
Estimation of an Image Biomarker for Distant Recurrence Prediction in NSCLC Using Proliferation-Related Genes

Abstract
:1. Introduction
2. Results
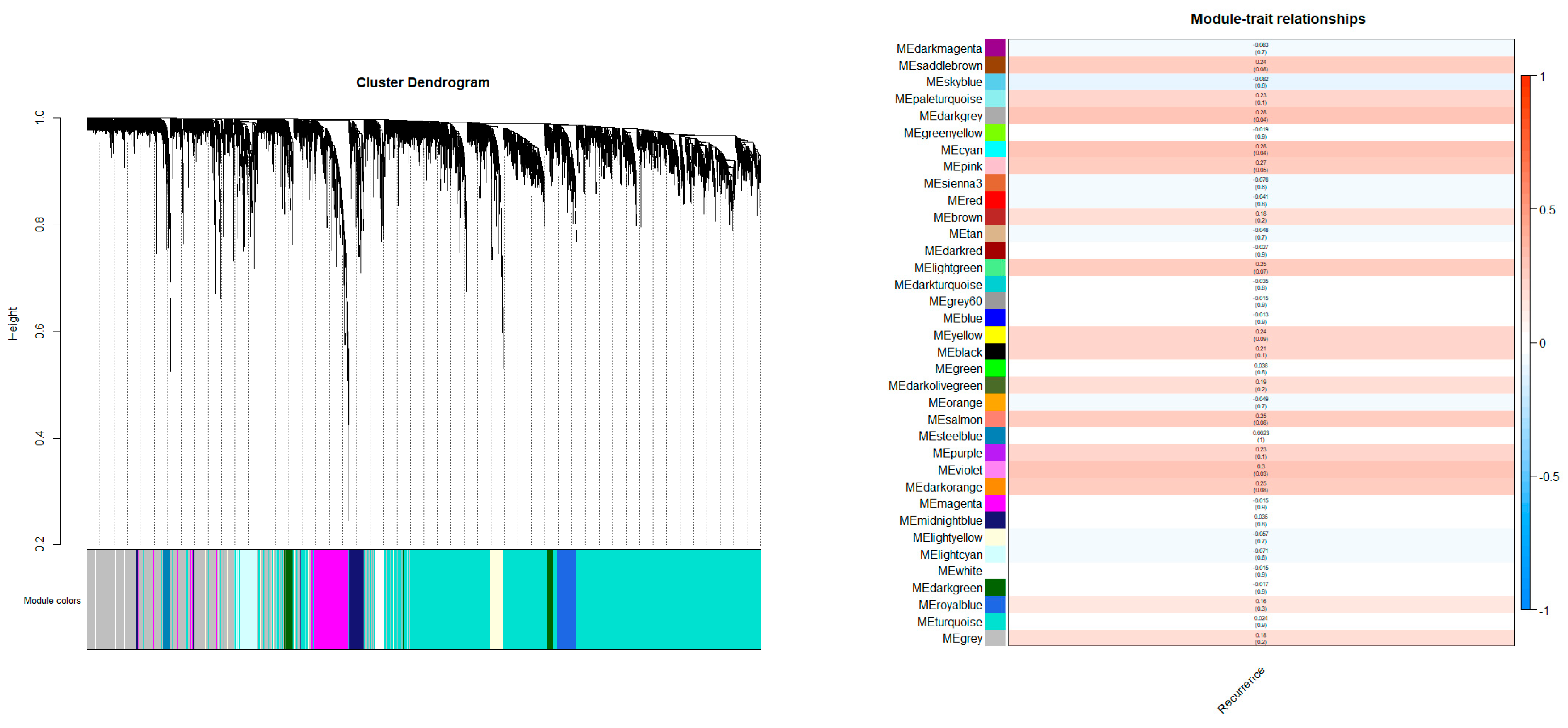
2.1. Gene Modulation and Hub Gene Assay
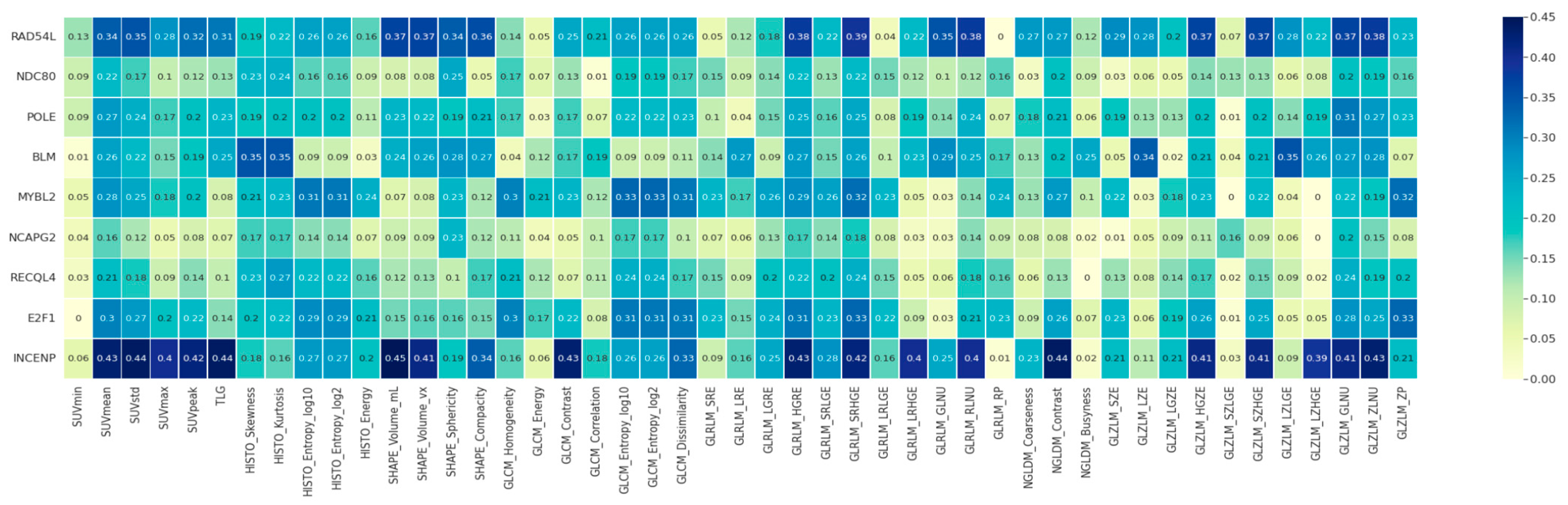
2.2. Hub Gene and Image Feature Associations
2.3. Creation of the Prediction Model
3. Discussion
4. Materials and Methods
4.1. NSCLC NGS Data Processing
4.2. Weighted Gene Co-Expression Networks and Modules Associated with Clinical Traits
4.3. Hub Gene Analysis
4.4. 18F-FDG PET Imaging
4.5. Hub Gene and Image Feature Correlation
4.6. RF Prediction Model Construction
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, Z.; Fillmore, C.M.; Hammerman, P.S.; Kim, C.F.; Wong, K.K. Non-small-cell lung cancers: A heterogeneous set of diseases. Nat. Rev. Cancer 2014, 14, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.; Wang, Z.; Zhang, J.; Zhou, H.; Jin, L.; Bai, R.; Weng, Y.J.C.P. Adam17, a target of Mir-326, promotes emt-induced cells invasion in lung adenocarcinoma. Cell. Physiol. Biochem. 2015, 36, 1175–1185. [Google Scholar] [CrossRef] [PubMed]
- Zito Marino, F.Z.; Bianco, R.; Accardo, M.; Ronchi, A.; Cozzolino, I.; Morgillo, F.; Rossi, G.; Franco, R. Molecular heterogeneity in lung cancer: From mechanisms of origin to clinical implications. Int. J. Med. Sci. 2019, 16, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Burrell, R.A.; McGranahan, N.; Bartek, J.; Swanton, C.J.N. The causes and consequences of genetic heterogeneity in cancer evolution. Nature 2013, 501, 338–345. [Google Scholar] [CrossRef]
- Kamel, H.F.M.; Al-Amodi, H.S.A.B. Exploitation of gene expression and cancer biomarkers in paving the path to era of personalized medicine. Genom. Proteom. Bioinform. 2017, 15, 220–235. [Google Scholar] [CrossRef]
- Ambardar, S.; Gupta, R.; Trakroo, D.; Lal, R.; Vakhlu, J. High throughput sequencing: An overview of sequencing chemistry. Indian J. Microbiol. 2016, 56, 394–404. [Google Scholar] [CrossRef]
- O’Brien, B.; van der Putten, W. Quantification of risk-benefit in interventional radiology. Radiat. Prot. Dosim. 2008, 129, 59–62. [Google Scholar] [CrossRef]
- Sala, E.; Mema, E.; Himoto, Y.; Veeraraghavan, H.; Brenton, J.D.; Snyder, A.; Weigelt, B.; Vargas, H.A. Unravelling tumour heterogeneity using next-generation imaging: Radiomics, radiogenomics, and habitat imaging. Clin. Radiol. 2017, 72, 3–10. [Google Scholar] [CrossRef]
- Zhong, G.X.; Feng, S.D.; Shen, R.; Wu, Z.Y.; Chen, F.; Zhu, X. The clinical significance of the ezrin gene and circulating tumor cells in osteosarcoma. Onco Targets Ther. 2017, 10, 527–533. [Google Scholar] [CrossRef]
- Yoon, H.J.; Sohn, I.; Cho, J.H.; Lee, H.Y.; Kim, J.H.; Choi, Y.L.; Kim, H.; Lee, G.; Lee, K.S.; Kim, J. Decoding tumor phenotypes for ALK, ROS1, and RET fusions in lung adenocarcinoma using a radiomics approach. Medicine 2015, 94, e1753. [Google Scholar] [CrossRef]
- Bianconi, F.; Palumbo, I.; Fravolini, M.L.; Chiari, R.; Minestrini, M.; Brunese, L.; Palumbo, B. Texture analysis on [18F]FDG PET/CT in non-small-cell lung cancer: Correlations between PET features, CT features, and histological types. Mol. Imaging Biol. 2019, 21, 1200–1209. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.B.; Harders, S.W.; Ganeshan, B.; Thygesen, J.; Torp Madsen, H.H.; Rasmussen, F. CT texture analysis can help differentiate between malignant and benign lymph nodes in the mediastinum in patients suspected for lung cancer. Acta Radiol. 2016, 57, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Nakajo, M.; Nakajo, M.; Nakayama, H.; Jinguji, M.; Nakabeppu, Y.; Higashi, M.; Nakamura, Y.; Sato, M.; Yoshiura, T. Dexamethasone suppression FDG PET/CT for differentiating between true- and false-positive pulmonary and mediastinal lymph node metastases in non-small cell lung cancer: A pilot study of FDG PET/CT after oral administration of dexamethasone. Radiology 2016, 279, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Ravanelli, M.; Agazzi, G.M.; Ganeshan, B.; Roca, E.; Tononcelli, E.; Bettoni, V.; Caprioli, A.; Borghesi, A.; Berruti, A.; Maroldi, R.; et al. CT texture analysis as predictive factor in metastatic lung adenocarcinoma treated with tyrosine kinase inhibitors (TKIs). Eur. J. Rad. 2018, 109, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Piñeiro-Fiel, M.; Moscoso, A.; Lado-Cacheiro, L.; Pombo-Pasín, M.; Rey-Bretal, D.; Gómez-Lado, N.; Mondelo-García, C.; Silva-Rodríguez, J.; Pubul, V.; Sánchez, M.; et al. Is FDG-PET texture analysis related to intratumor biological heterogeneity in lung cancer? Eur. Radiol. 2021, 31, 4156–4165. [Google Scholar] [CrossRef]
- Imai, M.A.; Oda, Y.; Oda, M.; Nakanishi, I.; Kawahara, E. Overexpression of E2F1 associated with LOH at RB locus and hyperphosphorylation of RB in non-small cell lung carcinoma. J. Cancer Res. Clin. Oncol. 2004, 130, 320–326. [Google Scholar] [CrossRef]
- Chicklore, S.; Goh, V.; Siddique, M.; Roy, A.; Marsden, P.K.; Cook, G.J. Quantifying tumour heterogeneity in 18F-FDG PET/CT imaging by texture analysis. Eur. J. Nucl. Med. Mol. Imaging 2013, 40, 133–140. [Google Scholar] [CrossRef]
- Valladares, A.; Beyer, T.; Rausch, I.J. Physical imaging phantoms for simulation of tumor heterogeneity in PET, CT, and MRI: An overview of existing designs. Med. Phys. 2020, 47, 2023–2037. [Google Scholar] [CrossRef]
- Grimm, L.J.; Mazurowski, M.A. Breast cancer radiogenomics: Current status and future directions. Acad. Radiol. 2020, 27, 39–46. [Google Scholar] [CrossRef]
- Fisher, R.; Pusztai, L.; Swanton, C. J Cancer heterogeneity: Implications for targeted therapeutics. Br. J. Cancer 2013, 108, 479–485. [Google Scholar] [CrossRef] [Green Version]
- Spillane, J.B.; Henderson, M.A. Cancer stem cells: A review. ANZ J. Surg. 2007, 77, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.W.; Prindle, M.J.; Loeb, L.A. Implications of genetic heterogeneity in cancer. Ann. N. Y. Acad. Sci. 2012, 1267, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Fedotov, S.; Iomin, A. Migration and proliferation dichotomy in tumor-cell invasion. Phys. Rev. Lett. 2007, 98, 118101. [Google Scholar] [CrossRef]
- Uramoto, H.; Tanaka, F. Prediction of recurrence after complete resection in patients with NSCLC. Anticancer Res. 2012, 32, 3953–3960. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Goto, T.; Hirotsu, Y.; Amemiya, K.; Mochizuki, H.; Omata, M. Understanding intratumor heterogeneity and evolution in nsclc and potential new therapeutic approach. Cancers 2018, 10, 212. [Google Scholar] [CrossRef]
- Kramer, G.M.; Frings, V.; Hoetjes, N.; Hoekstra, O.S.; Smit, E.F.; de Langen, A.J.; Boellaard, R.J. Repeatability of quantitative whole-body 18F-FDG PET/CT uptake measures as function of uptake interval and lesion selection in non-small cell lung cancer patients. J. Nucl. Med. 2016, 57, 1343–1349. [Google Scholar] [CrossRef]
- Gorgoulis, V.G.; Zacharatos, P.; Mariatos, G.; Kotsinas, A.; Bouda, M.; Kletsas, D.; Asimacopoulos, P.J.; Agnantis, N.; Kittas, C.; Papavassiliou, A.G. Transcription factor E2F-1 acts as a growth-promoting factor and is associated with adverse prognosis in non-small cell lung carcinomas. J. Pathol. 2020, 198, 142–156. [Google Scholar] [CrossRef]
- Pei, G.; Chen, L.; Zhang, W. WGCNA application to proteomic and metabolomic data analysis. Methods Enzymol. 2017, 585, 135–158. [Google Scholar]
- Langfelder, P.; Zhang, B.; Horvath, S.J.B. Defining clusters from a hierarchical cluster tree: The dynamic tree cut package for R. Bioinformatics 2008, 24, 719–720. [Google Scholar] [CrossRef]
- Nioche, C.; Orlhac, F.; Boughdad, S.; Reuzé, S.; Goya-Outi, J.; Robert, C.; Pellot-Barakat, C.; Soussan, M.; Frouin, F.; Buvat, I.J. LIFEx: A freeware for radiomic feature calculation in multimodality imaging to accelerate advances in the characterization of tumor heterogeneity. Cancer Res. 2018, 78, 4786–4789. [Google Scholar] [CrossRef] [PubMed]
- Byun, B.H.; Kong, C.B.; Park, J.; Seo, Y.; Lim, I.; Choi, C.W.; Cho, W.H.; Jeon, D.G.; Koh, J.S.; Lee, S.Y.; et al. Initial metabolic tumor volume measured by 18F-FDG PET/CT can predict the outcome of osteosarcoma of the extremities. J. Nucl. Med. 2013, 54, 1725–1732. [Google Scholar] [CrossRef] [Green Version]
- Sheen, H.; Kim, W.; Byun, B.H.; Kong, C.B.; Song, W.S.; Cho, W.H.; Lim, I.; Lim, S.M.; Woo, S.K. Metastasis risk prediction model in osteosarcoma using metabolic imaging phenotypes: A multivariable radiomics model. PLoS One 2019, 14, e0225242. [Google Scholar] [CrossRef] [PubMed]
- Way, G.P.; Allaway, R.J.; Bouley, S.J.; Fadul, C.E.; Sanchez, Y.; Greene, C.S. A machine learning classifier trained on cancer transcriptomes detects NF1 inactivation signal in glioblastoma. BMC Genom. 2017, 18, 127. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Deng, Q.; Liang, W.; Zou, X. An efficient feature selection strategy based on multiple support vector machine technology with gene expression data. BioMed Res. Int. 2018, 2018, 7538204. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Characteristic | Result | Rate |
|---|---|---|
| Average age | 69.28 | |
| Sex | ||
| Male | 44 | 83 |
| Female | 9 | 17 |
| Histology | ||
| Adenocarcinoma | 36 | 68 |
| Non-small cell lung cancer (not otherwise specified) | 2 | 4 |
| Squamous cell carcinoma | 15 | 28 |
| Smoking status | ||
| Current | 8 | 15 |
| Former | 37 | 70 |
| Non-smoker | 8 | 15 |
| EGFR mutation | ||
| Wild | 38 | 72 |
| Mutation | 6 | 11 |
| Unknown | 9 | 17 |
| KRAS mutation | ||
| Wild | 32 | 60 |
| Mutation | 12 | 23 |
| Unknown | 9 | 17 |
| T stage | ||
| T1a | 10 | 19 |
| T1b | 13 | 25 |
| T2a | 17 | 32 |
| T2b | 5 | 9 |
| T3 | 5 | 9 |
| T4 | 2 | 4 |
| Tis | 1 | 2 |
| N stage | ||
| N0 | 41 | 77 |
| N1 | 5 | 9 |
| N2 | 7 | 13 |
| Recurrence | ||
| No recurrence | 34 | 64 |
| Distant recurrence | 19 | 36 |
| Random Forest | Image Texture Features | Hub Genes | Correlation of Genes and Image Texture | Four Image Texture Features |
|---|---|---|---|---|
| Precision | 0.692 | 0.8 | 0.802 | 0.832 |
| Recall | 0.733 | 0.783 | 0.792 | 0.75 |
| AUC | 0.729 | 0.808 | 0.912 | 0.779 |
| Accuracy | 0.59 | 0.767 | 0.783 | 0.738 |
| Feature Family | Features |
|---|---|
| Intensity histogram | Maximum standard uptake value (SUVmax) |
| Mean standard uptake value (SUVmean) | |
| Standard deviation (SUV_SD) | |
| Total lesion glycolysis (TLG) | |
| Metabolic tumor volume (MTV) | |
| 1st entropy | |
| Gray-level co-occurrence matrix (GLCM) | Energy |
| Contrast | |
| Entropy | |
| Homogeneity | |
| Dissimilarity | |
| Neighboring gray-level dependence matrix (NGLDM) | Contrast |
| Coarseness | |
| Busyness | |
| Small number emphasis (SNE) | |
| Gray-level run length matrix (GLRLM) | Short run emphasis (SRE) |
| Long run emphasis (LRE) | |
| Gray-level non-uniformity (GLNU) | |
| Run length non-uniformity (RLNU) | |
| Low gray-level run emphasis (SRLGE) | |
| High gray-level run emphasis (SGHGE) | |
| Gray-level size zone matrix (GLSZM) | Small zone emphasis (SAE) |
| Large zone emphasis (LAE) | |
| Gray-level non-uniformity (GLN) | |
| Zone size non-uniformity (SZN) | |
| Low gray-level zone emphasis (LGLZE) | |
| High gray level zone emphasis (HGLZE) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ju, H.M.; Kim, B.-C.; Lim, I.; Byun, B.H.; Woo, S.-K. Estimation of an Image Biomarker for Distant Recurrence Prediction in NSCLC Using Proliferation-Related Genes. Int. J. Mol. Sci. 2023, 24, 2794. https://doi.org/10.3390/ijms24032794
Ju HM, Kim B-C, Lim I, Byun BH, Woo S-K. Estimation of an Image Biomarker for Distant Recurrence Prediction in NSCLC Using Proliferation-Related Genes. International Journal of Molecular Sciences. 2023; 24(3):2794. https://doi.org/10.3390/ijms24032794
Chicago/Turabian StyleJu, Hye Min, Byung-Chul Kim, Ilhan Lim, Byung Hyun Byun, and Sang-Keun Woo. 2023. "Estimation of an Image Biomarker for Distant Recurrence Prediction in NSCLC Using Proliferation-Related Genes" International Journal of Molecular Sciences 24, no. 3: 2794. https://doi.org/10.3390/ijms24032794
APA StyleJu, H. M., Kim, B. -C., Lim, I., Byun, B. H., & Woo, S. -K. (2023). Estimation of an Image Biomarker for Distant Recurrence Prediction in NSCLC Using Proliferation-Related Genes. International Journal of Molecular Sciences, 24(3), 2794. https://doi.org/10.3390/ijms24032794