formerly Obese Mice. Diabetes 2017, 66, 392–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirakawa, K.; Endo, J.; Katsumata, Y.; Yamamoto, T.; Kataoka, M.; Isobe, S.; Yoshida, N.; Fukuda, K.; Sano, M. Negative legacy of obesity. PLoS ONE 2017, 12, e0186303. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, J.; Evers, N.; Awazawa, M.; Nicholls, H.T.; Brönneke, H.S.; Dietrich, A.; Mauer, J.; Blüher, M.; Brüning, J.C. Obesogenic memory can confer long-term increases in adipose tissue but not liver inflammation and insulin resistance after weight loss. Mol. Metab. 2016, 5, 328–339. [Google Scholar] [CrossRef]
- Wang, X.; Ge, A.; Cheng, M.; Guo, F.; Zhao, M.; Zhou, X.; Liu, L.; Yang, N. Increased hypothalamic inflammation associated with the susceptibility to obesity in rats exposed to high-fat diet. Exp. Diabetes Res. 2012, 2012, 847246. [Google Scholar] [CrossRef] [Green Version]
- de Castro, J.M.; Stein, D.J.; Medeiros, H.R.; de Oliveira, C.; Torres, I.L.S. Nicotinamide Riboside Neutralizes Hypothalamic Inflammation and Increases Weight Loss Without Altering Muscle Mass in Obese Rats Under Calorie Restriction: A Preliminary Investigation. Front. Nutr. 2021, 8, 648893. [Google Scholar] [CrossRef]
- Patkar, P.P.; Hao, Z.; Mumphrey, M.B.; Townsend, R.L.; Berthoud, H.R.; Shin, A.C. Unlike calorie restriction, Roux-en-Y gastric bypass surgery does not increase hypothalamic AgRP and NPY in mice on a high-fat diet. Int. J. Obes. 2019, 43, 2143–2150. [Google Scholar] [CrossRef]
- Barreto-Vianna, A.R.; Aguila, M.B.; Mandarim-de-Lacerda, C.A. Effects of liraglutide in hypothalamic arcuate nucleus of obese mice. Obesity 2016, 24, 626–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Haase, N.; Haange, S.B.; Sucher, R.; Münzker, J.; Jäger, E.; Schischke, K.; Seyfried, F.; von Bergen, M.; Hankir, M.K.; et al. Roux-en-Y gastric bypass contributes to weight loss-independent improvement in hypothalamic inflammation and leptin sensitivity through gut-microglia-neuron-crosstalk. Mol. Metab. 2021, 48, 101214. [Google Scholar] [CrossRef]
- Frank, A.P.; Zechner, J.F.; Clegg, D.J. Gastric Bypass Surgery but not Caloric Restriction Improves Reproductive Function in Obese Mice. Obes. Surg. 2016, 26, 467–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.Y.; Mu, S.; Zhang, S.P.; Guo, W.; Li, Q.F.; Xiao, X.Q.; Zhang, J.; Wang, Z.H. Roux-en-Y gastric bypass surgery suppresses hypothalamic PTP1B protein level and alleviates leptin resistance in obese rats. Exp. Ther. Med. 2017, 14, 2536–2542. [Google Scholar] [CrossRef] [Green Version]
- Manning, S.; Pucci, A.; Batterham, R.L. Roux-en-Y gastric bypass: Effects on feeding behavior and underlying mechanisms. J. Clin. Investig. 2015, 125, 939–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hankir, M.K.; Rullmann, M.; Seyfried, F.; Preusser, S.; Poppitz, S.; Heba, S.; Gousias, K.; Hoyer, J.; Schütz, T.; Dietrich, A.; et al. Roux-en-Y gastric bypass surgery progressively alters radiologic measures of hypothalamic inflammation in obese patients. JCI Insight 2019, 4, e131329. [Google Scholar] [CrossRef]
- Kratz, M.; Hagman, D.K.; Kuzma, J.N.; Foster-Schubert, K.E.; Chan, C.P.; Stewart, S.; van Yserloo, B.; Westbrook, E.O.; Arterburn, D.E.; Flum, D.R.; et al. Improvements in glycemic control after gastric bypass occur despite persistent adipose tissue inflammation. Obesity 2016, 24, 1438–1445. [Google Scholar] [CrossRef] [Green Version]
- Billeter, A.T.; Vittas, S.; Israel, B.; Scheurlen, K.M.; Hidmark, A.; Fleming, T.H.; Kopf, S.; Büchler, M.W.; Müller-Stich, B.P. Gastric bypass simultaneously improves adipose tissue function and insulin-dependent type 2 diabetes mellitus. Langenbecks Arch. Surg. 2017, 402, 901–910. [Google Scholar] [CrossRef]
- Neff, K.J.; O’Donohoe, P.K.; le Roux, C.W. Anti-inflammatory effects of gastric bypass surgery and their association with improvement in metabolic profile. Expert Rev. Endocrinol. Metab. 2015, 10, 435–446. [Google Scholar] [CrossRef]
- Bendotti, G.; Montefusco, L.; Lunati, M.E.; Usuelli, V.; Pastore, I.; Lazzaroni, E.; Assi, E.; Seelam, A.J.; El Essawy, B.; Jang, J.; et al. The anti-inflammatory and immunological properties of GLP-1 Receptor Agonists. Pharmacol. Res. 2022, 182, 106320. [Google Scholar] [CrossRef]
- O’Neil, P.M.; Birkenfeld, A.L.; McGowan, B.; Mosenzon, O.; Pedersen, S.D.; Wharton, S.; Carson, C.G.; Jepsen, C.H.; Kabisch, M.; Wilding, J.P.H. Efficacy and safety of semaglutide compared with liraglutide and placebo for weight loss in patients with obesity: A randomised, double-blind, placebo and active controlled, dose-ranging, phase 2 trial. Lancet 2018, 392, 637–649. [Google Scholar] [CrossRef] [PubMed]
- Phillips, A.; Clements, J.N. Clinical review of subcutaneous semaglutide for obesity. J. Clin. Pharm. Ther. 2022, 47, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Kanoski, S.E.; Ong, Z.Y.; Fortin, S.M.; Schlessinger, E.S.; Grill, H.J. Liraglutide, leptin and their combined effects on feeding: Additive intake reduction through common intracellular signalling mechanisms. Diabetes Obes. Metab. 2015, 17, 285–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Z.; Gao, Y.; Lieu, L.; Afrin, S.; Cao, J.; Michael, N.J.; Dong, Y.; Sun, J.; Guo, H.; Williams, K.W. Direct and indirect effects of liraglutide on hypothalamic POMC and NPY/AgRP neurons—Implications for energy balance and glucose control. Mol. Metab. 2019, 28, 120–134. [Google Scholar] [CrossRef]
- Tam, C.S.; Lecoultre, V.; Ravussin, E. Novel strategy for the use of leptin for obesity therapy. Expert Opin. Biol. Ther. 2011, 11, 1677–1685. [Google Scholar] [CrossRef] [Green Version]
- Coker, C.R.; White, M.; Singal, A.; Bingaman, S.S.; Paul, A.; Arnold, A.C.; Silberman, Y. Minocycline Reduces Hypothalamic Microglia Activation and Improves Metabolic Dysfunction in High Fat Diet-Induced Obese Mice. Front. Physiol. 2022, 13, 933706. [Google Scholar] [CrossRef]
- Vaughn, A.C.; Cooper, E.M.; DiLorenzo, P.M.; O’Loughlin, L.J.; Konkel, M.E.; Peters, J.H.; Hajnal, A.; Sen, T.; Lee, S.H.; de La Serre, C.B.; et al. Energy-dense diet triggers changes in gut microbiota, reorganization of gut-brain vagal communication and increases body fat accumulation. Acta Neurobiol. Exp. 2017, 77, 18–30. [Google Scholar] [CrossRef]

| Cytokine | Description | Changes with Obesity |
|---|---|---|
| Leptin | Pro-inflammatory adipokine released by fat cells. Stimulates T cells, macrophages, and neutrophils to release pro-inflammatory cytokines [73]. Leptin is essential for normal T-cell proliferation and its deficiency causes thymus atrophy and severe immune dysfunction [73,74]. Surprisingly, leptin improves survival during sepsis [75]. | Levels directly related to fat stores; increase with obesity. WAT inflammation was strongly reduced when LepR was knocked out in leukocytes in DIO mice [76]. |
| Adiponectin | An anti-inflammatory adipokine that modulates a number of metabolic processes [77]. | Adiponectin circulating levels are usually inversely proportional to the level of visceral adiposity; therefore, obese individuals have very low levels of adiponectin [78]. The adiponectin–leptin ratio is often considered a functional marker of inflammation associated with obesity [77]. |
| Resistin | Also known as adipose tissue-specific secretory factor, it plays a role in the pathogenesis of atherosclerosis by enhancing the synthesis of hepatic LDL [79]. | Elevated with non-morbid obesity. Acts locally on leukocytes, located in the WAT, to induce the release of pro-inflammatory cytokines [80]. |
| Tumor necrosis factor α (TNF) | TNF is a necessary and sufficient mediator of inflammation, acutely released by macrophages, T cells, and natural killer cells during infection [81]. TNF is also released by adipocytes [82]. | Its plasma levels are generally high in obese individuals, especially in those presenting visceral obesity rather than subcutaneous obesity [83]. However, it is not clear whether its levels decrease after weight loss [84]. |
| IL-6 | A proinflammatory cytokine produced by immune, endothelial, and muscle cells as well as adipocytes [70]. Surprisingly, an anti-IL-6 antibody therapy, used for the treatment of rheumatoid arthritis, causes weight gain [85] | Plasma levels are correlated with BMI and especially with adipose tissue mass [86]. Surgery- induced weight loss is associated with a significant decrease in IL-6 levels [87]. |
| Monocyte chemoattractant protein-1 (MCP-1) | Key chemokine that regulates migration and infiltration of monocytes and macrophages [88]. | Circulating MCP-1 levels do not differ between lean and obese individuals [89]; however, levels of this chemokine increase selectively in the WAT of obese adults [90]. MCP-1 levels in plasma significantly decrease after Roux-en-Y Gastric Bypass (RYGB) surgery or a low calorie diet [70,91]. |
| Interleukin 8 (IL-8) | Pro-inflammatory and chemoattractant cytokine [92]. | Systemic levels are closely correlated with BMI, waist circumference, and other obesity-related parameters [93]. Weight loss is not always associated with a decrease in IL-8 plasma levels; in fact, low calorie diet-induced weight loss is associated with an increase in IL-8 levels [94], whereas weight loss induced by RYGB produces a decrease in IL-8 levels [95]. |
| Interleukin 10 (IL-10) | Anti-inflammatory cytokine released by M2 macrophages, Th2 T cells, neutrophils, and adipocytes [96]. | Systemic IL-10 levels are generally inversely correlated with BMI and body fat percentage [70,97]. Nevertheless, plasma IL-10 levels have been reported to be elevated in obese women; however, obese women with lower IL-10 levels were more prone to develop metabolic syndrome [98]. |
| C-reactive protein (CRP) | Released by hepatocytes in response to trauma, infection, or injury [99]. | CRP levels are significantly higher in obese individuals than in lean subjects [86] and decrease with diet or weight loss induced by surgical intervention [100,101], making it a good marker of meta-inflammation. |
| Transforming growth factor β (TGF-β) | Cytokine released by all leukocytes [102]. | Regardless of the location of fat mass, obesity is associated with enhanced levels of TGF-β [103], while weight loss decreases TGF- β circulating levels [104]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martelli, D.; Brooks, V.L. Leptin Increases: Physiological Roles in the Control of Sympathetic Nerve Activity, Energy Balance, and the Hypothalamic–Pituitary–Thyroid Axis. Int. J. Mol. Sci. 2023, 24, 2684. https://doi.org/10.3390/ijms24032684
Martelli D, Brooks VL. Leptin Increases: Physiological Roles in the Control of Sympathetic Nerve Activity, Energy Balance, and the Hypothalamic–Pituitary–Thyroid Axis. International Journal of Molecular Sciences. 2023; 24(3):2684. https://doi.org/10.3390/ijms24032684
Chicago/Turabian StyleMartelli, Davide, and Virginia L. Brooks. 2023. "Leptin Increases: Physiological Roles in the Control of Sympathetic Nerve Activity, Energy Balance, and the Hypothalamic–Pituitary–Thyroid Axis" International Journal of Molecular Sciences 24, no. 3: 2684. https://doi.org/10.3390/ijms24032684
APA StyleMartelli, D., & Brooks, V. L. (2023). Leptin Increases: Physiological Roles in the Control of Sympathetic Nerve Activity, Energy Balance, and the Hypothalamic–Pituitary–Thyroid Axis. International Journal of Molecular Sciences, 24(3), 2684. https://doi.org/10.3390/ijms24032684