
CLEC12A Binds to Legionella pneumophila but Has No Impact on the Host’s Antibacterial Response

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
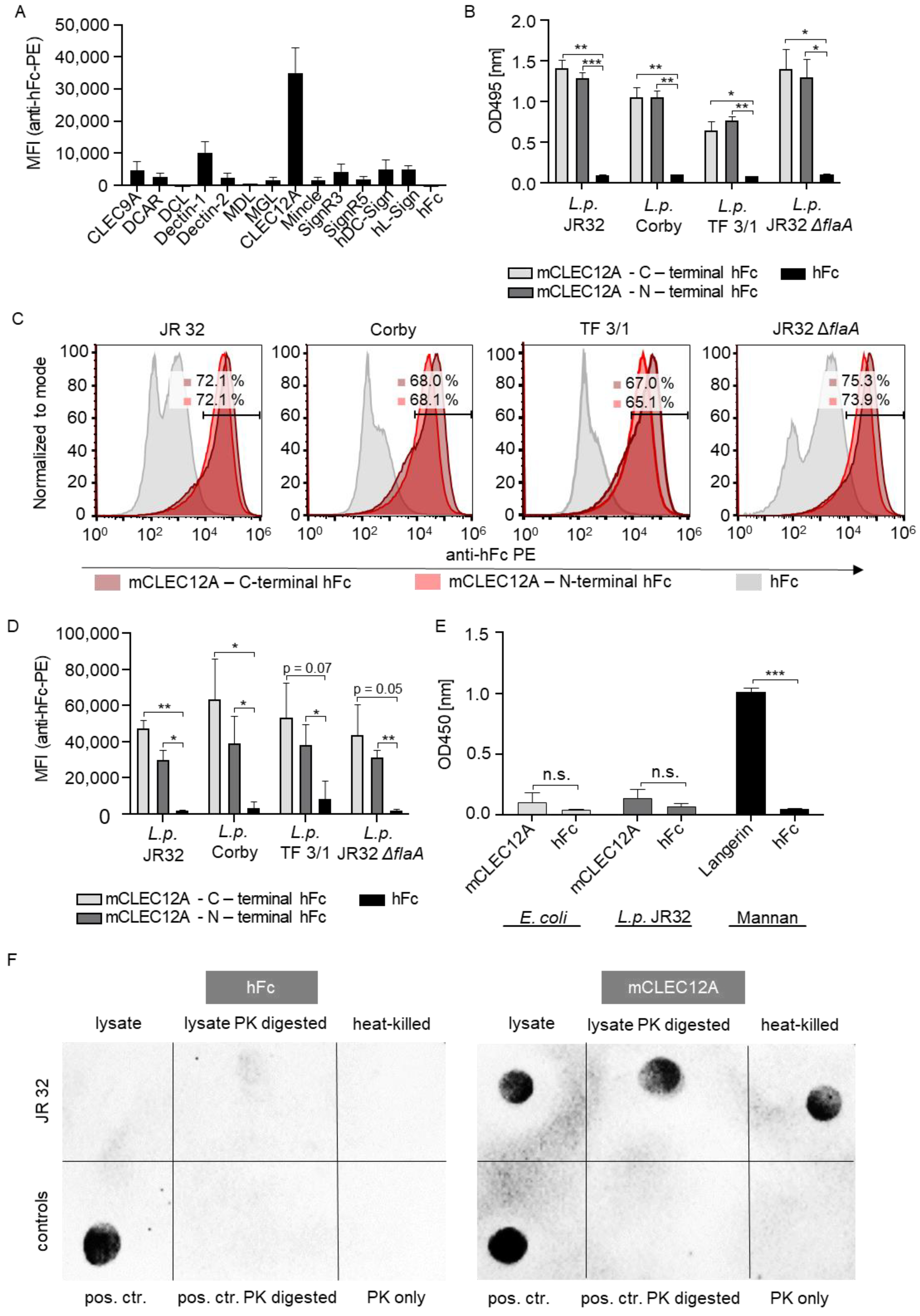
2.1. The CLR CLEC12A Recognizes L. pneumophila
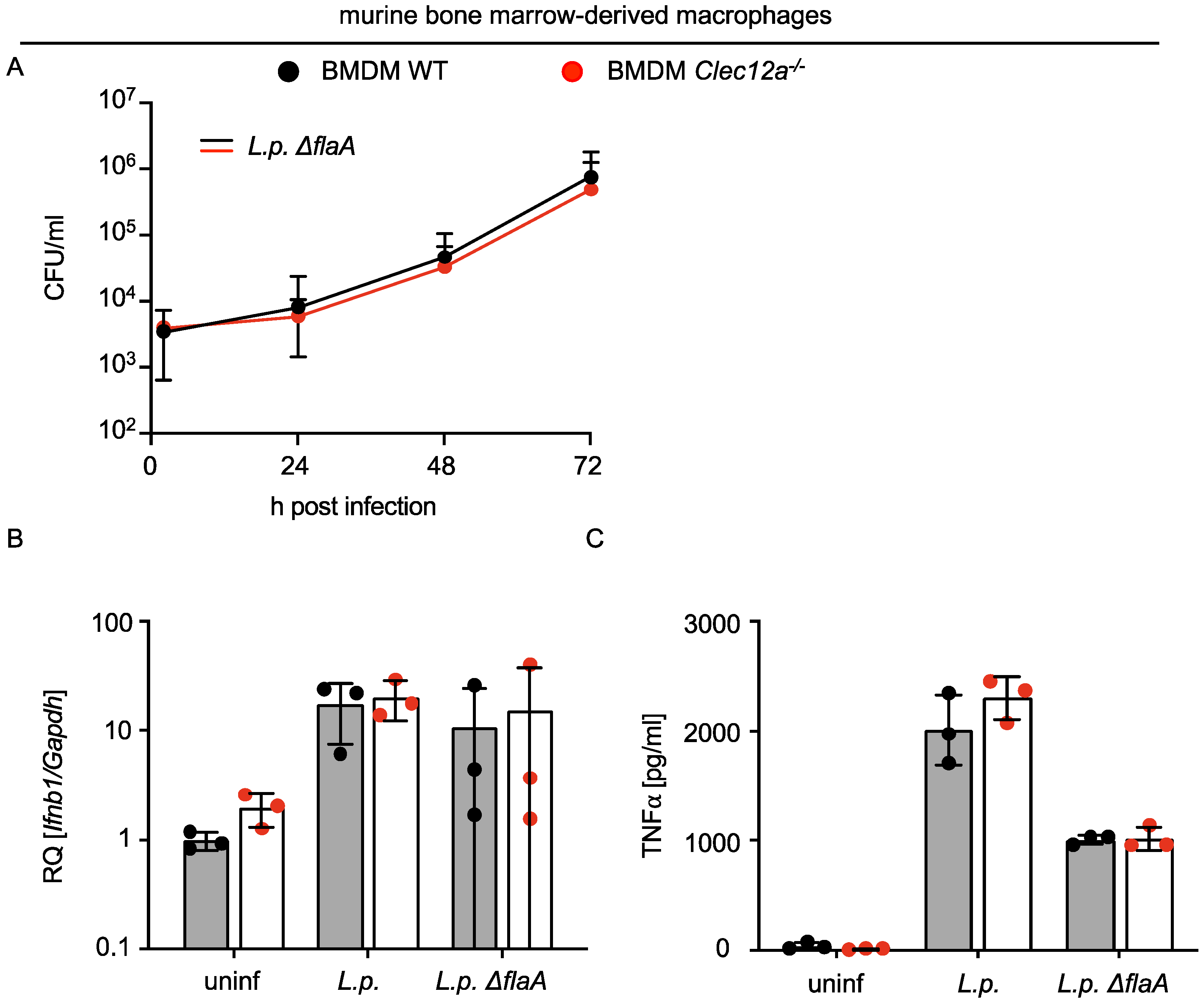
2.2. CLEC12A Does Not Affect the Replication of L. pneumophila in Murine Macrophages and Infection-Induced Cytokine Responses
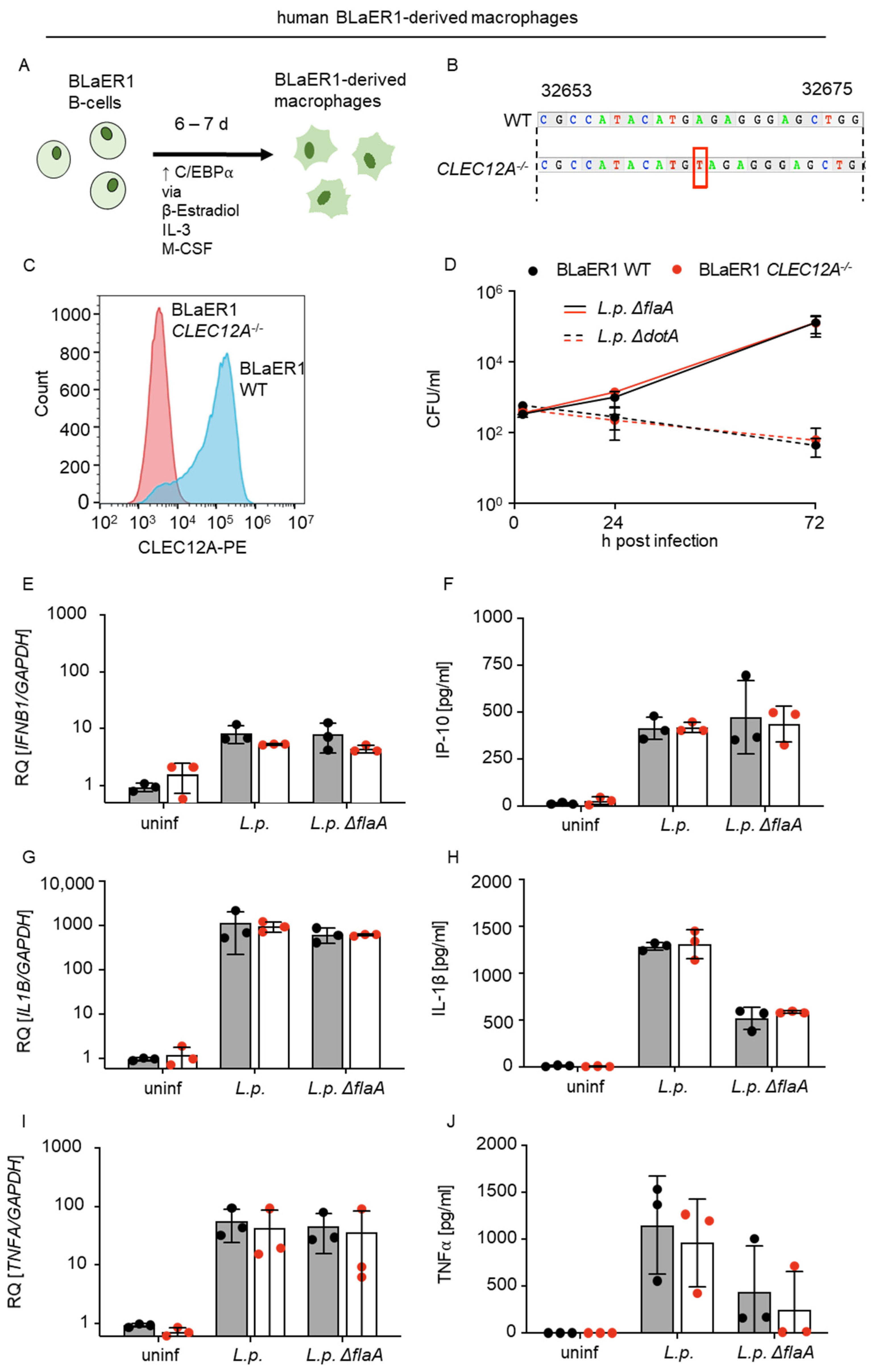
2.3. The Limited Role of Human CLEC12A in L. pneumophila Infection
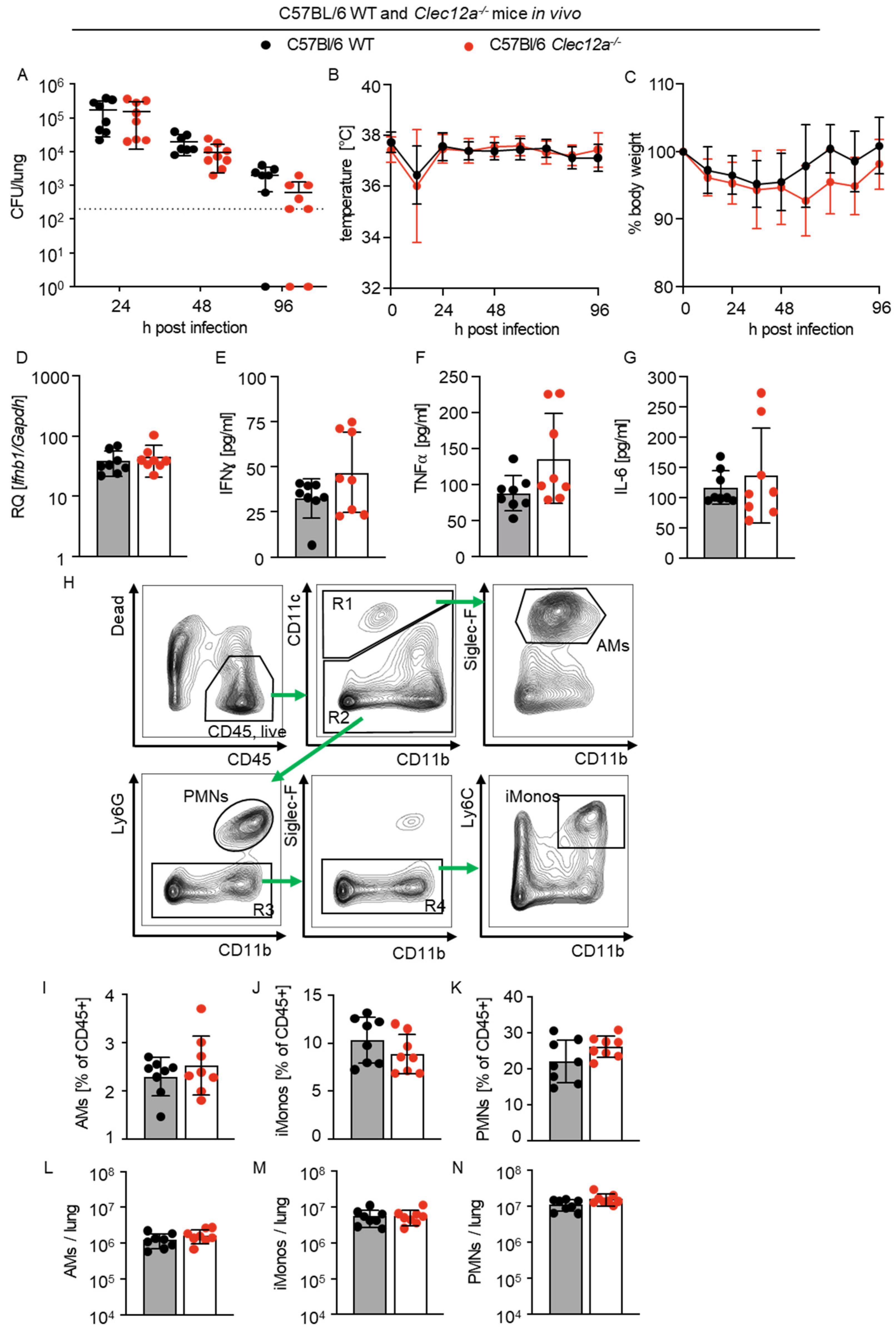
2.4. Role of CLEC12A in Pulmonary L. pneumophila Infection In Vivo
3. Discussion
4. Materials and Methods
4.1. Ethical Statement
4.2. Bacteria and Culturing
4.3. Mice
4.4. Murine Model of Legionnaires’ Disease
4.5. CLR-Fc Fusion Proteins
4.6. ELISA-Based Binding Studies
4.7. Flow Cytometry-Based Binding Studies
4.8. LPS Extraction
4.9. Immunoblotting
4.10. Isolation and Differentiation of Murine BMMs
4.11. Isolation of Human Alveolar Macrophages
4.12. Human BLaER1 Cell Transdifferentiation and Generation of a Human BLaER1
CLEC12A−/− Line
4.13. Short-Term Infection of Murine BMMs and Human BLaER1 Cells
4.14. Quantitative RT-PCR
4.15. ELISA
4.16. Flow Cytometry Assay
4.17. Intracellular Bacterial Replication Assay
4.18. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fields, B.S. The Molecular Ecology of Legionellae. Trends Microbiol. 1996, 4, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Cunha, B.A.; Burillo, A.; Bouza, E. Legionnaires’ Disease. Lancet 2016, 387, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Phin, N.; Parry-Ford, F.; Harrison, T.; Stagg, H.R.; Zhang, N.; Kumar, K.; Lortholary, O.; Zumla, A.; Abubakar, I. Epidemiology and Clinical Management of Legionnaires’ Disease. Lancet Infect. Dis. 2014, 14, 1011–1021. [Google Scholar] [CrossRef]
- Isberg, R.R.; O’Connor, T.; Heidtman, M. The Legionella pneumophila Replication Vacuole: Making a Cozy Niche inside Host Cells. Nat. Rev. Microbiol. 2009, 7, 13–24. [Google Scholar] [CrossRef] [Green Version]
- Horwitz, M.A. Formation of a Novel Phagosome by the Legionnaires’ Disease Bacterium (Legionella pneumophila) in Human Monocytes. J. Exp. Med. 1983, 158, 1319–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubber, A.; Roy, C.R. Modulation of Host Cell Function by Legionella pneumophila Type IV Effectors. Annu. Rev. Cell Dev. Biol. 2010, 26, 261–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asrat, S.; de Jesús, D.A.; Hempstead, A.D.; Ramabhadran, V.; Isberg, R.R. Bacterial Pathogen Manipulation of Host Membrane Trafficking. Annu. Rev. Cell Dev. Biol. 2014, 30, 79–109. [Google Scholar] [CrossRef]
- Naujoks, J.; Lippmann, J.; Suttorp, N.; Opitz, B. Innate Sensing and Cell-Autonomous Resistance Pathways in Legionella pneumophila Infection. Int. J. Med. Microbiol. 2018, 308, 161–167. [Google Scholar] [CrossRef]
- Ruiz-Moreno, J.S.; Hamann, L.; Shah, J.A.; Verbon, A.; Mockenhaupt, F.P.; Puzianowska-Kuznicka, M.; Naujoks, J.; Sander, L.E.; Witzenrath, M.; Cambier, J.C.; et al. The Common HAQ STING Variant Impairs CGAS-Dependent Antibacterial Responses and Is Associated with Susceptibility to Legionnaires’ Disease in Humans. PLoS Pathog. 2018, 14, e1006829. [Google Scholar] [CrossRef]
- Massis, L.M.; Zamboni, D.S. Innate Immunity to Legionella pneumophila. Front. Microbiol. 2011, 2, 109. [Google Scholar] [CrossRef]
- Liu, X.; Shin, S. Viewing Legionella pneumophila Pathogenesis through an Immunological Lens. J. Mol. Biol. 2019, 431, 4321–4344. [Google Scholar] [CrossRef] [PubMed]
- Naujoks, J.; Tabeling, C.; Dill, B.D.; Hoffmann, C.; Brown, A.S.; Kunze, M.; Kempa, S.; Peter, A.; Mollenkopf, H.-J.; Dorhoi, A.; et al. IFNs Modify the Proteome of Legionella-Containing Vacuoles and Restrict Infection Via IRG1-Derived Itaconic Acid. PLoS Pathog. 2016, 12, e1005408. [Google Scholar] [CrossRef] [Green Version]
- Ziltener, P.; Reinheckel, T.; Oxenius, A. Neutrophil and Alveolar Macrophage-Mediated Innate Immune Control of Legionella pneumophila Lung Infection via TNF and ROS. PLoS Pathog. 2016, 12, e1005591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skerrett, S.J.; Bagby, G.J.; Schmidt, R.A.; Nelson, S. Antibody-Mediated Depletion of Tumor Necrosis Factor-a Impairs Pulmonary Host Defenses to Legionella pneumophila. J. Infect. 1997, 176, 1019–1028. [Google Scholar] [CrossRef] [Green Version]
- Zamboni, D.S.; Kobayashi, K.S.; Kohlsdorf, T.; Ogura, Y.; Long, E.M.; Vance, R.E.; Kuida, K.; Mariathasan, S.; Dixit, V.M.; Flavell, R.A.; et al. The Birc1e Cytosolic Pattern-Recognition Receptor Contributes to the Detection and Control of Legionella pneumophila Infection. Nat. Immunol. 2006, 7, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Molofsky, A.B.; Byrne, B.G.; Whitfield, N.N.; Madigan, C.A.; Fuse, E.T.; Tateda, K.; Swanson, M.S. Cytosolic Recognition of Flagellin by Mouse Macrophages Restricts Legionella pneumophila Infection. J. Exp. Med. 2006, 203, 1093–1104. [Google Scholar] [CrossRef] [Green Version]
- Lippmann, J.; Müller, H.C.; Naujoks, J.; Tabeling, C.; Shin, S.; Witzenrath, M.; Hellwig, K.; Kirschning, C.J.; Taylor, G.A.; Barchet, W.; et al. Dissection of a Type I Interferon Pathway in Controlling Bacterial Intracellular Infection in Mice: Role of Type I IFNs in L. pneumophila Infection. Cell. Microbiol. 2011, 13, 1668–1682. [Google Scholar] [CrossRef] [Green Version]
- Prado Acosta, M.; Lepenies, B. Bacterial Glycans and Their Interactions with Lectins in the Innate Immune System. Biochem. Soc. Trans. 2019, 47, 1569–1579. [Google Scholar] [CrossRef]
- Sancho, D.; Reis e Sousa, C. Signaling by Myeloid C-Type Lectin Receptors in Immunity and Homeostasis. Annu. Rev. Immunol. 2012, 30, 491–529. [Google Scholar] [CrossRef] [Green Version]
- Brown, G.D.; Willment, J.A.; Whitehead, L. C-Type Lectins in Immunity and Homeostasis. Nat. Rev. Immunol. 2018, 18, 374–389. [Google Scholar] [CrossRef]
- Marshall, A.S.J.; Willment, J.A.; Pyż, E.; Dennehy, K.M.; Reid, D.M.; Dri, P.; Gordon, S.; Wong, S.Y.C.; Brown, G.D. Human MICL (CLEC12A) Is Differentially Glycosylated and Is down-Regulated Following Cellular Activation. Eur. J. Immunol. 2006, 36, 2159–2169. [Google Scholar] [CrossRef] [Green Version]
- Fischer, S.; Stegmann, F.; Gnanapragassam, V.S.; Lepenies, B. From Structure to Function—Ligand Recognition by Myeloid C-Type Lectin Receptors. Comput. Struct. Biotechnol. J. 2022, 20, 5790–5812. [Google Scholar] [CrossRef] [PubMed]
- Pyż, E.; Huysamen, C.; Marshall, A.S.J.; Gordon, S.; Taylor, P.R.; Brown, G.D. Characterisation of Murine MICL (CLEC12A) and Evidence for an Endogenous Ligand. Eur. J. Immunol. 2008, 38, 1157–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redelinghuys, P.; Whitehead, L.; Augello, A.; Drummond, R.A.; Levesque, J.-M.; Vautier, S.; Reid, D.M.; Kerscher, B.; Taylor, J.A.; Nigrovic, P.A.; et al. MICL Controls Inflammation in Rheumatoid Arthritis. Ann. Rheum. Dis. 2016, 75, 1386–1391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sagar, D.; Singh, N.P.; Ginwala, R.; Huang, X.; Philip, R.; Nagarkatti, M.; Nagarkatti, P.; Neumann, K.; Ruland, J.; Andrews, A.M.; et al. Antibody Blockade of CLEC12A Delays EAE Onset and Attenuates Disease Severity by Impairing Myeloid Cell CNS Infiltration and Restoring Positive Immunity. Sci. Rep. 2017, 7, 2707. [Google Scholar] [CrossRef] [Green Version]
- Neumann, K.; Castiñeiras-Vilariño, M.; Höckendorf, U.; Hannesschläger, N.; Lemeer, S.; Kupka, D.; Meyermann, S.; Lech, M.; Anders, H.-J.; Kuster, B.; et al. Clec12a Is an Inhibitory Receptor for Uric Acid Crystals That Regulates Inflammation in Response to Cell Death. Immunity 2014, 40, 389–399. [Google Scholar] [CrossRef] [Green Version]
- Raulf, M.-K.; Johannssen, T.; Matthiesen, S.; Neumann, K.; Hachenberg, S.; Mayer-Lambertz, S.; Steinbeis, F.; Hegermann, J.; Seeberger, P.H.; Baumgärtner, W.; et al. The C-Type Lectin Receptor CLEC12A Recognizes Plasmodial Hemozoin and Contributes to Cerebral Malaria Development. Cell Rep. 2019, 28, 30–38.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Begun, J.; Lassen, K.G.; Jijon, H.B.; Baxt, L.A.; Goel, G.; Heath, R.J.; Ng, A.; Tam, J.M.; Kuo, S.-Y.; Villablanca, E.J.; et al. Integrated Genomics of Crohn’s Disease Risk Variant Identifies a Role for CLEC12A in Antibacterial Autophagy. Cell Rep. 2015, 11, 1905–1918. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, N.; Tomiyasu, N.; Torigoe, S.; Mizuno, S.; Fukano, H.; Ishikawa, E.; Katano, H.; Hoshino, Y.; Matsuo, K.; Takahashi, M.; et al. Mycobacterial Mycolic Acids Trigger Inhibitory Receptor Clec12A to Suppress Host Immune Responses. Tuberculosis 2023, 138, 102294. [Google Scholar] [CrossRef]
- Maglinao, M.; Eriksson, M.; Schlegel, M.K.; Zimmermann, S.; Johannssen, T.; Götze, S.; Seeberger, P.H.; Lepenies, B. A Platform to Screen for C-Type Lectin Receptor-Binding Carbohydrates and Their Potential for Cell-Specific Targeting and Immune Modulation. J. Control. Release 2014, 175, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Mayer, S.; Moeller, R.; Monteiro, J.T.; Ellrott, K.; Josenhans, C.; Lepenies, B. C-Type Lectin Receptor (CLR)–Fc Fusion Proteins As Tools to Screen for Novel CLR/Bacteria Interactions: An Exemplary Study on Preselected Campylobacter Jejuni Isolates. Front. Immunol. 2018, 9, 213. [Google Scholar] [CrossRef] [Green Version]
- Ricci, M.L.; Fillo, S.; Ciammaruconi, A.; Lista, F.; Ginevra, C.; Jarraud, S.; Girolamo, A.; Barbanti, F.; Rota, M.C.; Lindsay, D.; et al. Genome Analysis of Legionella pneumophila ST23 from Various Countries Reveals Highly Similar Strains. Life Sci. Alliance 2022, 5, e202101117. [Google Scholar] [CrossRef]
- Lück, P.C.; Freier, T.; Steudel, C.; Knirel, Y.A.; Lüneberg, E.; Zähringer, U.; Helbig, J.H. A Point Mutation in the Active Site of Legionella pneumophila O-Acetyltransferase Results in Modified Lipopolysaccharide but Does Not Influence Virulence. Int. J. Med. Microbiol. 2001, 291, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Zhu, W.; Hu, B.-J.; Qu, J.-M.; Luo, Z.-Q. Induction of Rapid Cell Death by an Environmental Isolate of Legionella pneumophila in Mouse Macrophages. Infect. Immun. 2013, 81, 3077–3088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lück, C.; Helbig, J.H. Characterization of Legionella Lipopolysaccharide. In Legionella; Buchrieser, C., Hilbi, H., Eds.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2013; Volume 954, pp. 381–390. ISBN 978-1-62703-160-8. [Google Scholar]
- Rapino, F.; Robles, E.F.; Richter-Larrea, J.A.; Kallin, E.M.; Martinez-Climent, J.A.; Graf, T. C/EBPα Induces Highly Efficient Macrophage Transdifferentiation of B Lymphoma and Leukemia Cell Lines and Impairs Their Tumorigenicity. Cell Rep. 2013, 3, 1153–1163. [Google Scholar] [CrossRef] [Green Version]
- Gaidt, M.M.; Ebert, T.S.; Chauhan, D.; Schmidt, T.; Schmid-Burgk, J.L.; Rapino, F.; Robertson, A.A.B.; Cooper, M.A.; Graf, T.; Hornung, V. Human Monocytes Engage an Alternative Inflammasome Pathway. Immunity 2016, 44, 833–846. [Google Scholar] [CrossRef] [Green Version]
- Gaidt, M.M.; Rapino, F.; Graf, T.; Hornung, V. Modeling Primary Human Monocytes with the Trans–Differentiation Cell Line BLaER1. In Innate Immune Activation; De Nardo, D., De Nardo, C.M., Eds.; Methods in Molecular Biology; Springer New York: New York, NY, USA, 2018; Volume 1714, pp. 57–66. ISBN 978-1-4939-7518-1. [Google Scholar]
- Li, K.; Neumann, K.; Duhan, V.; Namineni, S.; Hansen, A.L.; Wartewig, T.; Kurgyis, Z.; Holm, C.K.; Heikenwalder, M.; Lang, K.S.; et al. The Uric Acid Crystal Receptor Clec12A Potentiates Type I Interferon Responses. Proc. Natl. Acad. Sci. USA 2019, 116, 18544–18549. [Google Scholar] [CrossRef] [Green Version]
- Omotade, T.O.; Roy, C.R. Legionella pneumophila Excludes Autophagy Adaptors from the Ubiquitin-Labeled Vacuole in Which It Resides. Infect. Immun. 2020, 88, e00793-19. [Google Scholar] [CrossRef] [PubMed]
- Choy, A.; Dancourt, J.; Mugo, B.; O’Connor, T.J.; Isberg, R.R.; Melia, T.J.; Roy, C.R. The Legionella Effector RavZ Inhibits Host Autophagy Through Irreversible Atg8 Deconjugation. Science 2012, 338, 1072–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabes, A.; Zimmermann, S.; Reppe, K.; Lang, R.; Seeberger, P.H.; Suttorp, N.; Witzenrath, M.; Lepenies, B.; Opitz, B. The C-Type Lectin Receptor Mincle Binds to Streptococcus pneumoniae but Plays a Limited Role in the Anti-Pneumococcal Innate Immune Response. PLoS ONE 2015, 10, e0117022. [Google Scholar] [CrossRef]
- Behler-Janbeck, F.; Takano, T.; Maus, R.; Stolper, J.; Jonigk, D.; Tort Tarrés, M.; Fuehner, T.; Prasse, A.; Welte, T.; Timmer, M.S.M.; et al. C-Type Lectin Mincle Recognizes Glucosyl-Diacylglycerol of Streptococcus pneumoniae and Plays a Protective Role in Pneumococcal Pneumonia. PLoS Pathog. 2016, 12, e1006038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowalczyk, B.; Chmiel, E.; Palusinska-Szysz, M. The Role of Lipids in Legionella-Host Interaction. Int. J. Mol. Sci. 2021, 22, 1487. [Google Scholar] [CrossRef] [PubMed]
- Johannssen, T.; Lepenies, B. Identification and Characterization of Carbohydrate-Based Adjuvants. In Carbohydrate-Based Vaccines; Lepenies, B., Ed.; Methods in Molecular Biology; Springer New York: New York, NY, USA, 2015; Volume 1331, pp. 173–187. ISBN 978-1-4939-2873-6. [Google Scholar]
- Raulf, M.-K.; Lepenies, B.; Strube, C. Toxocara Canis and Toxocara Cati Somatic and Excretory-Secretory Antigens Are Recognised by C-Type Lectin Receptors. Pathogens 2021, 10, 321. [Google Scholar] [CrossRef] [PubMed]
- Berg, J.; Zscheppang, K.; Fatykhova, D.; Tönnies, M.; Bauer, T.T.; Schneider, P.; Neudecker, J.; Rückert, J.C.; Eggeling, S.; Schimek, M.; et al. Tyk2 as a Target for Immune Regulation in Human Viral/Bacterial Pneumonia. Eur. Respir. J. 2017, 50, 1601953. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klatt, A.-B.; Diersing, C.; Lippmann, J.; Mayer-Lambertz, S.; Stegmann, F.; Fischer, S.; Caesar, S.; Fiocca Vernengo, F.; Hönzke, K.; Hocke, A.C.; et al. CLEC12A Binds to Legionella pneumophila but Has No Impact on the Host’s Antibacterial Response. Int. J. Mol. Sci. 2023, 24, 3891. https://doi.org/10.3390/ijms24043891
Klatt A-B, Diersing C, Lippmann J, Mayer-Lambertz S, Stegmann F, Fischer S, Caesar S, Fiocca Vernengo F, Hönzke K, Hocke AC, et al. CLEC12A Binds to Legionella pneumophila but Has No Impact on the Host’s Antibacterial Response. International Journal of Molecular Sciences. 2023; 24(4):3891. https://doi.org/10.3390/ijms24043891
Chicago/Turabian StyleKlatt, Ann-Brit, Christina Diersing, Juliane Lippmann, Sabine Mayer-Lambertz, Felix Stegmann, Swantje Fischer, Sandra Caesar, Facundo Fiocca Vernengo, Katja Hönzke, Andreas C. Hocke, and et al. 2023. "CLEC12A Binds to Legionella pneumophila but Has No Impact on the Host’s Antibacterial Response" International Journal of Molecular Sciences 24, no. 4: 3891. https://doi.org/10.3390/ijms24043891
APA StyleKlatt, A. -B., Diersing, C., Lippmann, J., Mayer-Lambertz, S., Stegmann, F., Fischer, S., Caesar, S., Fiocca Vernengo, F., Hönzke, K., Hocke, A. C., Ruland, J., Witzenrath, M., Lepenies, B., & Opitz, B. (2023). CLEC12A Binds to Legionella pneumophila but Has No Impact on the Host’s Antibacterial Response. International Journal of Molecular Sciences, 24(4), 3891. https://doi.org/10.3390/ijms24043891