

Alcohol, Inflammation, and Microbiota in Alcoholic Liver Disease


 ,
, 
 , ,
, ,  , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
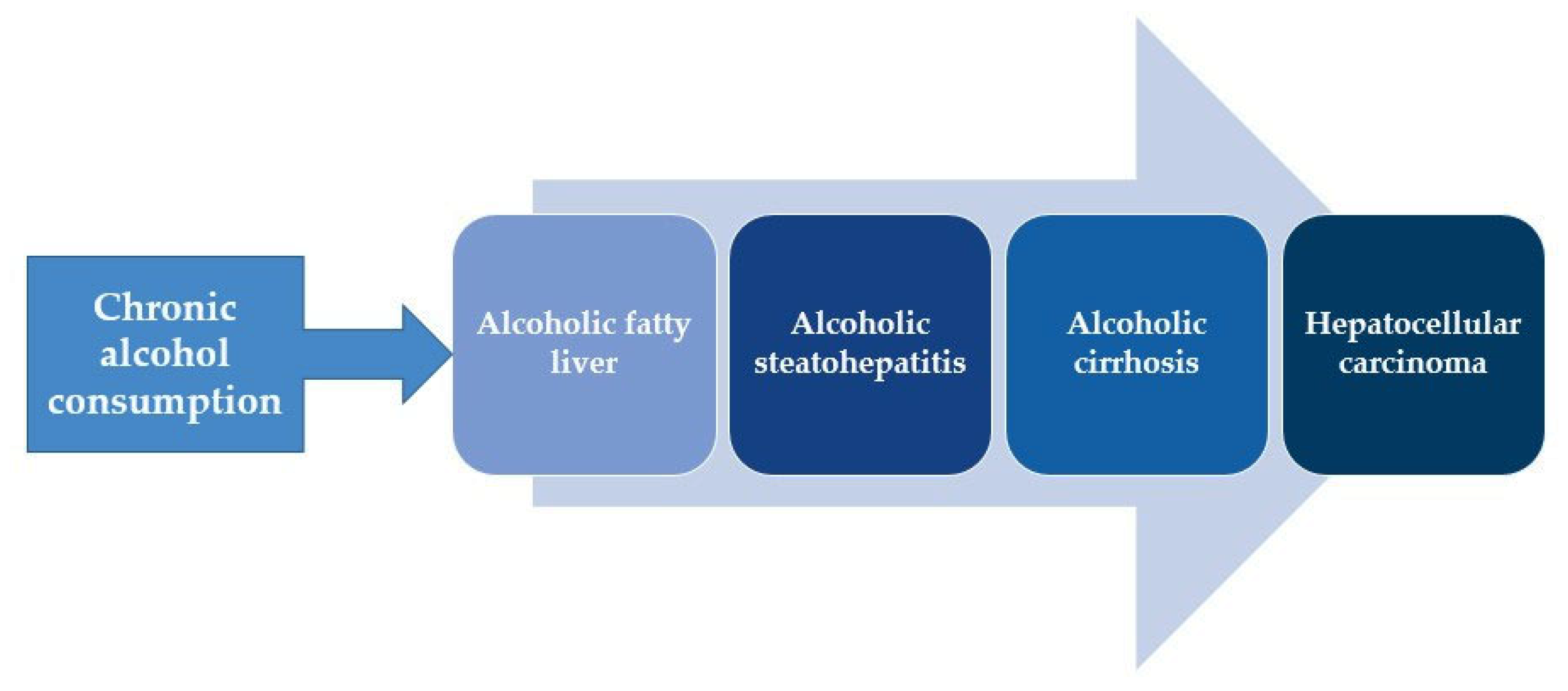
:1. Introduction
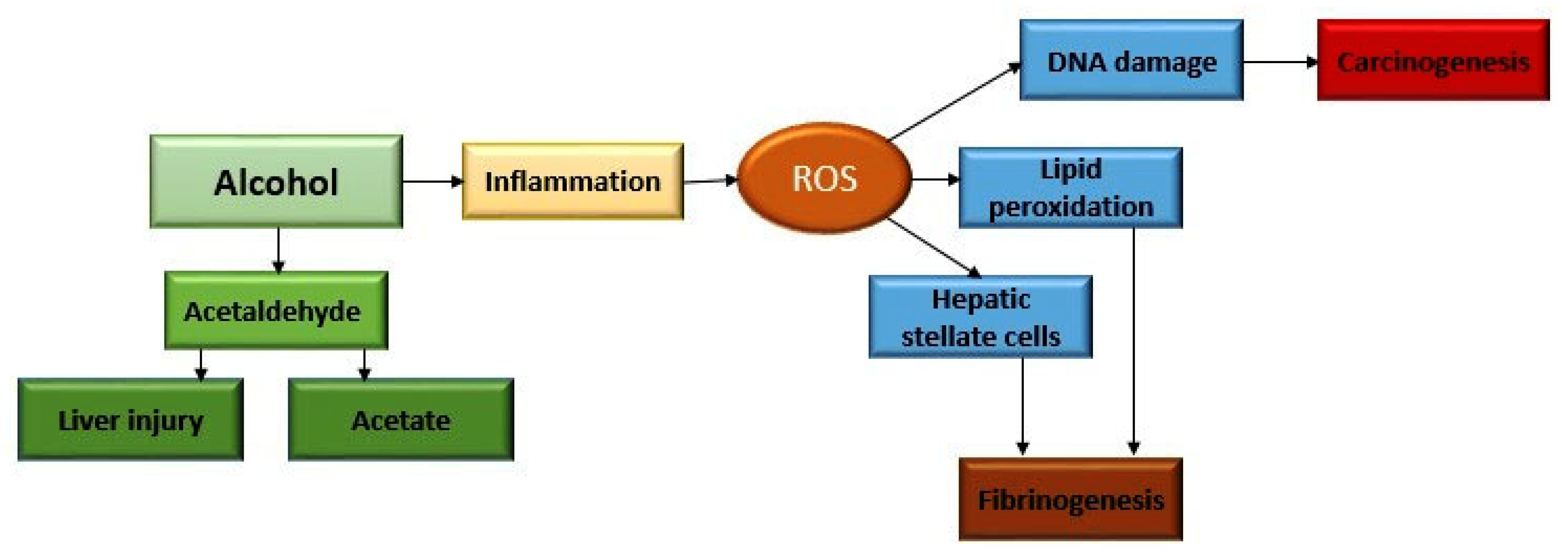
2. Alcohol Metabolism
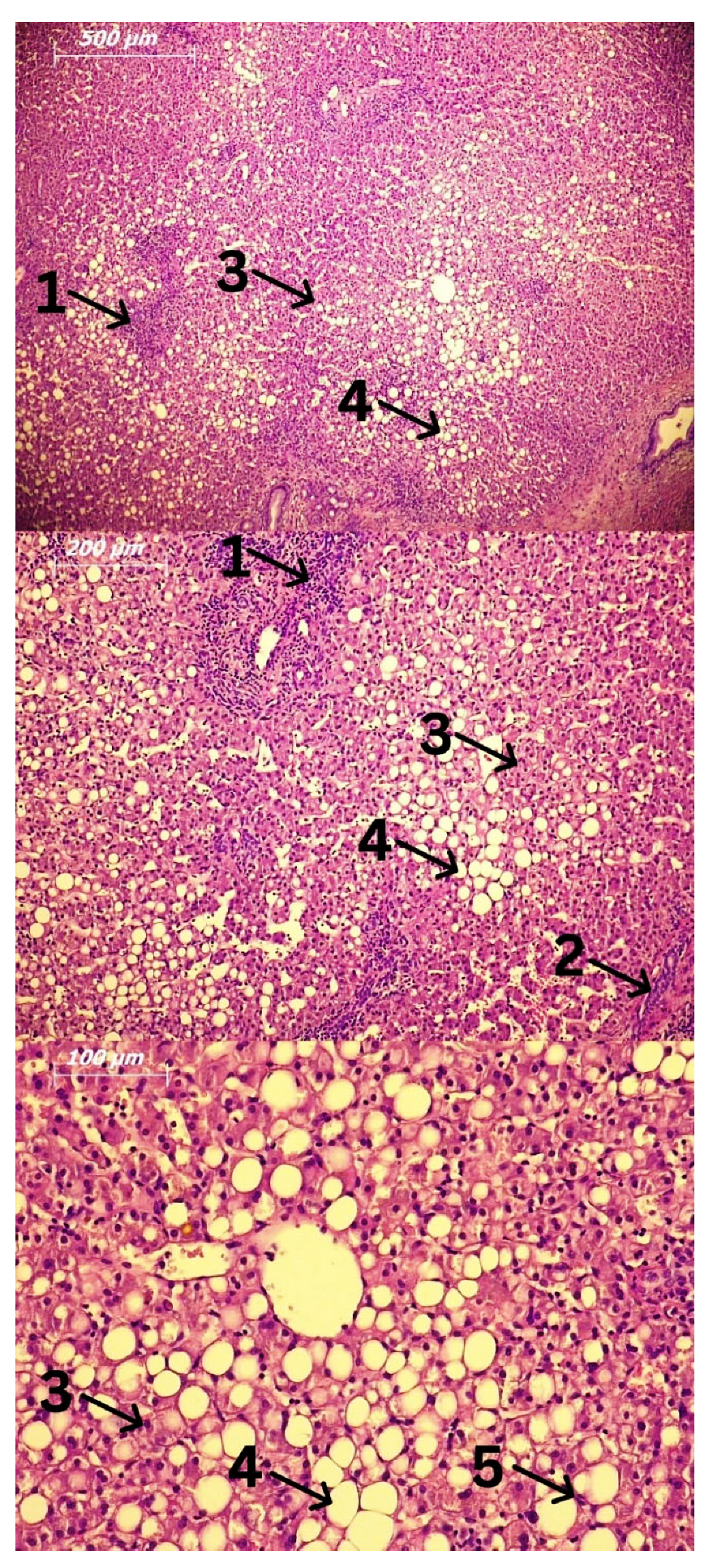
3. Cells Involved in the Pathogenesis of Alcoholic Liver Disease (ALD) and Alcoholic Steatohepatitis (ASH)
3.1. Kupffer and Circulating Monocytes
3.2. Neutrophils
3.3. T Lymphocytes
3.4. Hepatocytes and Hepatic Stellate Cells
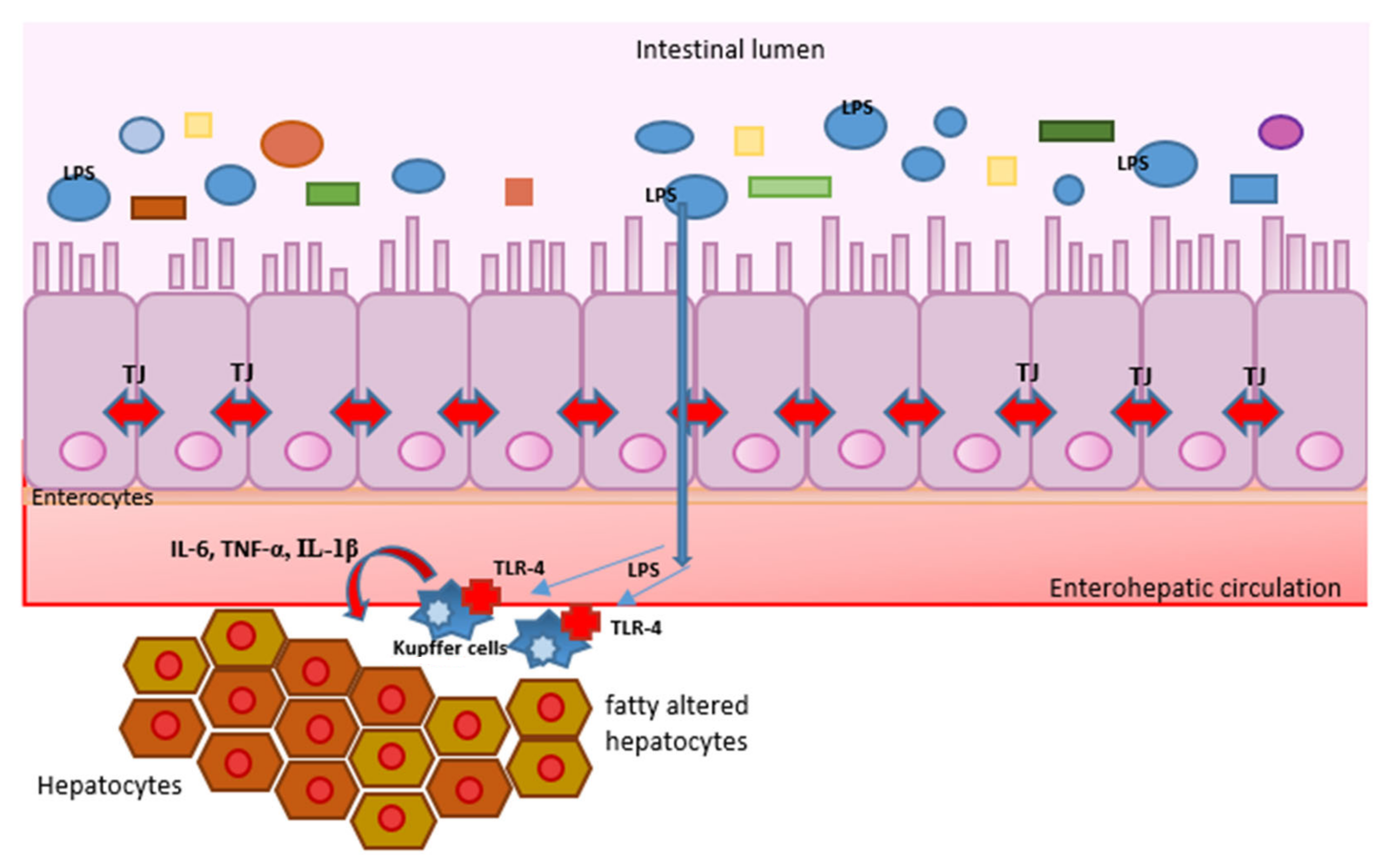
4. Pathogen-Associated Molecular Patterns (PAMPs), Damage-Associated Molecular Patterns (DAMPs), Toll-like Receptors (TLRs), and Inflammation
5. What Is the Role of Intestinal Microbiota in the Mentioned Processes?
6. Therapeutic Interventions in Alcoholic Liver Disease (ALD) Involving the Microbiota
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Meroni, M.; Longo, M.; Dongiovanni, P. Alcohol or Gut Microbiota: Who Is the Guilty? Int. J. Mol. Sci. 2019, 20, 4568. [Google Scholar] [CrossRef] [PubMed]
- Drill, V.A. Hepatotoxic agents; mechanism of action and dietary interrelationship. Pharmacol. Rev. 1952, 4, 1–42. [Google Scholar] [PubMed]
- Neuman, M.G.; Seitz, H.K.; French, S.W.; Malnick, S.; Tsukamoto, H.; Cohen, L.B.; Hoffman, P.; Tabakoff, B.; Fasullo, M.; Nagy, L.; et al. Alcoholic-Hepatitis, Links to Brain and Microbiome: Mechanisms, Clinical and Experimental Research. Biomedicines 2020, 8, 63. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Bataller, R.; Cortez-Pinto, H.; Gao, B.; Gual, A.; Lackner, C.; Mathurin, P.; Mueller, S.; Szabo, G.; Tsukamoto, H. Alcoholic liver disease. Nat. Rev. Dis. Primers 2018, 4, 16. [Google Scholar] [CrossRef]
- Rehm, J.; Taylor, B.; Mohapatra, S.; Irving, H.; Baliunas, D.; Patra, J.; Roerecke, M. Alcohol as a risk factor for liver cirrhosis: A systematic review and meta-analysis. Drug Alcohol Rev. 2010, 29, 437–445. [Google Scholar] [CrossRef]
- Brar, G.; Tsukamoto, H. Alcoholic and non-alcoholic steatohepatitis: Global perspective and emerging science. J. Gastroenterol. 2019, 54, 218–225. [Google Scholar] [CrossRef]
- World Health Organisation. Global Status Report on Alcohol and Health 2014; World Health Organisation: Geneva, Switzerland, 2014. [Google Scholar]
- Shield, K.D.; Rylett, M.; Rehm, J. Public Health Successes and Missed Opportunities: Trends in Alcohol Consumption and Attributable Mortality in the WHO European Region, 1990–2014; World Health Organization: Geneva, Switzerland, 2016. [Google Scholar]
- Becker, U.; Deis, A.; Sørensen, T.I.; Grønbaek, M.; Borch-Johnsen, K.; Müller, C.F.; Schnohr, P.; Jensen, G. Prediction of risk of liver disease by alcohol intake, sex, and age: A prospective population study. Hepatology 1996, 23, 1025–1029. [Google Scholar] [CrossRef]
- Shimizu, I.; Kamochi, M.; Yoshikawa, H.; Nakayama, Y. Trends in Alcoholic Liver Disease Research: Clinical and Scientific Aspects; Shimizu, I., Ed.; InTech: Rijeka, Croatia, 2012; pp. 23–40. [Google Scholar]
- Erol, A.; Karpyak, V.M. Sex and gender-related differences in alcohol use and its consequences: Contemporary knowledge and future research considerations. Drug Alcohol Depend. 2015, 156, 1–13. [Google Scholar] [CrossRef]
- Colantoni, A.; Emanuele, M.A.; Kovacs, E.J.; Villa, E.; Van Thiel, D.H. Hepatic estrogen receptors and alcohol intake. Mol. Cell. Endocrinol. 2002, 193, 101–104. [Google Scholar] [CrossRef]
- Améen, C.; Oscarsson, J. Sex difference in hepatic microsomal triglyceride transfer protein expression is determined by the growth hormone secretory pattern in the rat. Endocrinology 2003, 144, 3914–3921. [Google Scholar] [CrossRef] [Green Version]
- Naveau, S.; Giraud, V.; Borotto, E.; Aubert, A.; Capron, F.; Chaput, J.C. Excess weight is a risk factor for alcoholic liver disease. Hepatology 1997, 25, 108–111. [Google Scholar] [CrossRef]
- Strnad, P.; Buch, S.; Hamesch, K.; Fischer, J.; Rosendahl, J.; Schmelz, R.; Brueckner, S.; Brosch, M.; Heimes, C.V.; Woditsch, V.; et al. Heterozygous carriage of the alpha1-antitrypsin Z variant rs28929474 predisposes to the development of cirrhosis in the presence of alcohol misuse and non-alcohol-related fatty liver disease. J. Hepatol. 2017, 66, S177. [Google Scholar] [CrossRef]
- Chen, C.J.; Liang, K.Y.; Chang, A.S.; Chang, Y.C.; Lu, S.N.; Liaw, Y.F.; Chang, W.Y.; Sheen, M.C.; Lin, T.M. Effects of hepatitis B virus, alcohol drinking, cigarette smoking and familial tendency on hepatocellular carcinoma. Hepatology 1991, 13, 398–406. [Google Scholar] [CrossRef]
- Fletcher, L.M.; Powel, L.W. Hemochromatosis and alcoholic liver disease. Alcohol 2003, 30, 131–136. [Google Scholar] [CrossRef]
- Mueller, S.; Millonig, G.; Seitz, H.K. Alcoholic liver disease and hepatitis C: A frequently underestimated combination. World J. Gastroenterol. 2009, 15, 3462–3471. [Google Scholar] [CrossRef]
- Teli, M.; James, O.; Burt, A.; Bennett, M.; Day, C. The natural history of nonalcoholic fatty liver: A follow-up study. Hepatology 1995, 22, 1714–1719. [Google Scholar] [CrossRef]
- Stickel, F. Alcoholic cirrhosis and hepatocellular carcinoma. Adv. Exp. Med. Biol. 2015, 815, 113–130. [Google Scholar] [CrossRef]
- Fattovich, G.; Stroffolini, T.; Zagni, I.; Donato, F. Hepatocellular carcinoma in cirrhosis: Incidence and risk factors. Gastroenterology 2004, 127, S35–S50. [Google Scholar] [CrossRef]
- Ganne-Carrie, N.; Chaffaut, C.; Bourcier, V.; Archambeaud, I.; Perarnau, J.M.; Oberti, F.; Roulot, D.; Moreno, C.; Louvet, A.; Dao, T.; et al. Estimate of hepatocellular carcinoma incidence in patients with alcoholic cirrhosis. J. Hepatol. 2018, 69, 1274–1283. [Google Scholar] [CrossRef]
- Teschke, R. Alcoholic steatohepatitis (ASH) and alcoholic hepatitis (AH): Cascade of events, clinical aspects, and pharmacotherapy options. Expert Opin. Pharmacother. 2018, 19, 779–793. [Google Scholar] [CrossRef]
- Lieber, C.S. Metabolic effects of acetaldehyde. Biochem. Soc. Trans. 1988, 16, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S.; Rubin, E.; DeCarli, L.M. Hepatic microsomal ethanol oxidizing system (MEOS): Differentiation from alcohol dehydrogenase and NADPH oxidase. Biochem. Biophys. Res. Commun. 1970, 40, 858–865. [Google Scholar] [CrossRef] [PubMed]
- Gouillon, Z.; Lucas, D.; Li, J.; Hagbjork, A.L.; French, B.A.; Fu, P.; Fang, C.; Ingelman-Sundberg, M.; Donohue, T.M., Jr.; French, S.W. Inhibition of ethanol-induced liver disease in the intragastric feeding rat model by chlormethiazole. Proc. Soc. Exp. Biol. Med. 2000, 224, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Lian, F.; Chavez, P.R.G.; Chung, J.; Ling, W.; Qin, H.; Seitz, H.K.; Wang, X. Cytochrome P450 2E1 inhibition prevents hepatic carcinogenesis induced by diethylnitrosamine in alcohol-fed rats. Hepatobiliary Surg. Nutr. 2012, 1, 5–18. [Google Scholar] [CrossRef]
- Lieber, C.; DeCarli, L.M. Ethanol oxidation by hepatic microsomes: Adaptive increase after ethanol feeding. Science 1968, 162, 917–918. [Google Scholar] [CrossRef]
- Seitz, H.K.; Mueller, S. Alcoholic liver disease. Clin. Hepatol. 2010, 2, 1111–1151. [Google Scholar]
- Neuman, M. Cytokines- Central Factors in Alcoholic Liver. Alcohol Res. Health 2003, 27, 307–316. [Google Scholar]
- Lieber, C.S.; DeCarli, L.M. Hepatotoxicity of ethanol. J. Hepatol. 1991, 12, 394–401. [Google Scholar] [CrossRef]
- Wang, M.; You, Q.; Lor, K.; Chen, F.; Gao, B.; Ju, C. Chronic alcohol ingestion modulates hepatic macrophage populations and functions in mice. J. Leukoc. Biol. 2014, 96, 657–665. [Google Scholar] [CrossRef]
- Nagy, L.E. The role of innate immunity in alcoholic liver disease. Alcohol Res. 2015, 37, 237–250. [Google Scholar]
- Wang, M.; Frasch, S.C.; Li, G.; Feng, D.; Gao, B.; Xu, L.; Ir, D.; Frank, D.N.; Bratton, D.L.; Ju, C. Role of gp91(phox) in hepatic macrophage programming and alcoholic liver disease. Hepatol. Commun. 2017, 1, 765–779. [Google Scholar] [CrossRef]
- Lazaro, R.; Wu, R.; Lee, S.; Zhu, N.L.; Chen, C.L.; French, S.W.; Xu, J.; Machida, K.; Tsukamoto, H. Osteopontin deficiency does not prevent but promotes alcoholic neutrophilic hepatitis in mice. Hepatology 2015, 61, 129–140. [Google Scholar] [CrossRef]
- Altamirano, J.; Miquel, R.; Katoonizadeh, A.; Abraldes, J.G.; Duarte-Rojo, A.; Louvet, A.; Augustin, S.; Mookerjee, R.P.; Michelena, J.; Smyrk, T.C. A histologic scoring system for prognosis of patients with alcoholic hepatitis. Gastroenterology 2014, 146, e1231–e1236. [Google Scholar] [CrossRef]
- Rajkovic, I.A.; Williams, R. Abnormalities of neutrophil phagocytosis, intracellular killing and metabolic activity in alcoholic cirrhosis and hepatitis. Hepatology 1986, 6, 252–262. [Google Scholar] [CrossRef]
- Singh, V.; Sharma, A.K.; Narasimhan, R.L.; Bhalla, A.; Sharma, N.; Sharma, R. Granulocyte colony-stimulating factor in severe alcoholic hepatitis: A randomized pilot study. Am. J. Gastroenterol. 2014, 109, 1417–1423. [Google Scholar] [CrossRef]
- Chedid, A.; Mendenhall, C.L.; Moritz, T.E.; French, S.W.; Chen, T.S.; Morgan, T.R.; Roselle, G.A.; Nemchausky, B.A.; Tamburro, C.H.; Schiff, E.R.; et al. Cell-mediated hepatic injury in alcoholic liver disease. Veterans Affairs Cooperative Study Group 275. Gastroenterology 1993, 105, 254–266. [Google Scholar] [CrossRef]
- Liaskou, E.; Klemsdal Henriksen, E.K.; Holm, K.; Kaveh, F.; Hamm, D.; Fear, J.; Viken, M.K.; Hov, J.R.; Melum, E.; Robins, H.; et al. High-throughput T-cell receptor sequencing across chronic liver diseases reveals distinct disease-associated repertoires. Hepatology 2016, 63, 1608–1619. [Google Scholar] [CrossRef]
- Sutti, S.; Bruzzi, S.; Albano, E. The role of immune mechanisms in alcoholic and nonalcoholic steatohepatitis: A 2015 update. Expert Rev. Gastroenterol. Hepatol. 2016, 10, 243–253. [Google Scholar] [CrossRef]
- Barnes, M.A.; McMullen, M.R.; Roychowdhury, S.; Pisano, S.G.; Liu, X.; Stavitsky, A.B.; Bucala, R.; Nagy, L.E. Macrophage migration inhibitory factor contributes to ethanol-induced liver injury by mediating cell injury, steatohepatitis, and steatosis. Hepatology 2013, 57, 1980–1991. [Google Scholar] [CrossRef]
- Marin, V.; Poulsen, K.; Odena, G.; McMullen, M.R.; Altamirano, J.; Sancho-Bru, P.; Tiribelli, C.; Caballeria, J.; Rosso, N.; Bataller, R.; et al. Hepatocyte-derived macrophage migration inhibitory factor mediates alcohol-induced liver injury in mice and patients. J. Hepatol. 2017, 67, 1018–1025. [Google Scholar] [CrossRef]
- Li, W.; Amet, T.; Xing, Y.; Yang, D.; Liangpunsakul, S.; Puri, P.; Kamath, P.S.; Sanyal, A.J.; Shah, V.H.; Katz, B.P.; et al. Alcohol abstinence ameliorates the dysregulated immune profiles in patients with alcoholic hepatitis: A prospective observational study. Hepatology 2017, 66, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Masuhima, K.; Morishita, K.; Yoshimura, T.; Lavu, S.; Kobayashi, Y.; Lew, W.; Appella, E.; Kung, H.F.; Leonard, E.J.; Oppenheim, J.J. Molecular cloning of a human monocyte-derived neutrophil chemotactic factor (MONCF) and the induction of MDNCF mRNA by interleukin 1 and tumor necrosis factor. J. Exp. Med. 1988, 167, 1883–1893. [Google Scholar] [CrossRef]
- Feniger-Barish, R.; Belkin, D.; Zaslaver, A.; Gal, S.; Dori, M.; Ran, M.; Ben-Baruch, A. GCP-2-induced internalization of IL-8 receptors: Hierarohical relationships between GCP-2 and other ELR (+)-CXC chemokines and mechanisms regulating CXCR2, internalization and recycling. Blood 2002, 95, 1551. [Google Scholar] [CrossRef]
- Xu, R.; Huang, H.; Zhang, Z.; Wang, F.S. The role of neutrophils in the development of liver disease. Cell. Mol. Immunol. 2014, 11, 224–231. [Google Scholar] [CrossRef]
- Wieser, V.; Adolph, T.E.; Enrich, B.; Kuliopulos, A.; Kaser, A.; Tilg, H. Reversal of murine alcoholic steatohepatitis by pepducin-based functional blockade of interlink-8 receptors. Gut 2017, 66, 930–938. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Radaeva, S.; Sun, R.; Jaruga, B.; Nguyen, V.T.; Tian, Z.; Gao, B. Natural killer cells ameliorate liver fibrosis by killing activated stellate cells in NKG2D- dependent and tumor necrosis factor-related apoptosis-inducing ligand-dependent manners. Gastroenterology 2006, 130, 435–452. [Google Scholar] [CrossRef]
- Jeong, W.I.; Park, O.; Gao, B. Abrogation of the antifibrotic effects of natural killer cells/interferon-γ contributes to alcohol acceleration of liver fibrosis. Gastroenterology 2008, 134, 248–258. [Google Scholar] [CrossRef]
- Gao, B.; Ahmad, M.F.; Nagy, L.E.; Tsukamoto, H. Inflammatory pathways in alcoholic steatohepatitis. J. Hepatol. 2019, 70, 249–259. [Google Scholar] [CrossRef]
- Glaser, T.; Baiocchi, L.; Zhou, T.; Francis, H.; Lenci, I.; Grassi, G.; Kennedy, L.; Liangpunsakul, S.; Glaser, S.; Alpini, G.; et al. Pro-inflammatory signalling and gut-liver axis in non-alcoholic and alcoholic steatohepatitis: Differences and similarities along the path. J. Cell. Mol. Med. 2020, 24, 5955–5965. [Google Scholar] [CrossRef]
- Sharifnia, T.; Antoun, J.; Verriere, T.G.; Suarez, G.; Wattacheril, J.; Wilson, K.T.; Peek, R.M., Jr.; Abumrad, N.N.; Flynn, C.R. Hepatic TLR4 signaling in obese NAFLD. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G270–G278. [Google Scholar] [CrossRef]
- Schwabe, R.F.; Seki, E.; Brenner, D.A. Toll-like receptor signaling in the liver. Gastroenterology 2006, 130, 1886–1900. [Google Scholar] [CrossRef]
- Pone, E.J. Analysis by Flow Cytometry of B-Cell Activation and Antibody Responses Induced by Toll-like Receptors. Methods Mol. Biol. 2016, 1390, 229–248. [Google Scholar] [CrossRef]
- Thurman, R.G., II. Alcoholic liver injury involves activation of Kupffer cells by endotoxin. Am. J. Physiol. 1998, 275, G605–G611. [Google Scholar] [CrossRef]
- Wang, S.; Pacher, P.; De Lisle, R.C.; Huang, H.; Ding, W.X.A. Mechanistic review of cell death in alcohol-induced liver injury. Alcohol. Clin. Exp. Res. 2016, 40, 1215–1223. [Google Scholar] [CrossRef]
- Roh, Y.S.; Zhang, B.; Loomba, R.; Seki, E. TLR2 and TLR9 contribute to alcohol-mediated liver injury through induction of CXCL1 and neutrophil infiltration. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G30–G41. [Google Scholar] [CrossRef]
- Petrasek, J.; Bala, S.; Csak, T.; Lippai, D.; Kodys, K.; Menashy, V.; Barrieau, M.; Min, S.Y.; Kurt-Jones, E.A.; Szabo, G. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J. Clin. Investig. 2012, 122, 3476–3489. [Google Scholar] [CrossRef]
- Iracheta-Vellve, A.; Petrasek, J.; Satishchandran, A.; Gyongyosi, B.; Saha, B.; Kodys, K.; Fitzgerald, K.A.; Kurt-Jones, E.A.; Szabo, G. Inhibition of sterile danger signals, uric acid and ATP, prevents inflammasome activation and protects from alcoholic steatohepatitis in mice. J. Hepatol. 2015, 63, 1147–1155. [Google Scholar] [CrossRef]
- Petrasek, J.; Iracheta-Vellve, A.; Saha, B.; Satishchandran, A.; Kodys, K.; Fitzgerald, K.A.; Kurt-Jones, E.A.; Szabo, G. Metabolic danger signals, uric acid and ATP, mediate inflammatory cross-talk between hepatocytes and immune cells in alcoholic liver disease. J. Leukoc. Biol. 2015, 98, 249–256. [Google Scholar] [CrossRef]
- Iracheta-Vellve, A.; Petrasek, J.; Gyogyosi, B.; Bala, S.; Csak, T.; Kodys, K.; Szabo, G. Interleukin-1 inhibition facilitates recovery from liver injury and promotes regeneration of hepatocytes in alcoholic hepatitis in mice. Liver Int. 2017, 37, 968–973. [Google Scholar] [CrossRef]
- Han, Y.P.; Yan, C.; Zhou, L.; Qin, L.; Tsukamoto, H. A matrix metalloproteinase-9 activation cascade by hepatic stellate cells in trans-differentiation in the three-dimensional extracellular matrix. J. Biol. Chem. 2007, 282, 12928–12939. [Google Scholar] [CrossRef]
- Khanova, E.; Wu, R.; Wang, W.; Yan, R.; Chen, Y.; French, S.W.; Llorente, C.; Pan, S.Q.; Yang, Q.; Li, Y.; et al. Pyroptosis by caspase11/4-gasdermin-D pathway in alcoholic hepatitis in mice and patients. Hepatology 2017, 67, 1737–1753. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The human microbiome project. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Cope, K.; Risby, T.; Diehl, A.M. Increased gastrointestinal ethanol production in obese mice: Implications for fatty liver disease pathogenesis. Gastroenterology 2000, 119, 1340–1347. [Google Scholar] [CrossRef]
- Lu, J.; Wang, Y.; Xu, M.; Fei, Q.; Gu, Y.; Luo, Y.; Wu, H. Efficient biosynthesis of 3-hydroxypropionic acid from ethanol in metabolically engineered Escherichia coli. Bioresour. Technol. 2022, 363, 127907. [Google Scholar] [CrossRef]
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609. [Google Scholar] [CrossRef]
- Duan, Y.; Llorente, C.; Lang, S.; Brandl, K.; Chu, H.; Jiang, L.; White, R.C.; Clarke, T.H.; Nguyen, K.; Torralba, M.; et al. Bacteriophage targeting of gut bacterium attenuates alcoholic liver disease. Nature 2019, 575, 505–511. [Google Scholar] [CrossRef]
- Lang, S.; Duan, Y.; Liu, J.; Torralba, M.G.; Kuelbs, C.; Ventura-Cots, M.; Abraldes, J.G.; Bosques-Padilla, F.; Verna, E.C.; Brown, R.S., Jr.; et al. Intestinal fungal dysbiosis and systemic immune response to fungi in patients with alcoholic hepatitis. Hepatology 2020, 71, 522–538. [Google Scholar] [CrossRef]
- Lang, S.; Schnabl, B. Microbiota and Fatty Liver Disease—The Known, the Unknown, and the Future. Cell Host Microbe 2020, 28, 233–244. [Google Scholar] [CrossRef]
- Leclercq, S.; Matamoros, S.; Cani, P.D.; Neyrinck, A.M.; Jamar, F.; Starkel, P.; Windey, K.; Tremaroli, V.; Backhed, F.; Verbeke, K.; et al. Intestinal permeability, gut-bacterial dysbiosis, and behavioral markers of alcohol-dependence severity. Proc. Natl. Acad. Sci. USA 2014, 111, E4485–E4493. [Google Scholar] [CrossRef]
- Wang, L.; Fouts, D.E.; Starkel, P.; Hartmann, P.; Chen, P.; Llorente, C.; DePew, J.; Moncera, K.; Ho, S.B.; Brenner, D.A.; et al. Intestinal REG3 lectins protect against alcoholic steatohepatitis by reducing mucosa-associated microbiota and preventing bacterial translocation. Cell Host Microbe 2016, 19, 227–239. [Google Scholar] [CrossRef]
- Jandhyala, S.M.; Talukdar, R.; Subramanyam, C.; Vuyyuru, H.; Sasikala, M.; Nageshwar Reddy, D. Role of the normal gut microbiota. World J. Gastroenterol. 2015, 21, 8787–8803. [Google Scholar] [CrossRef]
- Eckburg, P.B.; Bik, E.M.; Bernstein, C.N.; Purdom, E.; Dethlefsen, L.; Sargent, M.; Gill, S.R.; Nelson, K.E.; Relman, D.A. Diversity of the human intestinal microbial flora. Science 2005, 308, 1635–1638. [Google Scholar] [CrossRef]
- Ley, R.E.; Backhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 2005, 102, 11070–11075. [Google Scholar] [CrossRef]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial ecology: Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef]
- Wlodarska, M.; Kostic, A.D.; Xavier, R.J. An integrative view of microbiome-host interactions in inflammatory bowel diseases. Cell Host Microbe 2015, 17, 577–591. [Google Scholar] [CrossRef]
- Gerard, P. Gut microbiota and obesity. Cell Mol. Life Sci. 2016, 73, 147–162. [Google Scholar] [CrossRef]
- Bode, C.; Kolepke, R.; Schafer, K.; Bode, J.C. Breath hydrogen excretion in patients with alcoholic liver disease—Evidence of small intestinal bacterial overgrowth. Z. Gastroenterol. 1993, 31, 3–7. [Google Scholar]
- Yan, A.W.; Fouts, D.E.; Brandl, J.; Starkel, P.; Torralba, M.; Schott, E.; Tsukamoto, H.; Nelson, K.E.; Brenner, D.A.; Schnabl, B. Enteric dysbiosis associated with a mouse model of alcoholic liver disease. Hepatology 2011, 53, 96–105. [Google Scholar] [CrossRef]
- Candela, M.; Perna, F.; Carnevali, P.; Vitali, B.; Ciati, R.; Gionchetti, P.; Rizzello, F.; Campieri, M.; Brigidi, P. Interaction of probiotic Lactobacillus and Bifidobacterium strains with human intestinal epithelial cells: Adhesion properties, competition against enteropathogens and modulation of IL-8 production. Int. J. Food Microbiol. 2008, 125, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Sanna, S.; van Zuydam, N.R.; Mahajan, A.; Kurilshikov, A.; Vich Vila, A.; Vosa, U.; Mujagic, Z.; Masclee, A.A.M.; Jonkers, D.; Oosting, M.; et al. Causal relationships among the gut microbiome, short-chain fatty acids and metabolic diseases. Nat. Genet. 2019, 51, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Torralba, M.; Tan, J.; Embree, M.; Zengler, K.; Starkel, P.; van Pijkeren, J.P.; DePew, J.; Loomba, R.; Ho, S.B.; et al. Supplementation of saturated long-chain fatty acids maintains intestinal eubiosis and reduces ethanol-induced liver injury in mice. Gastroenterology 2015, 148, 203–214.e216. [Google Scholar] [CrossRef] [PubMed]
- Wahlstro, M.A.; Sayin, S.; Marschall, H.U.; Backhed, F. Intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metab. 2016, 24, 41–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molinero, N.; Ruiz, L.; Sanchez, B.; Margolles, A.; Delgado, S. Intestinal bacteria interplay with bile and cholesterol metabolism: Implications on host physiology. Front. Physiol. 2019, 10, 185. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, T.; Moschetta, A.; Lee, Y.K.; Peng, L.; Zhao, G.; Downes, M.; Yu, R.T.; Shelton, J.M.; Richardson, J.A.; Repa, J.J.; et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 3920–3925. [Google Scholar] [CrossRef]
- Babu, S.; Blauvelt, C.P.; Kumaraswami, V.; Nutman, T.B. Cutting edge: Diminished T cell TLR expression and function modulates the immune response in human filarial infection. J. Immunol. 2006, 176, 3885–3889. [Google Scholar] [CrossRef]
- Park, J.W.; Kim, S.E.; Lee, N.Y.; Kim, J.H.; Jung, J.H.; Jang, M.K.; Park, S.H.; Lee, M.S.; Kim, D.J.; Kim, H.S.; et al. Role of Microbiota-Derived Metabolites in Alcoholic and Non-Alcoholic Fatty Liver Diseases. Int. J. Mol. Sci. 2021, 23, 426. [Google Scholar] [CrossRef]
- Fuller, R. Probiotics in man and animals. J. Appl. Bacteriol. 1989, 66, 365–378. [Google Scholar]
- Forsyth, C.B.; Farhadi, A.; Jakate, S.M.; Tang, Y.; Shaikh, M.; Keshavarzian, A. Lactobacillus GG treatment ameliorates alcohol-induced intestinal oxidative stress, gut leakiness, and liver injury in a rat model of alcoholic steatohepatitis. Alcohol 2009, 43, 163–172. [Google Scholar] [CrossRef]
- Chang, B.; Sang, L.; Wang, Y.; Tong, J.; Zhang, D.; Wang, B. The protective effect of VSL#3 on intestinal permeability in a rat model of alcoholic intestinal injury. BMC Gastroenterol. 2013, 13, 151. [Google Scholar] [CrossRef]
- Mimee, M.; Citorik, R.J.; Lu, T.K. Microbiome therapeutics—Advances and challenges. Adv. Drug Deliv. Rev. 2016, 105, 44–54. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Salzman, N.H.; Acharya, C.; Sterling, R.K.; White, M.B.; Gavis, E.A.; Fagan, A.; Hayward, M.; Holtz, M.L.; Matherly, S.; et al. Fecal microbial transplant capsules are safe in hepatic encephalopathy: A phase 1, randomized, placebo-controlled trial. Hepatology 2019, 70, 1690–1703. [Google Scholar] [CrossRef]
- Philips, C.A.; Pande, A.; Shasthry, S.M.; Jamwal, K.D.; Khillan, V.; Chandel, S.S.; Kumar, G.; Sharma, M.K.; Maiwall, R.; Jindal, A.; et al. Healthy donor fecal microbiota transplantation in steroid-ineligible severe alcoholic hepatitis: A pilot study. Clin. Gastroenterol. Hepatol. 2017, 15, 600–602. [Google Scholar] [CrossRef]
- Mouries, J.; Brescia, P.; Silvestri, A.; Spadoni, I.; Sorribas, M.; Wiest, R.; Mileti, E.; Galbiati, M.; Invernizzi, P.; Adorini, L.; et al. Microbiota-driven gut vascular barrier disruption is a prerequisite for non-alcoholic steatohepatitis development. J. Hepatol. 2019, 71, 1216–1228. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.M.; Ratziu, V.; Loomba, R.; Rinella, M.; Anstee, Q.M.; Goodman, Z.; Bedossa, P.; Geier, A.; Beckebaum, S.; Newsome, P.N.; et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: Interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2020, 394, 2184–2196. [Google Scholar] [CrossRef] [Green Version]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dukić, M.; Radonjić, T.; Jovanović, I.; Zdravković, M.; Todorović, Z.; Kraišnik, N.; Aranđelović, B.; Mandić, O.; Popadić, V.; Nikolić, N.; et al. Alcohol, Inflammation, and Microbiota in Alcoholic Liver Disease. Int. J. Mol. Sci. 2023, 24, 3735. https://doi.org/10.3390/ijms24043735
Dukić M, Radonjić T, Jovanović I, Zdravković M, Todorović Z, Kraišnik N, Aranđelović B, Mandić O, Popadić V, Nikolić N, et al. Alcohol, Inflammation, and Microbiota in Alcoholic Liver Disease. International Journal of Molecular Sciences. 2023; 24(4):3735. https://doi.org/10.3390/ijms24043735
Chicago/Turabian StyleDukić, Marija, Tijana Radonjić, Igor Jovanović, Marija Zdravković, Zoran Todorović, Nemanja Kraišnik, Bojana Aranđelović, Olga Mandić, Višeslav Popadić, Novica Nikolić, and et al. 2023. "Alcohol, Inflammation, and Microbiota in Alcoholic Liver Disease" International Journal of Molecular Sciences 24, no. 4: 3735. https://doi.org/10.3390/ijms24043735
APA StyleDukić, M., Radonjić, T., Jovanović, I., Zdravković, M., Todorović, Z., Kraišnik, N., Aranđelović, B., Mandić, O., Popadić, V., Nikolić, N., Klašnja, S., Manojlović, A., Divac, A., Gačić, J., Brajković, M., Oprić, S., Popović, M., & Branković, M. (2023). Alcohol, Inflammation, and Microbiota in Alcoholic Liver Disease. International Journal of Molecular Sciences, 24(4), 3735. https://doi.org/10.3390/ijms24043735