
ERβ1 Sensitizes and ERβ2 Desensitizes ERα-Positive Breast Cancer Cells to the Inhibitory Effects of Tamoxifen, Fulvestrant and Their Combination with All-Trans Retinoic Acid


 , , , and
, , , and
Abstract
:1. Introduction
2. Results
2.1. Establishment of MCF7 Clones Stably Expressing Human ERβ1 or ERβ2 and Assessment of Their Effect on ERα Gene Expression and Transcriptional Activity
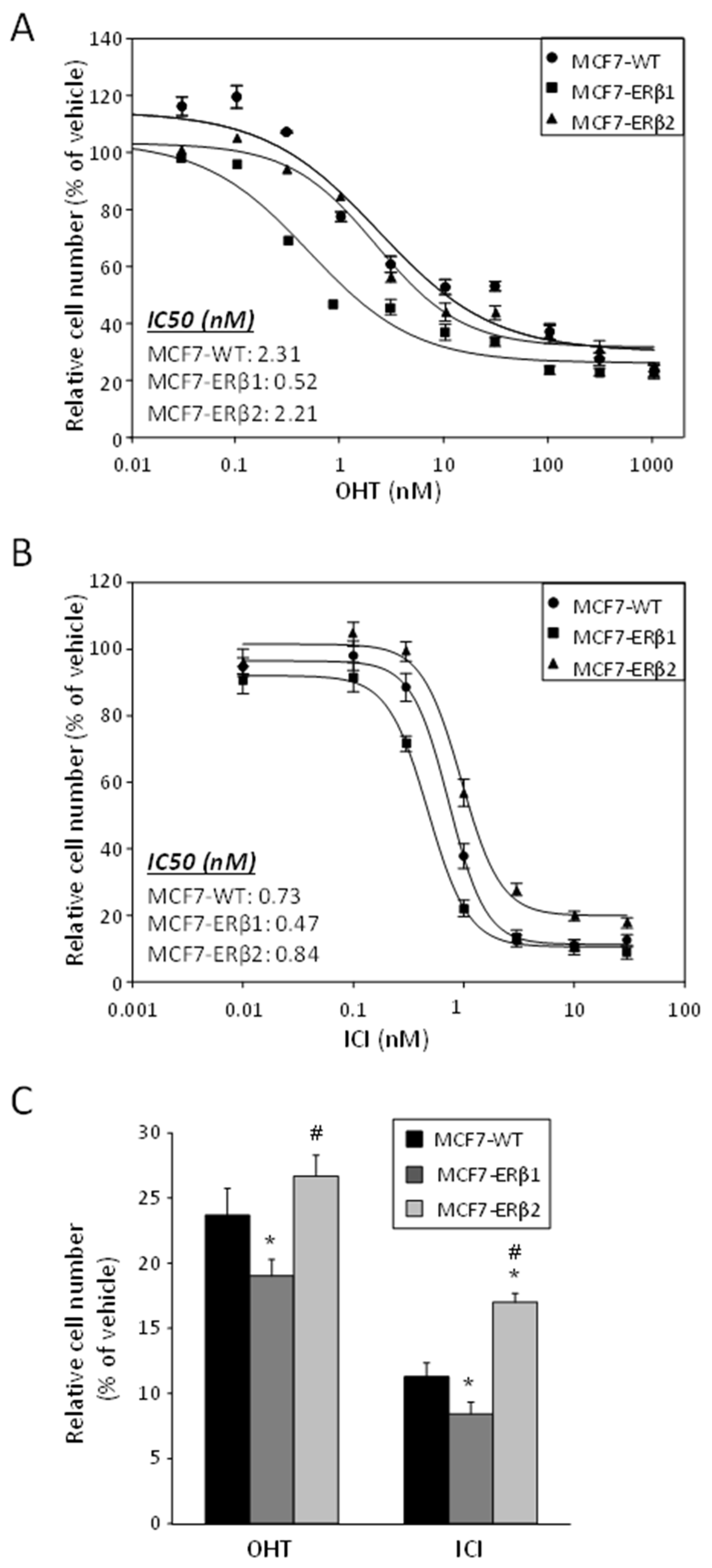
2.2. ERβ1 Sensitized and ERβ2 Desensitized, Respectively, MCF7 Cells to the Antiproliferative Effect of OHT or ICI, Whether Used Separately or in Combination with ATRA

2.3. OHT, ICI and ATRA Abolished the S-Phase Fraction of ERβ1-Expressing MCF7 Cells
2.4. ERβ1 Enhanced the Cytocidal Effects of ATRA Combinations with OHT or ICI

2.5. Global Transcriptional Profile of MCF7-WT, MCF7-ERβ1 and MCF7-ERβ2 Cells Treated with OHT+AΤRA
3. Discussion
4. Materials and Methods
4.1. Purchased Compounds, Chemicals and Plasmids
4.2. Cells, Establishment of MCF7 Cells Stably Expressing ERβ1 or ERβ2, Whole Cell Binding Assay and Western Blot Analysis
4.3. Crystal Violet Staining Assay, Flow Cytometry and Lactate Dehydrogenase (LDH) Assay
4.4. RNA Isolation, Reverse Transcription and Real-Time PCR (qPCR)
4.5. Illumina Array Analysis and Functional Interpretation
4.6. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Acheampong, T.; Kehm, R.D.; Terry, M.B.; Argov, E.L.; Tehranifar, P. Incidence Trends of Breast Cancer Molecular Subtypes by Age and Race/Ethnicity in the US From 2010 to 2016. JAMA Network Open 2020, 3, e2013226. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Gray, R.; Braybrooke, J.; Davies, C.; Taylor, C.; McGale, P.; Peto, R.; Pritchard, K.I.; Bergh, J.; Dowsett, M.; et al. EBCTCG. 20-Year Risks of Breast-Cancer Recurrence after Stopping Endocrine Therapy at 5 Years. N. Engl. J. Med. 2017, 377, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Richman, J.; Dowsett, M. Beyond 5 years: Enduring risk of recurrence in oestrogen receptor-positive breast cancer. Nat. Rev. Clin. Oncol. 2019, 16, 296–311. [Google Scholar] [CrossRef]
- Costantini, L.; Molinari, R.; Farinon, B.; Merendino, N. Retinoic Acids in the Treatment of Most Lethal Solid Cancers. J. Clin. Med. 2020, 9, 360. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Hu, Q.; Tao, X.; Xia, J.; Wu, T.; Cheng, B.; Wang, J. Retinoids in cancer chemoprevention and therapy: Meta-analysis of randomized controlled trials. Front. Genet. 2022, 13, 1065320. [Google Scholar] [CrossRef]
- Rochette-Egly, C.; Chambon, P. F9 embryocarcinoma cells: A cell autonomous model to study the functional selectivity of RARs and RXRs in retinoid signaling. Histol. Histopathol. 2001, 16, 909–922. [Google Scholar] [CrossRef]
- Ross-Innes, C.S.; Stark, R.; Holmes, K.A.; Schmidt, D.; Spyrou, C.; Russell, R.; Massie, C.E.; Vowler, S.L.; Eldridge, M.; Carroll, J.S. Cooperative interaction between retinoic acid receptor-alpha and estrogen receptor in breast cancer. Genes. Dev. 2010, 24, 171–182. [Google Scholar] [CrossRef]
- Hua, S.; Kittler, R.; White, K.P. Genomic antagonism between retinoic acid and estrogen signaling in breast cancer. Cell 2009, 137, 1259–1271. [Google Scholar] [CrossRef]
- Miro Estruch, I.; de Haan, L.H.J.; Melchers, D.; Houtman, R.; Louisse, J.; Groten, J.P.; Rietjens, I.M.C.M. The effects of all-trans retinoic acid on estrogen receptor signaling in the estrogen-sensitive MCF/BUS subline. J. Recept. Signal Transduct. Res. 2018, 38, 112–121. [Google Scholar] [CrossRef]
- Huang, S.; Chen, Y.; Liang, Z.M.; Li, N.N.; Liu, Y.; Zhu, Y.; Liao, D.; Zhou, X.Z.; Lu, K.P.; Yao, Y.; et al. Targeting Pin1 by All-Trans Retinoic Acid (ATRA) Overcomes Tamoxifen Resistance in Breast Cancer via Multifactorial Mechanisms. Front. Cell Dev. Biol. 2019, 7, 322. [Google Scholar] [CrossRef]
- Czeczuga-Semeniuk, E.; Lemancewicz, D.; Wołczyński, S. Estradiol and tamoxifen differently affects the inhibitory effects of vitamin A and their metabolites on the proliferation and expression of alpha2beta1 integrins in MCF-7 breast cancer cells. Adv. Med. Sci. 2009, 54, 91–98. [Google Scholar] [CrossRef]
- Rubin, M.; Fenig, E.; Rosenauer, A.; Menendez-Botet, C.; Achkar, C.; Bentel, J.M.; Yahalom, J.; Mendelsohn, J.; Miller, W.H., Jr. 9-Cis retinoic acid inhibits growth of breast cancer cells and down-regulates estrogen receptor RNA and protein. Cancer Res. 1994, 54, 6549–6556. [Google Scholar] [PubMed]
- Bentel, J.M.; Lebwohl, D.E.; Cullen, K.J.; Rubin, M.S.; Rosen, N.; Mendelsohn, J.; Miller, W.H., Jr. Insulin-like growth factors modulate the growth inhibitory effects of retinoic acid on MCF-7 breast cancer cells. J. Cell. Physiol. 1995, 165, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.I.; Kim, J.H.; Kang, C.W. Involvement of insulin-like growth factor-I secretion and all-trans-retinoic acid-induced decrement in viability in MCF-7 cells. Chemotherapy 2011, 57, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Salvatori, L.; Ravenna, L.; Caporuscio, F.; Principessa, L.; Coroniti, G.; Frati, L.; Russo, M.A.; Petrangeli, E. Action of retinoic acid receptor on EGFR gene transactivation and breast cancer cell proliferation: Interplay with the estrogen receptor. Biomed. Pharmacother. 2011, 65, 307–312. [Google Scholar] [CrossRef] [PubMed]
- del Rincón, S.V.; Rousseau, C.; Samanta, R.; Miller, W.H., Jr. Retinoic acid-induced growth arrest of MCF-7 cells involves the selective regulation of the IRS-1/PI 3-kinase/AKT pathway. Oncogene 2003, 22, 3353–3360. [Google Scholar] [CrossRef]
- Shang, Y.; Brown, M. Molecular determinants for the tissue specificity of SERMs. Science 2002, 295, 2465–2468. [Google Scholar] [CrossRef]
- Hodges, L.C.; Cook, J.D.; Lobenhofer, E.K.; Li, L.; Bennett, L.; Bushel, P.R.; Aldaz, C.M.; Afshari, C.A.; Walker, C.L. Tamoxifen functions as a molecular agonist inducing cell cycle-associated genes in breast cancer cells. Mol. Cancer Res. 2003, 1, 300–311. [Google Scholar]
- Frasor, J.; Chang, E.C.; Komm, B.; Lin, C.Y.; Vega, V.B.; Liu, E.T.; Miller, L.D.; Smeds, J.; Bergh, J.; Katzenellenbogen, B.S. Gene expression preferentially regulated by tamoxifen in breast cancer cells and correlations with clinical outcome. Cancer Res. 2006, 66, 7334–7340. [Google Scholar] [CrossRef]
- Chang, E.C.; Frasor, J.; Komm, B.; Katzenellenbogen, B.S. Impact of estrogen receptor beta on gene networks regulated by estrogen receptor alpha in breast cancer cells. Endocrinology 2006, 147, 4831–4842. [Google Scholar] [CrossRef]
- Kuiper, G.G.; Lemmen, J.G.; Carlsson, B.; Corton, J.C.; Safe, S.H.; van der Saag, P.T.; van der Burg, B.; Gustafsson, J.A. Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinology 1998, 139, 4252–4263. [Google Scholar] [CrossRef] [PubMed]
- Leung, Y.K.; Mak, P.; Hassan, S.; Ho, S.M. Estrogen receptor (ER)-beta isoforms: A key to understanding ER-beta signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 13162–13167. [Google Scholar] [CrossRef]
- Green, C.A.; Peter, M.B.; Speirs, V.; Shaaban, A.M. The potential role of ER beta isoforms in the clinical management of breast cancer. Histopathology 2008, 53, 374–380. [Google Scholar] [CrossRef]
- Zhao, C.; Matthews, J.; Tujague, M.; Wan, J.; Ström, A.; Toresson, G.; Lam, E.W.; Cheng, G.; Gustafsson, J.A.; Dahlman-Wright, K. Estrogen receptor beta2 negatively regulates the transactivation of estrogen receptor alpha in human breast cancer cells. Cancer Res. 2007, 67, 3955–3962. [Google Scholar] [CrossRef] [PubMed]
- Faria, M.; Karami, S.; Granados-Principal, S.; Dey, P.; Verma, A.; Choi, D.S.; Elemento, O.; Bawa-Khalfe, T.; Chang, J.C.; Strom, A.M.; et al. The ERβ4 variant induces transformation of the normal breast mammary epithelial cell line MCF-10A; the ERβ variants ERβ2 and ERβ5 increase aggressiveness of TNBC by regulation of hypoxic signaling. Oncotarget 2018, 9, 12201–12211. [Google Scholar] [CrossRef]
- Chen, D.; Wang, M.; Zhang, H.; Zhou, S.; Luo, C. Estrogen receptor β2 (ERβ2)-mediated upregulation of hsa_circ_0000732 promotes tumor progression via sponging microRNA-1184 in triple-negative breast cancer (TNBC). Inflamm. Res. 2022, 71, 255–266. [Google Scholar] [CrossRef]
- Honma, N.; Horii, R.; Iwase, T.; Saji, S.; Younes, M.; Takubo, K.; Matsuura, M.; Ito, Y.; Akiyama, F.; Sakamoto, G. Clinical importance of estrogen receptor-beta evaluation in breast cancer patients treated with adjuvant tamoxifen therapy. J. Clin. Oncol. 2008, 26, 3727–3734. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y. Estrogen Receptor β Expression and Its Clinical Implication in Breast Cancers: Favorable or Unfavorable? J. Breast Cancer 2022, 25, 75–93. [Google Scholar] [CrossRef]
- Chantzi, N.I.; Tiniakos, D.G.; Palaiologou, M.; Goutas, N.; Filippidis, T.; Vassilaros, S.D.; Dhimolea, E.; Mitsiou, D.J.; Alexis, M.N. Estrogen receptor beta 2 is associated with poor prognosis in estrogen receptor alpha-negative breast carcinoma. J. Cancer Res. Clin. Oncol. 2013, 139, 1489–1498. [Google Scholar] [CrossRef]
- Dhimolea, E.; Tiniakos, D.G.; Chantzi, Ν.Ι.; Goutas, N.; Vassilaros, S.D.; Mitsiou, D.J.; Alexis, Μ.N. Estrogen receptors β1 and β2 are associated with distinct responses of estrogen receptor α-positive breast carcinoma to adjuvant endocrine therapy. Cancer Lett. 2015, 358, 37–42. [Google Scholar] [CrossRef]
- Weaver, C.A.; Springer, P.A.; Katzenellenbogen, B.S. Regulation of pS2 gene expression by affinity labeling and reversibly binding estrogens and antiestrogens: Comparison of effects on the native gene and on pS2-chloramphenicol acetyltransferase fusion genes transfected into MCF-7 human breast cancer cells. Mol. Endocrinol. 1988, 2, 936–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Huang, Y.R.; Harrington, W.R.; Sheng, S.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S. Antagonists selective for estrogen receptor alpha. Endocrinology 2002, 143, 941–947. [Google Scholar] [CrossRef]
- Meyers, M.J.; Sun, J.; Carlson, K.E.; Marriner, G.A.; Katzenellenbogen, B.S.; Katzenellenbogen, J.A. Estrogen receptor-beta potency-selective ligands: Structure-activity relationship studies of diarylpropionitriles and their acetylene and polar analogues. J. Med. Chem. 2001, 44, 4230–4251. [Google Scholar] [CrossRef] [PubMed]
- Liston, D.R.; Davis, M. Clinically Relevant Concentrations of Anticancer Drugs: A Guide for Nonclinical Studies. Clin. Cancer Res. 2017, 23, 3489–3498. [Google Scholar] [CrossRef] [PubMed]
- Hata, A.N.L.; Engelman, J.A.; Faber, A.C. The BCL2 Family: Key Mediators of the Apoptotic Response to Targeted Anticancer Therapeutics. Cancer Discov. 2015, 5, 475–487. [Google Scholar] [CrossRef]
- Holmes, K.A.; Song, J.S.; Liu, X.S.; Brown, M.; Carroll, J.S. Nkx3-1 and LEF-1 function as transcriptional inhibitors of estrogen receptor activity. Cancer Res. 2008, 68, 7380–7385. [Google Scholar] [CrossRef]
- Arai, K.; Takano, S.; Teratani, T.; Ito, Y.; Yamada, T.; Nozawa, R. S100A8 and S100A9 overexpression is associated with poor pathological parameters in invasive ductal carcinoma of the breast. Curr. Cancer Drug Targets 2008, 8, 243–252. [Google Scholar] [CrossRef]
- Weichhaus, M.; Segaran, P.; Renaud, A.; Geerts, D.; Connelly, L. Osteoprotegerin expression in triple-negative breast cancer cells promotes metastasis. Cancer Med. 2014, 3, 1112–1125. [Google Scholar] [CrossRef]
- Kim, J.; Villadsen, R. The Expression Pattern of Epidermal Differentiation Marker Keratin 10 in the Normal Human Breast and Breast Cancer Cells. J. Histochem. Cytochem. 2020, 68, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Crighton, D.; Wilkinson, S.; O’Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006, 126, 121–134. [Google Scholar] [CrossRef]
- Schwartz, J.L.; Shajahan, A.N.; Clarke, R. The Role of Interferon Regulatory Factor-1 (IRF1) in Overcoming Antiestrogen Resistance in the Treatment of Breast Cancer. Int. J. Breast Cancer 2011, 912102. [Google Scholar] [CrossRef] [Green Version]
- Saito, M.; Goto, A.; Abe, N.; Saito, K.; Maeda, D.; Ohtake, T.; Murakami, Y.; Takenoshita, S. Decreased expression of CADM1 and CADM4 are associated with advanced stage breast cancer. Oncol. Lett. 2018, 15, 2401–2406. [Google Scholar] [CrossRef] [PubMed]
- Schüler-Toprak, S.; Häring, J.; Inwald, E.C.; Moehle, C.; Ortmann, O.; Treeck, O. Agonists and knockdown of estrogen receptor β differentially affect invasion of triple-negative breast cancer cells in vitro. BMC Cancer 2016, 16, 951. [Google Scholar] [CrossRef]
- Feng, L.; Ma, Y.; Sun, J.; Shen, Q.; Liu, L.; Lu, H.; Wang, F.; Yue, Y.; Li, J.; Zhang, S.; et al. YY1-MIR372-SQSTM1 regulatory axis in autophagy. Autophagy 2014, 10, 1442–1453. [Google Scholar] [CrossRef]
- Patra, K.C.; Wang, Q.; Bhaskar, P.T.; Miller, L.; Wang, Z.; Wheaton, W.; Chandel, N.; Laakso, M.; Muller, W.J.; Allen, E.L.; et al. Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell 2018, 24, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Qiu, T.; Cao, J.; Chen, W.; Wang, J.; Wang, Y.; Zhao, L.; Liu, M.; He, L.; Wu, G.; Li, H.; et al. 24-Dehydrocholesterol reductase promotes the growth of breast cancer stem-like cells through the Hedgehog pathway. Cancer Sci. 2020, 111, 3653–3664. [Google Scholar] [CrossRef]
- Gu, M.; Zheng, W.; Zhang, M.; Dong, X.; Zhao, Y.; Wang, S.; Jiang, H.; Liu, L.; Zheng, X. Downregulation of RAI14 inhibits the proliferation and invasion of breast cancer cells. J. Cancer 2019, 10, 6341–6348. [Google Scholar] [CrossRef]
- Yun, S.H.; Han, S.H.; Park, J.I. Peroxisome Proliferator-Activated Receptor γ and PGC-1α in Cancer: Dual Actions as Tumor Promoter and Suppressor. PPAR Res. 2018, 6727421. [Google Scholar] [CrossRef]
- De Nicola, F.; Catena, V.; Rinaldo, C.; Bruno, T.; Iezzi, S.; Sorino, C.; Desantis, A.; Camerini, S.; Crescenzi, M.; Floridi, A.; et al. HIPK2 sustains apoptotic response by phosphorylating Che-1/AATF and promoting its degradation. Cell Death Dis. 2014, 5, e1414. [Google Scholar] [CrossRef] [PubMed]
- Rataj, F.; Planel, S.; Denis, J.; Roelants, C.; Filhol, O.; Guyon, L.; Feige, J.J.; Cherradi, N. Targeting AU-rich element-mediated mRNA decay with a truncated active form of the zinc-finger protein TIS11b/BRF1 impairs major hallmarks of mammary tumorigenesis. Oncogene 2019, 38, 5174–5190. [Google Scholar] [CrossRef]
- Wang, L.L.; Meng, Q.H.; Jiao, Y.; Xu, J.Y.; Ge, C.M.; Zhou, J.Y.; Rosen, E.M.; Wang, H.C.; Fan, S.J. High-mobility group boxes mediate cell proliferation and radiosensitivity via retinoblastoma-interaction-dependent and -independent mechanisms. Cancer Biother. Radiopharm. 2012, 27, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Y.L.; Motomura, H.; Tamori, S.; Ozaki, A.; Onaga, C.; Hara, Y.; Sato, K.; Tahata, K.; Harada, Y.; Sasaki, K.; et al. High Expression of CD58 and ALDH1A3 Predicts a Poor Prognosis in Basal-like Breast Cancer. Anticancer Res. 2022, 42, 5223–5232. [Google Scholar] [CrossRef] [PubMed]
- Browne, A.L.; Charmsaz, S.; Varešlija, D.; Fagan, A.; Cosgrove, N.; Cocchiglia, S.; Purcell, S.; Ward, E.; Bane, F.; Hudson, L.; et al. Network analysis of SRC-1 reveals a novel transcription factor hub which regulates endocrine resistant breast cancer. Oncogene 2018, 37, 2008–2021. [Google Scholar] [CrossRef] [PubMed]
- Lasorella, A.; Benezra, R.; Iavarone, A. The ID proteins: Master regulators of cancer stem cells and tumour aggressiveness. Nat. Rev. Cancer 2014, 14, 77–91. [Google Scholar] [CrossRef]
- Okita, Y.; Chen, C.; Kato, M. Cell-surface GPNMB and induction of stemness. Oncotarget 2018, 9, 37289–37290. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhou, D.; Phung, S.; Masri, S.; Smith, D.; Chen, S. SGK3 is an estrogen-inducible kinase promoting estrogen-mediated survival of breast cancer cells. Mol. Endocrinol. 2011, 25, 72–82. [Google Scholar] [CrossRef]
- Iwasaki, M.; Homma, S.; Hishiya, A.; Dolezal, S.J.; Reed, J.C.; Takayama, S. BAG3 regulates motility and adhesion of epithelial cancer cells. Cancer Res. 2007, 67, 10252–10259. [Google Scholar] [CrossRef]
- Jinks, R.N.; Puffenberger, E.G.; Baple, E.; Harding, B.; Crino, P.; Fogo, A.B.; Wenger, O.; Xin, B.; Koehler, A.E.; McGlincy, M.H.; et al. Recessive nephrocerebellar syndrome on the Galloway-Mowat syndrome spectrum is caused by homozygous protein-truncating mutations of WDR73. Brain 2015, 138, 2173–2190. [Google Scholar] [CrossRef]
- Chen, H.; Zhou, L.; Li, J.; Hu, K. ALKBH family members as novel biomarkers and prognostic factors in human breast cancer. Aging 2022, 14, 6579–6593. [Google Scholar] [CrossRef]
- Ameri, K.; Maltepe, E. HIGD1A-mediated dormancy and tumor survival. Mol. Cell. Oncol. 2015, 2, e1030537. [Google Scholar] [CrossRef]
- Renda, I.; Bianchi, S.; Vezzosi, V.; Nori, J.; Vanzi, E.; Tavella, K.; Susini, T. Expression of FGD3 gene as prognostic factor in young breast cancer patients. Sci. Rep. 2019, 9, 15204. [Google Scholar] [CrossRef] [Green Version]
- Yunos, N.M.; Amin, N.D.M.; Jauri, M.H.; Ling, S.K.; Hassan, N.H.; Sallehudin, N.J. The In Vitro Anti-Cancer Activities and Mechanisms of Action of 9-Methoxycanthin-6-one from Eurycoma longifolia in Selected Cancer Cell Lines. Molecules 2022, 27, 585. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Kohno, T.; Ajima, R.; Sasaki, H.; Minna, J.D.; Fujiwara, T.; Tanaka, N.; Yokota, J. Homozygous deletion and reduced expression of the DOCK8 gene in human lung cancer. Int. J. Oncol. 2006, 28, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Obacz, J.; Brychtova, V.; Podhorec, J.; Fabian, P.; Dobes, P.; Vojtesek, B.; Hrstka, R. Anterior gradient protein 3 is associated with less aggressive tumors and better outcome of breast cancer patients. OncoTargets Ther. 2015, 8, 1523–1532. [Google Scholar] [CrossRef]
- Pietkiewicz, P.P.; Lutkowska, A.; Lianeri, M.; Jagodzinski, P.P. Tamoxifen epigenetically modulates CXCL12 expression in MCF-7 breast cancer cells. Biomed. Pharmacother. 2010, 64, 54–57. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Ma, J.; Chen, Q.; Hou, Z.; Luo, X.; Wang, G.; Wang, J.; Hu, J.; Cao, Z. SIX4 promotes metastasis through STAT3 activation in breast cancer. Am. J. Cancer Res. 2020, 10, 224–236. [Google Scholar]
- Chow, C.; Wong, N.; Fai To, K.; Wai Lo, K. Activin beta E (INHBE), a RASSF1A target gene is downregulated in hepatocellular carcinoma. Cancer Res. 2007, 67 (Suppl. 9), 3662. [Google Scholar]
- Harkin, D.P.; Bean, J.M.; Miklos, D.; Song, Y.H.; Truong, V.B.; Englert, C.; Christians, F.C.; Ellisen, L.W.; Maheswaran, S.; Oliner, J.D.; et al. Induction of GADD45 and JNK/SAPK-dependent apoptosis following inducible expression of BRCA1. Cell 1999, 97, 575–586. [Google Scholar] [CrossRef]
- Wang, Z.; Hou, J.; Lu, L.; Qi, Z.; Sun, J.; Gao, W.; Meng, J.; Wang, Y.; Sun, H.; Gu, H.; et al. Small ribosomal protein subunit S7 suppresses ovarian tumorigenesis through regulation of the PI3K/AKT and MAPK pathways. PLoS ONE 2013, 8, e79117. [Google Scholar] [CrossRef]
- Li, X.; Sun, G.; Wu, L.; Sun, G.; Cheng, Y.; Tao, J.; Li, Z.; Tang, W.; Wang, H. Upregulation of ADAR Promotes Breast Cancer Progression and Serves as a Potential Therapeutic Target. J. Oncol. 2021, 2021, 2012903. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.L.; Baxter, R.C. Signalling pathways of insulin-like growth factors (IGFs) and IGF binding protein-3. Growth Factors 2011, 29, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M.; Katoh, M. Precision medicine for human cancers with Notch signaling dysregulation (Review). Int. J. mol. Med. 2020, 45, 279–297. [Google Scholar] [CrossRef]
- Czapiewski, P.; Cornelius, M.; Hartig, R.; Kalinski, T.; Haybaeck, J.; Dittmer, A.; Dittmer, J.; Ignatov, A.; Nass, N. BCL3 expression is strongly associated with the occurrence of breast cancer relapse under tamoxifen treatment in a retrospective cohort study. Virchows Archiv. Int. J. Pathol. 2022, 480, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Yi, S.; Zhou, W. Tumorigenesis-related key genes in adolescents and young adults with HR(+)/HER2(-) breast cancer. Int. J. of clinical and exp. Int. J. Clin. Exp. Pathol. 2020, 13, 2701–2709. [Google Scholar] [PubMed]
- Perone, Y.; Farrugia, A.J.; Rodríguez-Meira, A.; Győrffy, B.; Ion, C.; Uggetti, A.; Chronopoulos, A.; Marrazzo, P.; Faronato, M.; Shousha, S.; et al. SREBP1 drives Keratin-80-dependent cytoskeletal changes and invasive behavior in endocrine-resistant ERα breast cancer. Nat. Commun. 2019, 10, 2115, Erratum in Nat. Commun. 2019, 10, 3791. [Google Scholar] [CrossRef]
- Bartella, V.; Rizza, P.; Barone, I.; Zito, D.; Giordano, F.; Giordano, C.; Catalano, S.; Mauro, L.; Sisci, D.; Panno, M.L.; et al. Estrogen receptor beta binds Sp1 and recruits a corepressor complex to the estrogen receptor alpha gene promoter. Breast Cancer Res. Treat. 2012, 134, 569–581. [Google Scholar] [CrossRef]
- Poola, I.; Abraham, J.; Baldwin, K.; Saunders, A.; Bhatnagar, R. Estrogen receptors beta4 and beta5 are full length functionally distinct ERβ isoforms: Cloning from human ovary and functional characterization. Endocrine 2005, 27, 227–238. [Google Scholar] [CrossRef]
- Song, D.; He, H.; Indukuri, R.; Huang, Z.; Stepanauskaite, L.; Sinha, I.; Haldosén, L.A.; Zhao, C.; Williams, C. ERα and ERβ Homodimers in the Same Cellular Context Regulate Distinct Transcriptomes and Functions. Front. Endocrinol. 2022, 13, 930227. [Google Scholar] [CrossRef]
- Hodges-Gallagher, L.; Valentine, C.D.; El Bader, S.; Kushner, P.J. Estrogen receptor beta increases the efficacy of antiestrogens by effects on apoptosis and cell cycling in breast cancer cells. Breast Cancer Res. Treat. 2008, 109, 241–250. [Google Scholar] [CrossRef]
- Powell, E.; Shanle, E.; Brinkman, A.; Li, J.; Keles, S.; Wisinski, K.B.; Huang, W.; Xu, W. Identification of estrogen receptor dimer selective ligands reveals growth-inhibitory effects on cells that co-express ERα and ERβ. PLoS ONE 2012, 7, e30993. [Google Scholar] [CrossRef]
- Bialesova, L.; Xu, L.; Gustafsson, J.Å.; Haldosen, L.A.; Zhao, C.; Dahlman-Wright, K. Estrogen receptor β2 induces proliferation and invasiveness of triple negative breast cancer cells: Association with regulation of PHD3 and HIF-1α. Oncotarget 2017, 8, 76622–76633. [Google Scholar] [CrossRef]
- Jacenik, D.; Cygankiewicz, A.I.; Krajewska, W.M. The G protein-coupled estrogen receptor as a modulator of neoplastic transformation. Mol. Cell. Endocrinology 2016, 429, 10–18. [Google Scholar] [CrossRef]
- Robertson, J.F. ICI 182,780 (Fulvestrant)--the first oestrogen receptor down-regulator--current clinical data. Brit. J. Cancer 2001, 85, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, S.; Inoue, S.; Watanabe, T.; Orimo, A.; Hosoi, T.; Ouchi, Y.; Muramatsu, M. Molecular cloning and characterization of human estrogen receptor betacx: A potential inhibitor of estrogen action in human. Nucleic Acids Res. 1998, 26, 3505–3512. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Wang, W.; Wang, M.; Wu, J.; Yao, F. DOK1/PPARgamma pathway mediates anti-tumor ability of all-trans retinoic acid in breast cancer MCF-7 cells. Biochem. Biophys. Res. Commun. 2017, 487, 189–193. [Google Scholar] [CrossRef]
- Paruthiyil, S.; Parmar, H.; Kerekatte, V.; Cunha, G.R.; Firestone, G.L.; Leitman, D.C. Estrogen receptor beta inhibits human breast cancer cell proliferation and tumor formation by causing a G2 cell cycle arrest. Cancer Res. 2004, 64, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Diel, P.; Smolnikar, K.; Michna, H. The pure antiestrogen ICI 182780 is more effective in the induction of apoptosis and down regulation of BCL-2 than tamoxifen in MCF-7 cells. Breast Cancer Res. Treat. 1999, 58, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Bursch, W.; Ellinger, A.; Kienzl, H.; Török, L.; Pandey, S.; Sikorska, M.; Walker, R.; Schulte Hermann, R. Active cell death induced by the anti-estrogens tamoxifen and ICI 164 384 in human mammary carcinoma cells (MCF-7) in culture: The role of autophagy. Carcinogenesis 1996, 17, 1595–1607. [Google Scholar] [CrossRef]
- Burow, M.E.; Weldon, C.B.; Tang, Y.; McLachlan, J.A.; Beckman, B.S. Oestrogen-mediated suppression of tumour necrosis factor alpha-induced apoptosis in MCF-7 cells: Subversion of Bcl-2 by anti-oestrogens. J. Steroid Biochem. Mol. Biol. 2001, 78, 409–418. [Google Scholar] [CrossRef]
- Li, Y.Z.; Li, C.J.; Pinto, A.V.; Pardee, A.B. Release of mitochondrial cytochrome C in both apoptosis and necrosis induced by beta-lapachone in human carcinoma cells. Mol. Med. 1999, 5, 232–239. [Google Scholar] [CrossRef]
- Chantzi, N.I.; Meligova, A.K.; Dhimolea, E.; Petrou, C.C.; Mitsiou, D.J.; Magafa, V.; Pechtelidou, A.; Florentin, I.; Kitraki, E.; Cordopatis, P.; et al. Insights into ectopic estrogen receptor expression, nucleocytoplasmic distribution and interaction with chromatin obtained with new antibodies to estrogen receptors α and β. Steroids 2011, 76, 974–985. [Google Scholar] [CrossRef]
- Fokialakis, N.; Lambrinidis, G.; Mitsiou, D.J.; Aligiannis, N.; Mitakou, S.; Skaltsounis, A.L.; Pratsinis, H.; Mikros, E.; Alexis, M.N. A new class of phytoestrogens; evaluation of the estrogenic activity of deoxybenzoins. Chem. Biol. 2004, 11, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Thai, Q.D.; Tchoumtchoua, J.; Makropoulou, M.; Boulaka, A.; Meligova, A.K.; Mitsiou, D.J.; Mitakou, S.; Michel, S.; Halabalaki, M.; Alexis, M.N.; et al. Phytochemical study and biological evaluation of chemical constituents of Platanus orientalis and Platanus × acerifolia buds. Phytochemistry 2016, 130, 170–181. [Google Scholar] [CrossRef]
- Panagiotidou, E.; Zerva, S.; Mitsiou, D.J.; Alexis, M.N.; Kitraki, E. Perinatal exposure to low-dose bisphenol A affects the neuroendocrine stress response in rats. J. Endocrinol. 2014, 220, 207–218. [Google Scholar] [CrossRef]
- Du, P.; Kibbe, W.A.; Lin, S.M. Lumi: A Pipeline for Processing Illumina Microarray. Bioinformatics 2008, 24, 1547–1548. [Google Scholar] [CrossRef]
- Parker, H.S.; Corrada Bravo, H.; Leek, J.T. Removing Batch Effects for Prediction Problems with Frozen Surrogate Variable Analysis. PeerJ 2014, 2, e561. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Koutsandreas, T.; Binenbaum, I.; Pilalis, E.; Valavanis, I.; Papadodima, O.; Chatziioannou, A. Analyzing and Visualizing Genomic Complexity for the Derivation of the Emergent Molecular Networks. Int. J. Monit. Surveill. Tech. Res. 2016, 4, 30–49. [Google Scholar] [CrossRef]
- The Gene Ontology Consortium; The Gene Ontology Resource: 20 Years and Still GOing Strong. Nucleic Acids Res. 2019, 47, D330–D338. [CrossRef]
- Köhler, S.; Carmody, L.; Vasilevsky, N.; Jacobsen, J.O.B.; Danis, D.; Gourdine, J.-P.; Gargano, M.; Harris, N.L.; Matentzoglu, N.; McMurry, J.A.; et al. Expansion of the Human Phenotype Ontology (HPO) Knowledge Base and Resources. Nucleic Acids Res. 2019, 47, D1018–D1027. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.L.; Eppig, J.T. The Mammalian Phenotype Ontology: Enabling Robust Annotation and Comparative Analysis. Wiley Interdiscip. Rev. Syst. Biol. Med. 2009, 1, 390–399. [Google Scholar] [CrossRef]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Kalliolias, G.D.; Ostaszewski, M.; Veyssiere, M.; Pilalis, E.; Gawron, P.; Mazein, A.; Bonnet, E.; Petit-Teixeira, E.; Niarakis, A. RA-Map: Building a State-of-the-Art Interactive Knowledge Base for Rheumatoid Arthritis. Database 2020, 2020, baaa017. [Google Scholar] [CrossRef] [PubMed]
- Vekris, A.; Pilalis, E.; Chatziioannou, A.; Petry, K.G. A Computational Pipeline for the Extraction of Actionable Biological Information From NGS-Phage Display Experiments. Front. Physiol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Maurel, M.; Obacz, J.; Avril, T.; Ding, Y.-P.; Papadodima, O.; Treton, X.; Daniel, F.; Pilalis, E.; Hörberg, J.; Hou, W.; et al. Control of Anterior GRadient 2 (AGR2) Dimerization Links Endoplasmic Reticulum Proteostasis to Inflammation. EMBO Mol. Med. 2019, 11, e10120. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | OHT | ICI | |
|---|---|---|---|
| IC50 (nM) | MCF7-WT | 2.26 ± 0.90 | 0.76 ± 0.06 |
| MCF7-ERβ1 | 0.59 ± 0.09 | 0.55 ± 0.03 | |
| MCF7-ERβ2 | 2.46 ± 0.35 | 0.96 ± 0.08 | |
| p value | MCF7-WT vs. MCF7-ERβ1 | 0.046 | 0.025 |
| MCF7-WT vs. MCF7-ERβ2 | 0.794 | 0.027 | |
| MCF7-ERβ1 vs. MCF7-ERβ2 | 0.030 | 0.000 |
| Cell Line | ATRA | |
|---|---|---|
| IC50 (nM) | MCF7-WT | 369 ± 62 |
| MCF7-ERβ1 | 89 ± 30 | |
| MCF7-ERβ2 | 649 ± 80 | |
| p value | MCF7-WT vs. MCF7-ERβ1 | 0.013 |
| MCF7-WT vs. MCF7-ERβ2 | 0.011 | |
| MCF7-ERβ1 vs. MCF7-ERβ2 | 0.000 |
| Gene Symbol 1 | Gene Name | Fold Change |
|---|---|---|
| RAI14 | retinoic acid induced 14 | 1.73 |
| IRF1 | interferon regulatory factor 1 | 1.72 |
| DHCR24 | 24-dehydrocholesterol reductase | 1.71 |
| HK2 | hexokinase 2 | 1.59 |
| SQSTM1 | sequestosome 1 | 1.57 |
| KRT4 | keratin 4 | 1.56 |
| NKX3-1 | NK3 homeobox 1 | 1.54 |
| DRAM1 | DNA damage regulated autophagy modulator 1 | 1.53 |
| S100A8 | S100 calcium binding protein A8 | 0.49 |
| TNFRSF11B | TNF receptor superfamily member 11b | 0.50 |
| KRT10 | keratin 10 | 0.56 |
| BCL2 | BCL2, apoptosis regulator | 0.62 |
| CADM1 | cell adhesion molecule 1 | 0.66 |
| Gene Symbol 1 | Gene Name | Fold Change |
|---|---|---|
| AGR3 | anterior gradient 3, protein disulphide isomerase family member | 2.86 |
| AKR1C3 | aldo-keto reductase family 1 member C3 | 2.21 |
| KRT13 | keratin 13 | 2.21 |
| ALDH1A3 | aldehyde dehydrogenase 1 family member A3 | 2.07 |
| MAG | myelin associated glycoprotein | 2.05 |
| NQO1 | NAD(P)H quinone dehydrogenase 1 | 1.83 |
| HMGB2 | high mobility group box 2 | 1.73 |
| SOX2 | SRY-box 2 | 1.73 |
| DOCK8 | dedicator of cytokinesis 8 | 1.71 |
| NDRG1 | N-myc downstream regulated 1 | 1.69 |
| ACAA2 | acetyl-CoA acyltransferase 2 | 1.67 |
| ACOX2 | acyl-CoA oxidase 2 | 1.65 |
| HMGB1 | high mobility group box 1 | 1.63 |
| ZFP36L1 | ZFP36 ring finger protein like 1 | 1.61 |
| ECT2 | epithelial cell transforming 2 | 1.58 |
| FGD3 | FYVE, RhoGEF and PH domain containing 3 | 1.58 |
| AGR2 | anterior gradient 2, protein disulphide isomerase family member | 1.55 |
| HIPK2 | homeodomain interacting protein kinase 2 | 1.53 |
| BCL6 | B-cell CLL/lymphoma 6 | 1.51 |
| PPARG | peroxisome proliferator activated receptor gamma | 1.50 |
| ASCL1 | achaete-scute family bHLH transcription factor 1 | 0.51 |
| ID3 | inhibitor of DNA binding 3, HLH protein | 0.58 |
| GPNMB | glycoprotein nmb | 0.61 |
| SGK3 | serum/glucocorticoid regulated kinase family member 3 | 0.62 |
| BAG3 | BCL2 associated athanogene 3 | 0.64 |
| ALKBH1 | alkB homolog 1, histone H2A dioxygenase | 0.65 |
| WDR74 | WD repeat domain 74 | 0.65 |
| HIGD1A | HIG1 hypoxia inducible domain family member 1A | 0.66 |
| Gene Symbol 1 | Gene Name | Fold Change |
|---|---|---|
| SIX4 | SIX homeobox 4 | 1.86 |
| KRT80 | keratin 80 | 1.60 |
| GPR37L1 | G protein-coupled receptor 37 like 1 | 1.60 |
| BCL3 | B-cell CLL/lymphoma 3 | 1.55 |
| JAG2 | jagged 2 | 1.53 |
| PLK2 | polo like kinase 2 | 1.53 |
| IGFBP3 | insulin like growth factor binding protein 3 | 1.52 |
| ADARB1 | adenosine deaminase, RNA specific B1 | 1.51 |
| CXCL12 | C-X-C motif chemokine ligand 12 | 0.49 |
| INHBE | inhibin beta E subunit | 0.50 |
| IFIT2 | interferon induced protein with tetratricopeptide repeats 2 | 0.60 |
| GADD45B | growth arrest and DNA damage inducible beta | 0.62 |
| RPS7 | ribosomal protein S7 | 0.64 |
| ERN1 | endoplasmic reticulum to nucleus signaling 1 | 0.64 |
| FCMR | Fc fragment of IgM receptor | 0.64 |
| KRT6B | keratin 6B | 0.64 |
| IFIT3 | interferon induced protein with tetratricopeptide repeats 3 | 0.65 |
| TGFA | transforming growth factor alpha | 0.66 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meligova, A.K.; Siakouli, D.; Stasinopoulou, S.; Xenopoulou, D.S.; Zoumpouli, M.; Ganou, V.; Gkotsi, E.-F.; Chatziioannou, A.; Papadodima, O.; Pilalis, E.; et al. ERβ1 Sensitizes and ERβ2 Desensitizes ERα-Positive Breast Cancer Cells to the Inhibitory Effects of Tamoxifen, Fulvestrant and Their Combination with All-Trans Retinoic Acid. Int. J. Mol. Sci. 2023, 24, 3747. https://doi.org/10.3390/ijms24043747
Meligova AK, Siakouli D, Stasinopoulou S, Xenopoulou DS, Zoumpouli M, Ganou V, Gkotsi E-F, Chatziioannou A, Papadodima O, Pilalis E, et al. ERβ1 Sensitizes and ERβ2 Desensitizes ERα-Positive Breast Cancer Cells to the Inhibitory Effects of Tamoxifen, Fulvestrant and Their Combination with All-Trans Retinoic Acid. International Journal of Molecular Sciences. 2023; 24(4):3747. https://doi.org/10.3390/ijms24043747
Chicago/Turabian StyleMeligova, Aggeliki K., Dimitra Siakouli, Sotiria Stasinopoulou, Despoina S. Xenopoulou, Maria Zoumpouli, Vassiliki Ganou, Eleni-Fani Gkotsi, Aristotelis Chatziioannou, Olga Papadodima, Eleftherios Pilalis, and et al. 2023. "ERβ1 Sensitizes and ERβ2 Desensitizes ERα-Positive Breast Cancer Cells to the Inhibitory Effects of Tamoxifen, Fulvestrant and Their Combination with All-Trans Retinoic Acid" International Journal of Molecular Sciences 24, no. 4: 3747. https://doi.org/10.3390/ijms24043747
APA StyleMeligova, A. K., Siakouli, D., Stasinopoulou, S., Xenopoulou, D. S., Zoumpouli, M., Ganou, V., Gkotsi, E.-F., Chatziioannou, A., Papadodima, O., Pilalis, E., Alexis, M. N., & Mitsiou, D. J. (2023). ERβ1 Sensitizes and ERβ2 Desensitizes ERα-Positive Breast Cancer Cells to the Inhibitory Effects of Tamoxifen, Fulvestrant and Their Combination with All-Trans Retinoic Acid. International Journal of Molecular Sciences, 24(4), 3747. https://doi.org/10.3390/ijms24043747