
Potassium Dehydroandrograpolide Succinate Targets NRP1 Mediated VEGFR2/VE-Cadherin Signaling Pathway to Promote Endothelial Barrier Repair

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. PDA Promotes Partial Carotid Artery Ligation-Induced Neointima Formation
2.2. PDA Promotes Vascular Endothelial Cell Proliferation
2.3. PDA Promotes Vascular Endothelial Cells Migration
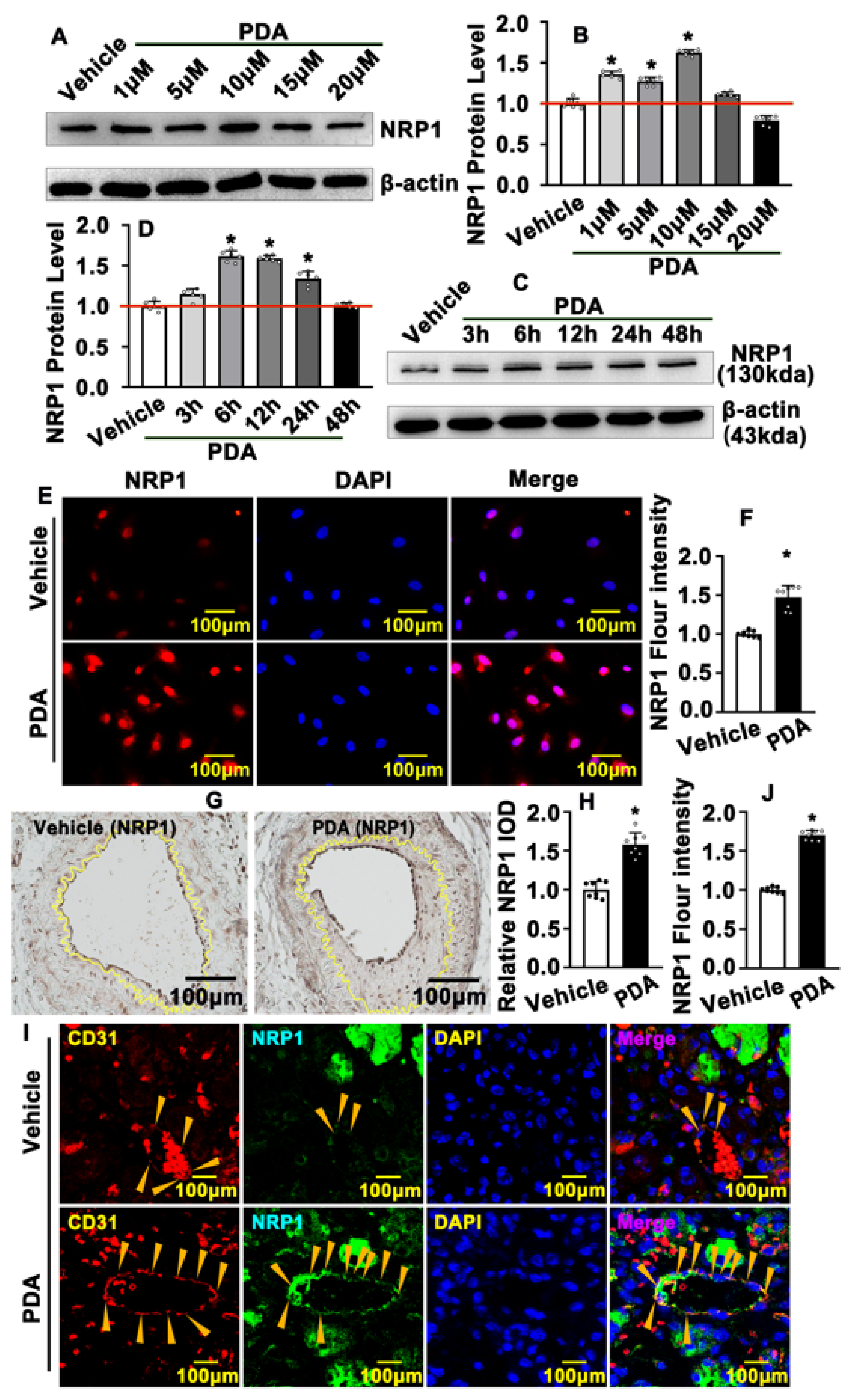
2.4. PDA Induces Vascular Endothelial Cell NRP1 Expression after Vascular Injury
2.5. PDA Activates VEGF Signaling Pathway after Vascular Injury
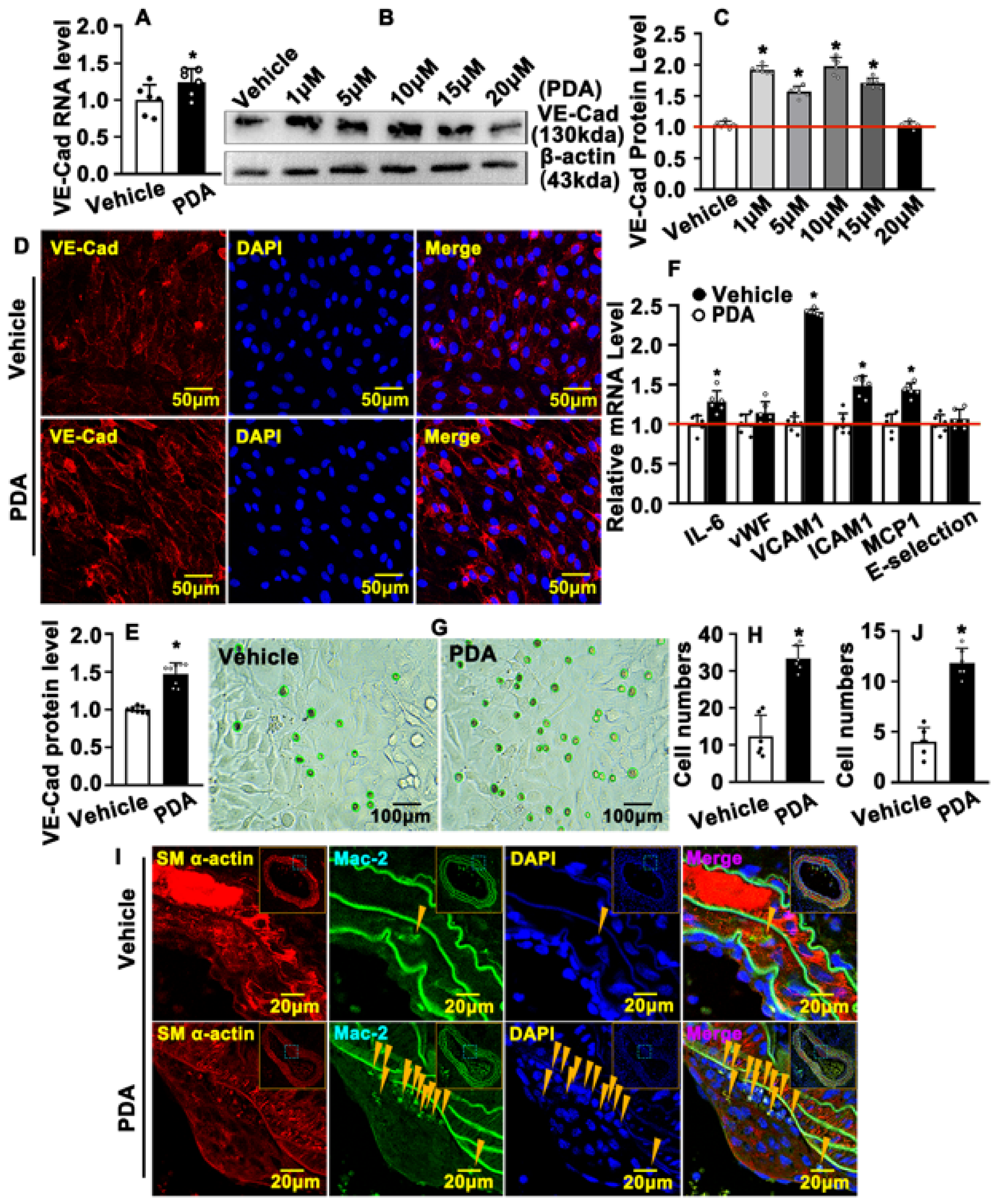
2.6. PDA Causes VE-Cad-Dependent Vascular Endothelial Barrier Repair
2.7. PDA Regulates the Interaction between NRP1, VEGFR and VE-Cad within Vascular Endothelial Cells
2.8. PDA Regulates Vascular Endothelial Barrier Function through the NRP1/VEGFR2/VE-Cad Signaling Pathway
3. Discussion
3.1. PDA Did Not Change HUVEC Viability
3.2. Diversity and Complexity of VE-Cad in Regulating Vascular Endothelial Barrier Function
3.3. Difference between Carotid Artery and Capillary Endothelial Cells
4. Materials and Methods
4.1. Ethics Statement regarding Animal Experiments
4.2. Partially Ligated Murine Carotid Artery model
4.3. Acute Skeletal Muscle Injury Model
4.4. Hematoxylin and Eosin (H&E) Staining, Immunohistochemistry Staining (IHC) and Immunofluorescence Staining (IF)
4.5. Cell Culture
4.6. HUVEC-C Cell Counting
4.7. CCK8 Cell Proliferation Assay
4.8. BRDU Incorporation Assay
4.9. Matrigel-Based Tube Formation Assay
4.10. Propidium Iodide (PI) Flow Cytometric Assay
4.11. Boyden Chamber Migration Assay
4.12. Spheroid Sprouting Assay
4.13. Quantitative Real-Time PCR Analysis
4.14. Protein Extraction and Western Blotting
4.15. Molecular Docking Simulation of PDA with NRP1, VE-Cad and VEGFR2
4.16. CO-Immunoprecipitation Assay
4.17. THP-1 Adhesion Assay
4.18. siRNA Transfection
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stevens, T.; Garcia, J.G.N.; Shasby, D.M.; Bhattacharya, J.; Malik, A.B. Mechanisms regulating endothelial cell barrier function. Am. J. Physiol. Cell. Mol. Physiol. 2000, 279, L419–L422. [Google Scholar] [CrossRef] [PubMed]
- Sluiter, T.; van Buul, J.; Huveneers, S.; Quax, P.; de Vries, M. Endothelial Barrier Function and Leukocyte Transmigration in Atherosclerosis. Biomedicines 2021, 9, 328. [Google Scholar] [CrossRef] [PubMed]
- Orsenigo, F.; Giampietro, C.; Ferrari, A.; Corada, M.; Galaup, A.; Sigismund, S.; Ristagno, G.; Maddaluno, L.; Koh, G.Y.; Franco, D.; et al. Phosphorylation of VE-cadherin is modulated by haemodynamic forces and contributes to the regulation of vascular permeability in vivo. Nat. Commun. 2012, 3, 1208. [Google Scholar] [CrossRef] [PubMed]
- Simmons, S.; Erfinanda, L.; Bartz, C.; Kuebler, W.M. Novel mechanisms regulating endothelial barrier function in the pulmonary microcirculation. J. Physiol. 2018, 597, 997–1021. [Google Scholar] [CrossRef] [PubMed]
- Mehta, L.; Ravindran, K.; Kuebler, W.M. Novel regulators of endothelial barrier function. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L924–L935. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Bolte, C.; Le, T.; Goda, C.; Xu, Y.; Kalin, T.V.; Kalinichenko, V.V. FOXF1 maintains endothelial barrier function and prevents edema after lung injury. Sci. Signal. 2016, 9, ra40. [Google Scholar] [CrossRef]
- Gimbrone, M.A., Jr.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef]
- Lawrie, A.; Francis, S.E. Frataxin and endothelial cell senescence in pulmonary hypertension. J. Clin. Investig. 2021, 131, e149721. [Google Scholar] [CrossRef]
- Bharadwaj, A.S.; Appukuttan, B.; Wilmarth, P.A.; Pan, Y.; Stempel, A.J.; Chipps, T.J.; Benedetti, E.E.; Zamora, D.O.; Choi, D.; David, L.L.; et al. Role of the retinal vascular endothelial cell in ocular disease. Prog. Retin. Eye Res. 2013, 32, 102–180. [Google Scholar] [CrossRef]
- Reymond, N.; d’Agua, B.B.; Ridley, A.J. Crossing the endothelial barrier during metastasis. Nat. Rev. Cancer 2013, 13, 858–870. [Google Scholar] [CrossRef]
- Esser, S.; Lampugnani, M.; Corada, M.; Dejana, E.; Risau, W. Vascular endothelial growth factor induces VE-cadherin tyrosine phosphorylation in endothelial cells. J. Cell Sci. 1998, 111, 1853–1865. [Google Scholar] [CrossRef] [PubMed]
- Leckband, D.; Sivasankar, S. Cadherin recognition and adhesion. Curr. Opin. Cell Biol. 2012, 24, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Grimsley-Myers, C.M.; Isaacson, R.H.; Cadwell, C.M.; Campos, J.; Hernandes, M.S.; Myers, K.R.; Seo, T.; Giang, W.; Griendling, K.K.; Kowalczyk, A.P. VE-cadherin endocytosis controls vascular integrity and patterning during development. J. Cell Biol. 2020, 219, e201909081. [Google Scholar] [CrossRef] [PubMed]
- Vestweber, D. VE-Cadherin: The major endothelial adhesion molecule controlling cellular junctions and blood vessel formation. Arter. Thromb. Vasc. Biol. 2008, 28, 223–232. [Google Scholar] [CrossRef]
- Lagendijk, A.K.; Hogan, B.M. VE-cadherin in Vascular Development: A coordinator of cell signaling and tissue morphogenesis. Curr. Top. Dev. Biol. 2015, 112, 325–352. [Google Scholar] [CrossRef]
- Corada, M.; Mariotti, M.; Thurston, G.; Smith, K.; Kunkel, R.; Brockhaus, M.; Lampugnani, M.G.; Martin-Padura, I.; Stoppacciaro, A.; Ruco, L.; et al. Vascular endothelial–cadherin is an important determinant of microvascular integrity in vivo. Proc. Natl. Acad. Sci. USA 1999, 96, 9815–9820. [Google Scholar] [CrossRef]
- Langer, V.; Vivi, E.; Regensburger, D.; Winkler, T.H.; Waldner, M.J.; Rath, T.; Schmid, B.; Skottke, L.; Lee, S.; Jeon, N.L.; et al. IFN-γ drives inflammatory bowel disease pathogenesis through VE-cadherin–directed vascular barrier disruption. J. Clin. Investig. 2019, 129, 4691–4707. [Google Scholar] [CrossRef]
- Mehta, V.; Fields, L.; Evans, I.M.; Yamaji, M.; Pellet-Many, C.; Jones, T.; Mahmoud, M.; Zachary, I. VEGF (Vascular Endothelial Growth Factor) Induces NRP1 (Neuropilin-1) Cleavage via ADAMs (a Disintegrin and Metalloproteinase) 9 and 10 to Generate Novel Carboxy-Terminal NRP1 Fragments That Regulate Angiogenic Signaling. Arter. Thromb. Vasc. Biol. 2018, 38, 1845–1858. [Google Scholar] [CrossRef]
- Fantin, A.; Herzog, B.; Mahmoud, M.; Yamaji, M.; Plein, A.; Denti, L.; Ruhrberg, C.; Zachary, I. Neuropilin 1 (NRP1) hypomorphism combined with defective VEGF-A binding reveals novel roles for NRP1 in developmental and pathological angiogenesis. Development 2014, 141, 556–562. [Google Scholar] [CrossRef]
- Pan, Q.; Chathery, Y.; Wu, Y.; Rathore, N.; Tong, R.K.; Peale, F.; Bagri, A.; Tessier-Lavigne, M.; Koch, A.W.; Watts, R.J. Neuropilin-1 Binds to VEGF121 and Regulates Endothelial Cell Migration and Sprouting. J. Biol. Chem. 2007, 282, 24049–24056. [Google Scholar] [CrossRef]
- Herzog, B.; Pellet-Many, C.; Britton, G.; Hartzoulakis, B.; Zachary, I.C. VEGF binding to NRP1 is essential for VEGF stimulation of endothelial cell migration, complex formation between NRP1 and VEGFR2, and signaling via FAK Tyr407 phosphorylation. Mol. Biol. Cell 2011, 22, 2766–2776. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, C.; Brash, J.T.; Fantin, A.; Ruhrberg, C. NRP1 function and targeting in neurovascular development and eye disease. Prog. Retin. Eye Res. 2016, 52, 64–83. [Google Scholar] [CrossRef] [PubMed]
- Lyu, Z.; Jin, H.; Yan, Z.; Hu, K.; Jiang, H.; Peng, H.; Zhuo, H. Effects of NRP1 on angiogenesis and vascular maturity in endothelial cells are dependent on the expression of SEMA4D. Int. J. Mol. Med. 2020, 46, 1321–1334. [Google Scholar] [CrossRef] [PubMed]
- Fantin, A.; Vieira, J.M.; Plein, A.; Denti, L.; Fruttiger, M.; Pollard, J.W.; Ruhrberg, C. NRP1 acts cell autonomously in endothelium to promote tip cell function during sprouting angiogenesis. Blood 2013, 121, 2352–2362. [Google Scholar] [CrossRef] [PubMed]
- Kohno, T.; Ishii, K.; Hirota, Y.; Honda, T.; Makino, M.; Kawasaki, T.; Nakajima, K.; Hattori, M. Reelin-Nrp1 Interaction Regulates Neocortical Dendrite Development in a Context-Specific Manner. J. Neurosci. 2020, 40, 8248–8261. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Wu, Z.; Xu, Y.; Xu, H.; Lou, G.; Jiang, Q.; Fan, W.; Liu, W.; Zheng, C.; Gao, Y.; et al. Andrographolide attenuates imbalance of gastric vascular homeostasis induced by ethanol through glycolysis pathway. Sci. Rep. 2019, 9, 4968. [Google Scholar] [CrossRef]
- Hu, W.; Wu, X.; Jin, Z.; Wang, Z.; Guo, Q.; Chen, Z.; Zhu, S.; Zhang, H.; Huo, J.; Zhang, L.; et al. Andrographolide Promotes Interaction Between Endothelin-Dependent EDNRA/EDNRB and Myocardin-SRF to Regulate Pathological Vascular Remodeling. Front. Cardiovasc. Med. 2022, 8, 783872. [Google Scholar] [CrossRef]
- Su, L.; Gao, Y.; Zhang, M.; Liu, Z.; Lin, Q.; Gong, L.; Guo, J.; Chen, L.; An, T.; Chen, J. Andrographolide and Its Derivative Potassium Dehydrographolide Succinate Suppress PRRSV Replication in Primary and Established Cells via Differential Mechanisms of Action. Virol. Sin. 2021, 36, 1626–1643. [Google Scholar] [CrossRef]
- Nam, D.; Ni, C.-W.; Rezvan, A.; Suo, J.; Budzyn, K.; Llanos, A.; Harrison, D.; Giddens, D.; Jo, H. Partial carotid ligation is a model of acutely induced disturbed flow, leading to rapid endothelial dysfunction and atherosclerosis. Am. J. Physiol. Circ. Physiol. 2009, 297, H1535–H1543. [Google Scholar] [CrossRef]
- Bi, J.; Zhang, J.; Ren, Y.; Du, Z.; Zhang, Y.; Liu, C.; Wang, Y.; Zhang, L.; Shi, Z.; Wu, Z.; et al. Exercise hormone irisin mitigates endothelial barrier dysfunction and microvascular leakage–related diseases. JCI Insight 2020, 5, e136277. [Google Scholar] [CrossRef]
- Guardiola, O.; Andolfi, G.; Tirone, M.; Iavarone, F.; Brunelli, S.; Minchiotti, G. Induction of Acute Skeletal Muscle Regeneration by Cardiotoxin Injection. J. Vis. Exp. 2017, 119, e54515. [Google Scholar] [CrossRef]
- Yamada, K.M.; Sixt, M. Mechanisms of 3D cell migration. Nat. Rev. Mol. Cell Biol. 2019, 20, 738–752. [Google Scholar] [CrossRef] [PubMed]
- Konukoglu, D.; Uzun, H. Endothelial Dysfunction and Hypertension. Adv. Exp. Med. Biol. 2017, 956, 511–540. [Google Scholar] [CrossRef] [PubMed]
- Bergé, M.; Allanic, D.; Bonnin, P.; de Montrion, C.; Richard, J.; Suc, M.; Boivin, J.-F.; Contrerès, J.-O.; Lockhart, B.P.; Pocard, M.; et al. Neuropilin-1 is upregulated in hepatocellular carcinoma and contributes to tumour growth and vascular remodelling. J. Hepatol. 2011, 55, 866–875. [Google Scholar] [CrossRef]
- Domingues, A.; Fantin, A. Neuropilin 1 Regulation of Vascular Permeability Signaling. Biomolecules 2021, 11, 666. [Google Scholar] [CrossRef]
- Fu, P.; Ramchandran, R.; Shaaya, M.; Huang, L.; Ebenezer, D.L.; Jiang, Y.; Komarova, Y.; Vogel, S.M.; Malik, A.B.; Minshall, R.D.; et al. Phospholipase D2 restores endothelial barrier function by promoting PTPN14-mediated VE-cadherin dephosphorylation. J. Biol. Chem. 2020, 295, 7669–7685. [Google Scholar] [CrossRef]
- Zhong, W.; Yang, W.; Qin, Y.; Gu, W.; Xue, Y.; Tang, Y.; Xu, H.; Wang, H.; Zhang, C.; Wang, C.; et al. 6-Gingerol stabilized the p-VEGFR2/VE-cadherin/β-catenin/actin complex promotes microvessel normalization and suppresses tumor progression. J. Exp. Clin. Cancer Res. 2019, 38, 285. [Google Scholar] [CrossRef]
- Flemming, S.; Burkard, N.; Renschler, M.; Vielmuth, F.; Meir, M.; Schick, M.A.; Wunder, C.; Germer, C.-T.; Spindler, V.; Waschke, J.; et al. Soluble VE-cadherin is involved in endothelial barrier breakdown in systemic inflammation and sepsis. Cardiovasc. Res. 2015, 107, 32–44. [Google Scholar] [CrossRef]
- Ševc, J.; Matiašová, A.; Smoleková, I.; Jendželovský, R.; Mikeš, J.; Tomášová, L.; Kútna, V.; Daxnerová, Z.; Fedoročko, P. Peroral administration of 5-bromo-2-deoxyuridine in drinking water is not a reliable method for labeling proliferating S-phase cells in rats. J. Pharmacol. Toxicol. Methods 2015, 74, 33–39. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z.; Wu, X.; Li, J.; Guo, Q.; Jin, Z.; Li, H.; Liang, B.; Hu, W.; Xu, H.; Shi, L.; et al. Potassium Dehydroandrograpolide Succinate Targets NRP1 Mediated VEGFR2/VE-Cadherin Signaling Pathway to Promote Endothelial Barrier Repair. Int. J. Mol. Sci. 2023, 24, 3096. https://doi.org/10.3390/ijms24043096
Wang Z, Wu X, Li J, Guo Q, Jin Z, Li H, Liang B, Hu W, Xu H, Shi L, et al. Potassium Dehydroandrograpolide Succinate Targets NRP1 Mediated VEGFR2/VE-Cadherin Signaling Pathway to Promote Endothelial Barrier Repair. International Journal of Molecular Sciences. 2023; 24(4):3096. https://doi.org/10.3390/ijms24043096
Chicago/Turabian StyleWang, Zheng, Xiao Wu, Jiali Li, Qiru Guo, Zhong Jin, Hongfei Li, Bing Liang, Wangming Hu, Huan Xu, Liangqin Shi, and et al. 2023. "Potassium Dehydroandrograpolide Succinate Targets NRP1 Mediated VEGFR2/VE-Cadherin Signaling Pathway to Promote Endothelial Barrier Repair" International Journal of Molecular Sciences 24, no. 4: 3096. https://doi.org/10.3390/ijms24043096
APA StyleWang, Z., Wu, X., Li, J., Guo, Q., Jin, Z., Li, H., Liang, B., Hu, W., Xu, H., Shi, L., Yang, L., & Wang, Y. (2023). Potassium Dehydroandrograpolide Succinate Targets NRP1 Mediated VEGFR2/VE-Cadherin Signaling Pathway to Promote Endothelial Barrier Repair. International Journal of Molecular Sciences, 24(4), 3096. https://doi.org/10.3390/ijms24043096