
S-Locus Genotyping in Japanese Plum by High Throughput Sequencing Using a Synthetic S-Loci Reference Sequence




Abstract
:1. Introduction
2. Results and Discussion
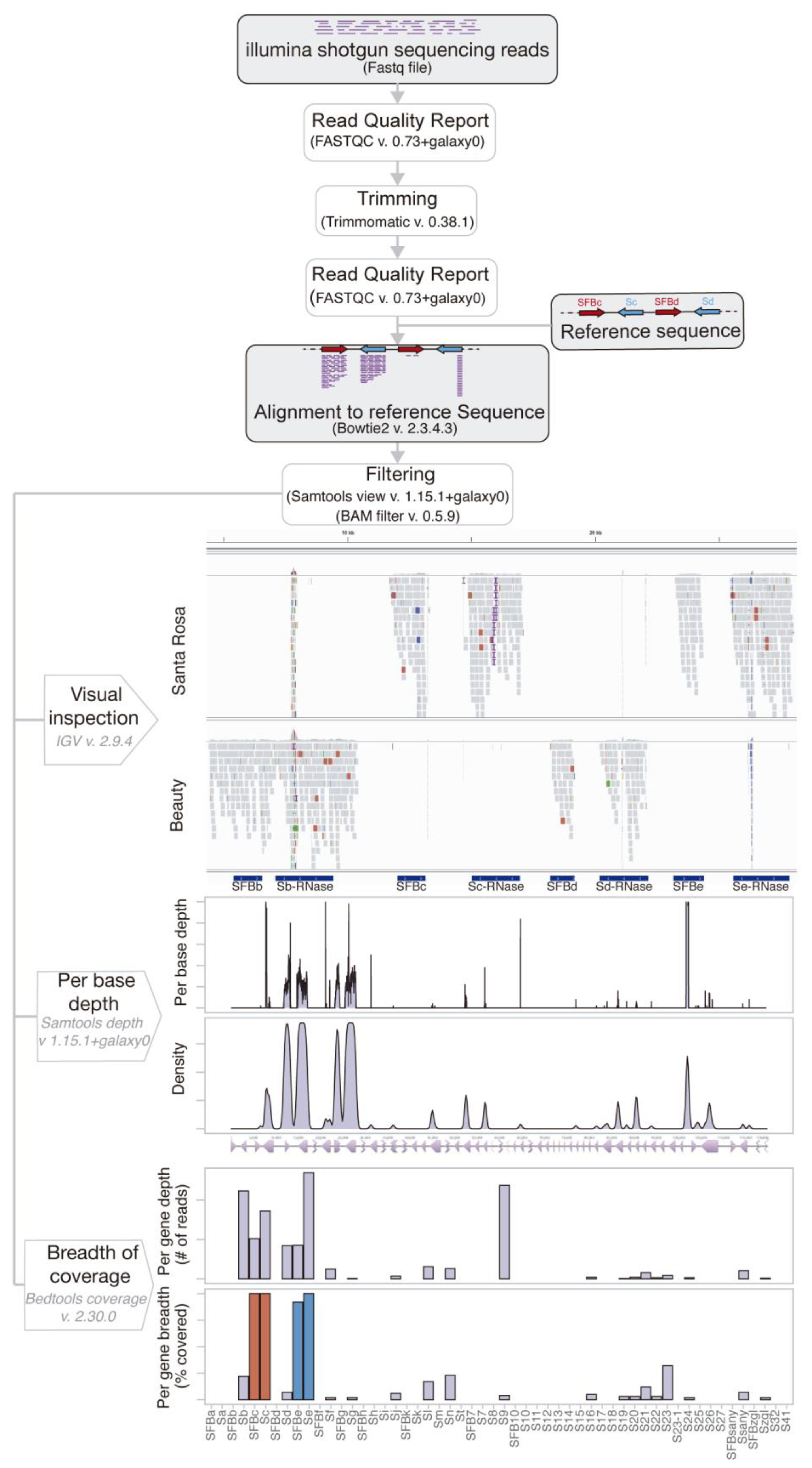
2.1. An S-Locus Based Synthetic Sequence

{kind=link}
{kind=link}
| Allele | Cultivar (S-locus Genotype) | Nucleotide Sequence | Protein Sequence | Coding Sequence (CDS) | Reference | ||
|---|---|---|---|---|---|---|---|
| ID | Length | ID | Length | ||||
| SFBa | Burmosa (SaSb) | AM746961.1 | 992 bp | CAN90151.1 | 331 aa | partial | [40] |
| SFBa | Ozarkpremier (SaSf) | DQ849087.1 | 977 bp | ABI15333.1 | 325 aa | partial | [45] |
| SFBa 1 | Sordum (SaSb) | AB252410.1 | 1131 bp | BAF42763.1 | 376 aa | complete | [48] |
| Sa-RNase 1 | Sordum (SaSb) | AB252411.1 | 1277 bp | BAF42764.1 | 226 aa | complete | [48] |
| S1-RNase | Red-beaut (SaSb) | AF433649.1 | 576 bp | AAP97311.1 | 95 aa | partial | [49] |
| SFBb | Black Golden | KJ396620.1 | 1131 bp | AHX39360.1 | 376 aa | complete | [50] |
| SFBb | Gaixiandali (SbSd) | DQ849088.1 | 978 bp | ABI15334.1 | 326 aa | partial | [45] |
| SFBb | Hamra Bedri | KJ396618.1 | 1131 bp | AHX39358.1 | 376 aa | complete | [50] |
| SFBb | Santa Rosa (ScSe) | KJ396607.1 2 | 1131 bp | AHX39347.1 | 376 aa | complete | [50] |
| SFBb 1 | Sordum (SaSb) | AB252412.1 | 1131 bp | BAF42765.1 | 376 aa | complete | [48] |
| Sb-RNase 1 | Sordum (SaSb) | AB252413.1 | 2332 bp | BAF42766.1 | 221 aa | complete | [48] |
| Sb-RNase | Unknown | DQ646488.1 | 2406 bp | ABG36934.1 | 159 aa | partial | [51] |
| SFBc | Ain Torkia | KJ396613.1 | 1128 bp | AHX39353.1 | 375 aa | complete | [50] |
| SFBc | Bedri1 (SeSh) | KJ396608.1 | 1128 bp | AHX39348.1 | 375 aa | complete | [43,50,52] |
| SFBc | Meiguili (ScSe) | DQ849084.1 | 1128 bp | ABI15330.1 | 375 aa | complete | [47] |
| SFBc 1 | Santa Rosa (ScSe) | AB280792.1 | 2124 bp | BAF91847.1 | 375 aa | complete | [53] |
| Sc-RNase | Oishiwasesumomo (ScSd) | AB084144.1 | 1781 bp | BAC20940.1 | 177 aa | partial | [41] |
| Sc-RNase | Santa Rosa (ScSe) | AB280791.1 | 2630 bp | BAF91846.1 | 230 aa | complete | [53] |
| Sc-RNase | Unknown | DQ646489.1 | 1920 bp | ABG36936.1 | 172 aa | partial | [51] |
| S4-RNase | Royal Zee (ScSe) | AF432418.1 | 1283 bp | AAP97308.1 | 95 aa | partial | [49] |
| SFBd 1 | Formosa (SbSd) | AM746962.1 | 992 bp | CAN90152.1 | 331 aa | partial | [40] |
| Sd-RNase 1 | Oishiwasesumomo (ScSd) | AB084145.1 | 1976 bp | BAC20941.1 | 169 aa | partial | [41] |
| SFBe | Aouina Hamra Bedria (SeS?) | KJ396612.1 | 1131 bp | AHX39352.1 | 376 aa | complete | [50] |
| SFBe | Black Diamant/ Black Diamond (SeSh) | KJ396605.1 | 1128 bp | AHX39345.1 | 375 aa | complete | [43,50] |
| SFBe | Cidre (SaSe) | KJ396611.1 | 1128 bp | AHX39351.1 | 375 aa | complete | [41,50] |
| SFBe | Meiguili (ScSe) | DQ849086.1 | 1036 bp | ABI15332.1 | 345 aa | partial | [45] |
| SFBe | Santa Rosa (ScSe) | AB280794.1 | 1248 bp | BAF91849.1 | 375 aa | partial | [53] |
| SFBe | Stanley | KJ396606.1 3 | 1128 bp | AHX39346.1 | 375 aa | complete | [50] |
| SFBe 1 | Unknown | DQ646490.1 | 1749 bp | ABG36937.1 | 373 aa | complete | [51] |
| Se-RNase 1 | Santa Rosa (ScSe) | AB280793.1 | 2622 bp | BAF91848.1 | 239 aa | complete | [53] |
| S5-RNase | Royal Zee (ScSe) | AF433647.1 | 1553 bp | AAP97309.1 | 95 aa | partial | [49] |
| SFBf 1 | Huangpili (S7Sf) | DQ849089.1 | 972 bp | ABI15335.1 | 324 aa | partial | [45] |
| SFBf | Janha | KJ396610.1 | 996 bp | AHX39350.1 | 331 aa | complete | [50] |
| SFBf | Meski Hamra | KJ396614.1 | 1086 bp | AHX39354.1 | 361 aa | complete | [50] |
| SFBf | Meski Kahla | KJ396617.1 | 1086 bp | AHX39357.1 | 361 aa | complete | [50] |
| SFBf | Unknown | DQ989578.1 | 1081 bp | ABM54900.1 | 360 aa | partial | [49] |
| SFBf | Zaghwenia (ScSf) | KJ396615.1 | 993 bp | AHX39355.1 | 330 aa | complete | [50,52] |
| Sf-RNase 1 | White Plum (SfSg) | AB084147.1 | 1554 bp | BAC20943.1 | 132 aa | partial | [41] |
| Sf-RNase mRNA | Huangpili (S7Sf) | DQ512911.1 | 762 bp | ABF61820.1 | 215 aa | partial | [54] |
| S6-RNase | Wikson | AF433648.1 | 1212 bp | AAP97310.1 | 95 aa | partial | [49] |
| SFBg | Bonnie (SgSh) | AM746963.1 | 992 bp | CAN90153.1 | 331 aa | partial | [40] |
| SFBg 1 | Unknown | DQ989579.1 | 1084 bp | ABM54901.1 | 361 aa | partial | [51] |
| Sg-RNase 1 | Bonnie (SgSh) | AM746950.1 | 1536 bp | CAN90140.1 | 169 aa | partial | [40] |
| Sg-RNase | Honey Rosa (SbSg) | AB093131.1 | 1266 bp | BAC75456.1 | 79 aa | partial | [41] |
| SFBh | Ain Tasstouria | KJ396616.1 | 1125 bp | AHX39356.1 | 374 aa | complete | [50] |
| SFBh | Bedri2 | KJ396619.1 | 1125 bp | AHX39359.1 | 374 aa | complete | [50] |
| SFBh | Jabounia Safra | KJ396609.1 | 1125 bp | AHX39349.1 | 374 aa | complete | [50] |
| SFBh 1 | Nvgelei (ScSh) | DQ849118.1 | 1131 bp | ABI15337.1 | 376 aa | complete | [45] |
| SFBh | Unspecified | DQ646491.1 | 1253 bp | ABG36938.1 | 374 aa | partial | [51] |
| Sh-RNase 1 | Kelsey (SfSh) | AB084148.1 | 1172 bp | BAC20944.1 | 175 aa | partial | [41] |
| Si-RNase 1 | Bakemonosumomo (SbSi) | AB084149.1 | 968 bp | BAC20945.1 | 170 aa | partial | [41] |
| Sj-RNase 1 | Tecumseh (SfSj) | AB093132.1 | 2346 bp | BAC75457.1 | 173 aa | partial | [41] |
| SFBk 1 | Wickson (SfSk) | DQ992485.1 | 1083 bp | ABM54902.1 | 361 aa | partial | [44,51] |
| Sk-RNase | Friar (ShSk) | DQ790372.1 | 374 bp | ABH07013.1 | 63 aa | partial | [55] |
| Sk-RNase 1 | Starkgold (SgSk) | AB093133.1 | 1035 bp | BAC75458.1 | 187 aa | partial | [41,56] |
| Sk-RNase | Wickson (SfSk) | EU113311.1 | 632 bp | ABW86860.1 | 149 aa | partial | [44,57] |
| S3-RNase | Unspecified | AF432417.1 | 467 bp | AAP97307.1 | 95 aa | partial | [58] |
| Sl-RNase 1 | Combination (SgSl) | AB093134 | 1533 bp | BAC75459.1 | 188 aa | partial | [41] |
| Sm-RNase 1 | Botan (SaSm) | AB093135.1 | 930 bp | BAC75460.1 | 171 aa | partial | [41] |
| Sn-RNase 1 | Superior (SaSn) | AB093136 | 843 bp | BAC75461.1 | 121 aa | partial | [41,56] |
| St-RNase 1 | Karari (SbSt) | AB573636.1 | 525 bp | BAJ13374.1 | 175 aa | partial | [59] |
| SFB7 1 | Huangpili (S7Sf) | DQ849085 | 975 bp | ABI15331.1 | 325 aa | partial | [45] |
| S7-RNase 1 | Huangpili (S7Sf) | AY781290.1 | 369 bp | AAV34703.1 | 94 aa | partial | [60] |
| S7-RNase mRNA | Huangpili (S7Sf) | DQ512912.1 | 761 bp | ABF61821.1 | 217 aa | partial | [51] |
| S8-RNase 1 | 82-1-109 | AY902455.1 | 450 bp | AAW83158.1 | 94 aa | partial | [60] |
| S8-RNase mRNA | 82-1-109 | DQ512913.1 | 750 bp | ABF61822.1 | 213 aa | partial | [51] |
| S9-RNase 1 | Tai Ping Guo | AY996051.1 | 815 bp | AAY15124.1 | 94 aa | partial | [60] |
| SFB10 1 | Mali (S10S14) | DQ849090.1 | 1041 bp | ABI15336.1 | 347 aa | partial | [45] |
| S10-RNase 1 | Hong Men Ling | DQ003310.1 | 880 bp | AAX98270.1 | 94 aa | partial | [60] |
| S11-RNase 1 | Zhushali | DQ512908.1 | 670 bp | ABF61817.1 | 119 aa | partial | [51] |
| S12-RNase 1 | Kongquedan (S12S13) | DQ512909.1 | 197 bp | ABF61818.1 | 65 aa | partial | [51] |
| S13-RNase 1 | Kongquedan (S12S13) | DQ512910.1 | 191 bp | ABF61819.1 | 63 aa | partial | [51] |
| S14-RNase 1 | Mali (S10S14) | EF177345.1 | 194 bp | ABM53475.1 | 37 aa | partial | [45] |
| S15-RNase 1 | Kuandiandali (S15S16) | EF177346.1 | 571 bp | ABM53476.1 | 39 aa | partial | [61] |
| S16-RNase 1 | Kuandiandali (S15S16) | EF177347.1 | 460 bp | ABM53477.1 | 39 aa | partial | [61] |
| S17-RNase 1 | Xinjiangsanhao (S17S19) | EF177348.1 | 261 bp | ABM53478.1 | 39 aa | partial | [61] |
| S18-RNase 1 | Danchengxingmei (S18S20) | EF177349.1 | 413 bp | ABM53479.1 | 38 aa | partial | [61] |
| S19-RNase 1 | Xinjiangsanhao (S17S19) | EU113259.1 | 690 bp | ABW88922.1 | 55 aa | partial | [62] |
| S20-RNase 1 | Danchengxingmei (S18S20) | EU113260.1 | 1925 bp | ABW88923.1 | 55 aa | partial | [62] |
| S21-RNase 1 | Zhenzhuli (S21S22) | EU113261.1 | 1601 bp | ABW88924.1 | 116 aa | partial | [62] |
| S22-RNase 1 | Zhenzhuli (S21S22) | EU113262.1 | 853 bp | ABW88925.1 | 117 aa | partial | [62] |
| S23-RNase 1 | Dameigui (S23S24) | EU113263.1 | 750 bp | ABW88926.1 | 54 aa | partial | [62] |
| S23-RNase 1 | Guangdonghongli | FJ377732.1 4 | 597 bp | ACI88749.1 | 64 aa | partial | [63] |
| S24-RNase 1 | Dameigui (S23S24) | EU113264.1 | 1155 bp | ABW88927.1 | 117 aa | partial | [62] |
| S25-RNase 1 | Unspecified | EU113265.1 | 920 bp | ABW88928.1 | 116 aa | partial | [62] |
| S26-RNase 1 | Unspecified | EU113266.1 | 1492 bp | ABW88929.1 | 117 aa | partial | [62] |
| S27-RNase 1 | Unspecified | EU113267.1 | 657 bp | ABW88930.1 | 54 aa | partial | [62] |
| S32-RNase 1 | Guali | GU574195.1 5 | 898 bp | ADD20972.1 | 100 aa | partial | [64] |
| S41-RNase 1 | Qingzhouli | GU968758.1 | 719 bp | ADF42685.1 | 121 aa | partial | [65] |
2.2. A Powerful S-Allele Calling Procedure

2.3. S-Allele Genotypes, Methodology Validation, and Incompatibility Groups

| I. G. | S-alleles | Cultivar | Origin 1 | Library | Reference |
|---|---|---|---|---|---|
| I | SaSb | Sordum | Japan | E | [35] |
| II | SbSc | Fortune | California, USA | G | [68] |
| Zaoshengyueguang | Japan | G | |||
| Dashizhongsheng | Japan | G | |||
| Taiyo | Japan | E | [41] | ||
| III | SbSf | Zhenyuanqiyuexiang | Guizhou China | G | |
| IV | SbSh | Friar | California, USA | G | [68] |
| Friar 2 | California, USA | G | [55] (ShSk) | ||
| Qiuji | Japan | G | [45] | ||
| Guofeng7 | Liaoning, China | G | |||
| C20 | Spain | G | |||
| Zhongly nº 6 | Northern China | G | |||
| V | SbSi | Fujianfurongli | Fujian, China | G | |
| VI | SfSh | Satsuma | California, USA | G | |
| VII | ScSh | Fulihong | Shaanxi, China | G | |
| Angeleno | California, USA | G | [43] | ||
| IX | SfSg | Jinshali | Yunnan, China | G | [45] |
| Cehengjixueli | Guizhou, China | G | |||
| XIV | SaSc | C46 | Spain | G | |
| XIX | SbSd | Abazhoumeiguili | Sichuan, China | G | |
| Beauty 2 | California, USA | G | [41] (ScSe) | ||
| XXI | SeSk | Wuxiangli | Hebei, China | G | |
| Changlixiangjiaoli | Hebei, China | G | |||
| Changlixiangbian | Hebei, China | G | |||
| XXIII | ScSd | Dashizaosheng | Japan | G | |
| Jingshang | Japan | G | |||
| Oishiwase | Japan | G | [41] | ||
| Oishiwase | Japan | E | [41] | ||
| XXIV | SeS11 | Jinxiqiuli | Liaoning, China | G | |
| Qiyuexiang | Liaoning, China | G | |||
| XXV | S15S16 | Pingguoli | Liaoning, China | G | [46] |
| XXVII 3 | SbS7 | Heyuansanhuali | Guangdong, China | G | |
| Qingpihongxin | Guangdong, China | G | |||
| Zaohuangli | Anhui, China | G | |||
| Jinganzhushali | Jiangxi, China | G | |||
| Jaiqingzi | Jiangsu, China | G | |||
| XXVII 3 | SbS8 | Huahongli | Yunnan, China | G | |
| Brace | California, USA | G | |||
| XXIX 3 | SeS10/32 | Shuili | Guangxi, China | G | |
| Dahuili | Henan, China | G | |||
| Xiangjiaoli | Liaoning, China | G | |||
| Qiuxiang | Liaoning, China | G | |||
| XXX 3 | SgSk | Yuanshuai | Japan | G | |
| XXXI 3 | ShS10/32 | Guofeng2 | Liaoning, China | G | |
| N2 Guofeng | Liaoning, China | G | |||
| XXXII 3 | SiS9 | Changkuangsanhuali | Guangdong, China | G | |
| Sanhua plum | Guangdong, China | G | |||
| XXXIII 3 | S10/32S11 | Tiankanmali | Sichuan, China | G | |
| Li He | China | G | |||
| Wushan plum | Chongging, China | G | |||
| Wanshuang plum | Chongging, China | G | |||
| XXXIV 3 | S11S20 | Yuhuangli | Shaanxi, China | G | |
| Huangli | Shandong, China | G | |||
| Lihe | Hebei, China | G | |||
| XXXV 3 | SbScSf | Guiyang | Japan | G | |
| Group 0 | SaSl | Hongshou | Japan | G | |
| SbS11 | Yanzhili | Fujian, China | G | ||
| SdSg | Fenghuali | Zhejiang, China | G | ||
| SeS12 | Huangguli | Zhejiang, China | G | ||
| SeS20 | Daqingke | Shandong, China | G | ||
| ShS15 | Guofeng17 | Liaoning, China | G | ||
| ShS7 | Zuili | Zhejiang, China | G | [46] | |
| SkS8 | Bullbank | California, USA | G | ||
| S7S20 | Pingdingxiang | Shandong, China | G | ||
| S8S9 | Wanshuhuanai | Fujian, China | G | ||
| S10/32S15 | Zhengzhouzaoli | Henan, China | G | ||
| S10/32Ssany | Sanyueli | Guangdong, China | G | ||
| Sanyueli | Guangdong, China | T | |||
| S10/32Szgl | Cuihongli | Sichuan, China | G | ||
| Cuihong plum | Sichuan, China | G | |||
| S11S16 | Saozouli | Guangxi, China | G | ||
| SeSkS10/32S11 | Xingyikongxinli | Guizhou, China | G | ||
| Cultivars in which only one S allele could be identified | |||||
| Sb | Cuipinwannai | Fujian, China | G | ||
| Sc | Guoli | Liaoning, China | G | ||
| Se | Tianmumili | Zhejiang, China | G | ||
| Se | Hongxinli | Anhui, China | G | ||
| Sh | Guomei | Liaoning, China | G | ||
| S10/32 | Yinhong plum | Sichuan, China | G | ||
| S10/32 | Fengtang plum | Guizhou, China | G | ||
| S11 | Tongkeli | Guangxi, China | G | ||
| S19 | Non-specified | Tibet, China | G | ||
| S21 | Damili | Guangdong, China | G | ||
| S22 | Zhushali | Yunnan, China | G | ||
| Cultivars in which no S allele could be identified | |||||
| Kuaishili | Guangdong, China | G | |||
| Lushanli | Jiangxi, China | G | |||
| Ni Ma Qu Ji Li | China | G | |||
| Wuyuecui | Sichuan, China | G | |||
| Self-compatible cultivars | |||||
| SC | SbSg | Honeyrosa | Japan | E | [42] |
| SC | ScSe | Santa Rosa | California, USA | G | [68] |
3. Materials and Methods
3.1. Building the Reference Synthetic S-Loci Sequence
3.2. Alignment to Reference Sequence
3.3. S-Allele Typing Methodology
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- de Nettancourt, D. Incompatibility in Angiosperms; Springer: Berlin/Heidelberg, Germany, 1977. [Google Scholar]
- Steinbachs, J.E.; Holsinger, K.E. S-RNase-medited Gametophytic Self-Incompatibility is Ancestral in Eudicots. Mol. Biol. Evol. 2002, 19, 825–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Nettancourt, D. Incompatibility and Incongruity in Wild and Cultivated Plants; Springer: Berlin/Heidelberg, Germany, 2001. [Google Scholar]
- Uyenoyama, M.K. Evolutionary Dynamics of Self-Incompatibility Alleles in Brassica. Genetics 2000, 156, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Wright, S. The distribution of self-sterility alleles in populations. Genetics 1939, 24, 538. [Google Scholar] [CrossRef]
- Wu, J.; Gu, C.; Khan, M.A.; Gao, Y.; Wang, C.; Korban, S.S.; Zhang, S. Molecular Determinants and Mechanisms of Gametophytic Self-Incompatibility in Fruit Trees of Rosaceae. CRC Crit. Rev. Plant Sci. 2013, 32, 53–68. [Google Scholar] [CrossRef]
- Ushijima, K.; Sassa, H.; Dandekar, A.M.; Gradziel, T.M.; Tao, R.; Hirano, H. Structural and Transcriptional Analysis of the Self-Incompatibility Locus of Almond: Identification of a Pollen-Expressed F-Box Gene with Haplotype-Specific Polymorphism. Plant Cell 2003, 15, 771–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, R.; Yamane, H.; Sassa, H.; Mori, H.; Gradziel, T.M.; Dandekar, A.M.; Sugiura, A. Identification of Stylar RNases Associated with Gametophytic Self-Incompatibility in Almond (Prunus dulcis). Plant Cell Physiol. 1997, 38, 304–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, R.; Yamane, H.; Sugiura, A.; Murayama, H.; Sassa, H.; Mori, H. Molecular Typing of S-alleles through Identification, Characterization and cDNA Cloning for S-RNases in Sweet Cherry. J. Am. Soc. Hortic. Sci. 1999, 124, 224–233. [Google Scholar] [CrossRef] [Green Version]
- McCubbin, A.G.; Kao, T. Molecular Recognition and Response in Pollen and Pistil Interactions. Annu. Rev. Cell Dev. Biol. 2000, 16, 333–364. [Google Scholar] [CrossRef]
- Castric, V.; Vekemans, X. Invited Review: Plant self-incompatibility in natural populations: A critical assessment of recent theoretical and empirical advances. Mol. Ecol. 2004, 13, 2873–2889. [Google Scholar] [CrossRef]
- Halász, J.; Molnár, A.B.; Ilhan, G.; Ercisli, S.; Hegedüs, A. Identification and Molecular Analysis of Putative Self-Incompatibility Ribonuclease Alleles in an Extreme Polyploid Species, Prunus laurocerasus L. Front. Plant Sci. 2021, 12, 715414. [Google Scholar] [CrossRef]
- Herrera, S.; Lora, J.; Hormaza, J.I.; Rodrigo, J. Pollination Management in Stone Fruit Crops. In Production Technology of Stone Fruits; Mir, M.M., Iqbal, U., Mir, S.A., Eds.; Springer: Singapore, 2021; pp. 75–102. [Google Scholar]
- Guerra, M.E.; Rodrigo, J. Japanese plum pollination: A review. Sci. Hortic. 2015, 197, 674–686. [Google Scholar] [CrossRef]
- Guerra, M.E.; Guerrero, B.I.; Casadomet, C.; Rodrigo, J. Self-(in)compatibility, S-RNase allele identification, and selection of pollinizers in new Japanese plum-type cultivars. Sci. Hortic. 2020, 261, 109022. [Google Scholar] [CrossRef]
- Busch, J.W.; Schoen, D.J. The evolution of self-incompatibility when mates are limiting. Trends Plant Sci. 2008, 1s, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Pickup, M.; Brandvain, Y.; Fraïsse, C.; Yakimowski, S.; Barton, N.H.; Dixit, T.; Lexer, C.; Cereghetti, E.; Field, D.L. Mating system variation in hybrid zones: Facilitation, barriers and asymmetries to gene flow. New Phytol. 2019, 224, 1035–1047. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.R. Techniques used in fruit-set experiments. In Towards Regulated Cropping; Williams, R.R., Wilson, D., Eds.; Grower Books: London, UK, 1970; pp. 57–61. [Google Scholar]
- Herrero, M.; Dickinson, H.G. Pollen tube growth following compatible and incompatible intraspecific pollinations in Petunia hybrida. Planta 1980, 148, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Rodrigo, J.; Herrero, M. Evaluation of pollination as the cause of erratic fruit set in apricot ‘Moniqui’. J. Hortic. Sci. 1996, 71, 801–805. [Google Scholar] [CrossRef]
- Sanzol, J.; Herrero, M. Identification of self-incompatibility alleles in pear cultivars (Pyrus communis L.). Euphytica 2002, 128, 325–331. [Google Scholar] [CrossRef]
- Guerra, M.E.; Wünsch, A.; López-Corrales, M.; Rodrigo, J. Lack of Fruit Set Caused by Ovule Degeneration in Japanese plum. J. Am. Soc. Hortic. Sci. 2011, 136, 375–381. [Google Scholar] [CrossRef] [Green Version]
- Bošković, R.; Tobutt, K.R. Correlation of Stylar Ribonuclease Zymograms with Incompatibility Alleles in Sweet Cherry. Euphytica 1996, 90, 245–250. [Google Scholar] [CrossRef]
- Sassa, H.; Nishio, T.; Kowyama, Y.; Hirano, H.; Koba, T.; Ikehashi, H. Self-incompatibility (S) alleles of the Rosaceae encode members of a distinct class of the T2/S ribonuclease superfamily. Mol. Gen. Genet. 1996, 250, 547–557. [Google Scholar]
- Halász, J.; Hegedüs, A. A critical evaluation of methods used for S-genotyping: From trees to DNA level. Int. J. Hortic. Sci. 2006, 12, 19–29. [Google Scholar] [CrossRef]
- Yamane, H.; Tao, R. Molecular Basis of Self-(in)compatibility and Current Status of S-genotyping in Rosaceous Fruit Trees. J. Jpn. Soc. Hortic. Sci. 2009, 78, 137–157. [Google Scholar] [CrossRef] [Green Version]
- Sutherland, B.G.; Robbins, T.P.; Tobutt, K.R. Primers amplifying a range of Prunus S-alleles. Plant Breed. 2004, 123, 582–584. [Google Scholar] [CrossRef]
- Delorme, V.; Gaude, T.; Heizmann, P.; Dumas, C. Use of Inmunochemical and SSCP analyses to test homozygosity at the S Locus of Brassica oleracea genotypes. Mol. Breed. 1995, 1, 237–244. [Google Scholar] [CrossRef]
- Jorgensen, R.A. Plant Genetics and Genomics: Crops and Models; Springer: Berlin/Heidelberg, Germany, 2014. [Google Scholar]
- Mable, B.K.; Hagmann, J.; Kim, S.T.; Adam, A.; Kilbride, E.; Weigel, D.; Stift, M. What causes mating system shifts in plants? Arabidopsis lyrata as a case study. Heredity 2017, 118, 52–63. [Google Scholar]
- Mable, B.K.; Brysting, A.K.; Jørgensen, M.H.; Carbonell, A.K.Z.; Kiefer, C.; Ruiz-Duarte, P.; Lagesen, K.; Koch, M.A. Adding Complexity to Complexity: Gene Family Evolution in Polyploids. Front. Ecol. Evol. 2018, 6, 114. [Google Scholar] [CrossRef] [Green Version]
- Tsuchimatsu, T.; Goubet, P.M.; Gallina, S.; Holl, A.C.; Fobis-Loisy, I.; Bergès, H.; Marande, W.; Prat, E.; Meng, D.; Long, Q.; et al. Patterns of Polymorphism at the Self-Incompatibility Locus in 1083 Arabidopsis thaliana Genomes. Mol. Biol. Evol. 2017, 34, 1878–1889. [Google Scholar] [CrossRef] [Green Version]
- De Franceschi, P.; Bianco, L.; Cestaro, A.; Dondini, L.; Velasco, R. Characterization of 25 full-length S-RNase alleles, including flanking regions, from a pool of resequenced apple cultivars. Plant Mol. Biol. 2018, 97, 279–296. [Google Scholar] [CrossRef] [Green Version]
- Genete, M.; Castric, V.; Vekemans, X. Genotyping and De Novo Discovery of Allelic Variants at the Brassicaceae Self-Incompatibility Locus from Short-Read Sequencing Data. Mol. Biol. Evol. 2020, 37, 1193–1201. [Google Scholar] [CrossRef]
- Yamane, H.; Tao, R.; Sigiura, A. Identification an cDNA Cloning for S-RNases in Self-incompatible Japanese Plum (Prunus salicina Lindl. cv. Sordum). Plant Biotechnol. 1999, 16, 389–396. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, M.J. Population Genetics of the Homomorphic Self-incompatibility Polymorphisms in Flowering Plants. Ann. Bot. 2000, 85, 221–226. [Google Scholar] [CrossRef] [Green Version]
- Igic, B.; Bohs, L.; Kohn, J.R. Historical inferences from the self-incompatibility locus. New Phytol. 2004, 161, 97–105. [Google Scholar] [CrossRef]
- Richman, A.D.; Kohn, J.R. Learning from rejection: The evolutionary biology of single-locus incompatibility. Trends Ecol. Evol. 1996, 11, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Ioerger, T.R.; Clark, A.G.; Kao, T. Polymorphism at the self-incompatibility locus in Solanaceae predates speciation. Proc. Natl. Acad. Sci. USA 1990, 87, 9732–9735. [Google Scholar] [CrossRef] [Green Version]
- Sutherland, B.G.; Tobutt, K.R.; Robbins, T.P. Trans-specific S-RNase and SFB alleles in Prunus self-incompatibility haplotypes. Mol. Genet. Genom. 2008, 279, 95–106. [Google Scholar] [CrossRef]
- Beppu, K.; Yamane, H.; Yaegaki, H.; Yamaguchi, M.; Kataoka, I.; Ryutaro, T. Diversity of S-RNase genes and S-haplotypes in Japanese plum (Prunus salicina Lindl.). J. Hortic. Sci. Biotechnol. 2002, 77, 658–664. [Google Scholar] [CrossRef]
- Beppu, K.; Takemoto, Y.; Yamane, H.; Yaegaki, H.; Yamaguchi, M.; Kataoka, I.; Tao, R. Determination of S-haplotypes of Japansese plum (Prunus salicina Lindl.) cultivars by PCR and cross-pollination test. J. Hortic. Sci. Biotechnol. 2003, 78, 315–318. [Google Scholar] [CrossRef]
- Sapir, G.; Stern, R.A.; Eisikowitch, D.; Goldway, M. Cloning of four new Japanese plum S-alleles and determination of the compatibility between cultivars by PCR analysis. J. Hortic. Sci. Biotechnol. 2004, 79, 223–227. [Google Scholar] [CrossRef]
- Sapir, G.; Stern, R.A.; Goldway, M.; Shafir, S. SFBs of Japanese Plum (Prunus salicina): Cloning Seven Alleles and Determining Their Linkage to the S-RNase Gene. HortScience 2007, 42, 1509–1512. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.L.; Huang, S.X.; Kitashiba, H.; Nishio, T. Identification of S-haplotype-specific F-box gene in Japanese plum (Prunus salicina Lindl.). Sex. Plant Reprod. 2007, 20, 1–8. [Google Scholar] [CrossRef]
- Zhang, S.J.; Huang, S.X.; Heng, W.; Wu, H.Q.; Wu, J.; Zhang, S.L. Identification of S-genotypes in 17 Chinese cultivars of Japanese plum (Prunus salicina Lindl.) and molecular characterisation of 13 novel S-alleles. J. Hortic. Sci. Biotechnol. 2008, 83, 635–640. [Google Scholar] [CrossRef]
- Ikeda, K.; Igic, B.; Ushijima, K.; Yamane, H.; Hauck, N.R.; Nakano, R.; Sassa, H.; Iezzoni, A.F.; Kohn, J.; Tao, R. Primary structural features of the S haplotype-specific F-box protein, SFB, in Prunus. Sex. Plant Reprod. 2004, 15, 235–243. [Google Scholar] [CrossRef]
- Tao, R.; Watari, A.; Hanada, T.; Habu, T.; Yaegaki, H.; Yamaguchi, M.; Yamane, H. Self-compatible peach (Prunus persica) has mutant versions of the S-haplotypes found in self-incompatible Prunus species. Plant Mol. Biol. 2007, 63, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Sapir, G.; Goldway, M. Prunus Salicina S-RNases. Nucleotide Sequences AF432418.1, AF433647.1, AF433648.1, AF433649.1. Available online: https://www.ncbi.nlm.nih.gov/nuccore (accessed on 28 December 2022).
- Abdallah, D.; Baraket, G.; Ben Tamarzizt, H.; Ben Mustapha, S.; Salhi Hannachi, A. Identification, Evolutionary Patterns and Intragenic Recombination of the Gametophytic Self Incompatibility Pollen Gene (SFB) in Tunisian Prunus Species (Rosaceae). Plant Mol. Biol. Report. 2016, 34, 339–352. [Google Scholar]
- Sapir, G.; Goldway, M. Identification of F-box Gene in Prunus Salicina. Nucleotide Sequences AF432417.1, AF433649.1, DQ646488.1, DQ646489.1, DQ646490.1, DQ646491.1, DQ989578.1, DQ989579.1, DQ992485.1. Available online: https://www.ncbi.nlm.nih.gov/nuccore (accessed on 28 December 2022).
- Abdallah, D.; Baraket, G.; Perez, V.; Ben Mustapha, S.; Salhi-Hannachi, A.; Hormaza, J.I. Analysis of self-incompatibility and genetic diversity in diploid and hexaploid plum genotypes. Front. Plant Sci. 2019, 10, 896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watari, A.; Hanada, T.; Yamane, H.; Esumi, T.; Tao, R.; Yaegaki, H.; Yamaguchi, M.; Beppu, K.; Kataoka, I. A Low Transcriptional Level of se-RNase in the Se-haplotype Confers Self-compatibility in Japanese Plum. J. Am. Soc. Hortic. Sci. 2007, 132, 396–406. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.L.; Huang, S.X.; Zhang, S.J.; Wu, J.; Wu, H.Q.; Heng, W.; Zhang, Y.Y. Prunus Salicina S-RNase Genes. Nucleotide Sequences DQ512908.1, DQ512909.1, DQ512910.1, DQ512911.1, DQ512912.1, DQ512913.1. Available online: https://www.ncbi.nlm.nih.gov/nuccore (accessed on 28 December 2022).
- Halász, J.; Hegedus, A.; Szabó, Z.; Nyéki, J.; Pedryc, A. DNA-based S-genotyping of Japanese Plum and Pluot Cultivars to Clarify Incompatibility Relationships. HortScience 2007, 42, 46–50. [Google Scholar] [CrossRef] [Green Version]
- Beppu, K.; Syogase, K.; Yamane, H.; Tao, R.; Kataoka, I. Inheritance of self-compatibility conferred by the Se-haplotype of Japanese plum and development of Se-RNase gene-specific PCR primers. J. Hortic. Sci. Biotechnol. 2010, 85, 215–218. [Google Scholar] [CrossRef]
- Sapir, G.; Goldway, M.; Stern, R.A.; Shafir, S. S-RNase Based S-genotyping of Japanese Plum (Prunus salicina Lindl.) Cultivars. Nucleotide Sequence EU113311.1. Available online: https://www.ncbi.nlm.nih.gov/nuccore (accessed on 28 December 2022).
- Sapir, G.; Goldway, M. Prunus salicina S-RNases. Nucleotide Sequence AF432417.1. Available online: https://www.ncbi.nlm.nih.gov/nuccore (accessed on 28 December 2022).
- Beppu, K.; Kumai, M.; Yamane, H.; Tao, R.; Kataoka, I. Molecular and genetic analyses of the S-haplotype of the self-compatible Japanese plum (Prunus salicina Lindl.) ‘Methley’. J. Hortic. Sci. Biotechnol. 2012, 87, 493–498. [Google Scholar] [CrossRef]
- Huang, S.X.; Zhang, S.L.; Zhang, Y.Y.; Wu, J.; Heng, W.; Zhao, C.P. Identification of New S Genotype in Prunus salicina. Nucleotide Sequences AY781290.1, AY902455.1, AY996051.1, DQ003310.1. Available online: https://www.ncbi.nlm.nih.gov/nuccore (accessed on 28 December 2022).
- Zhang, S.L.; Zhang, S.J.; Huang, S.X.; Wu, H.Q.; Wu, J. Identification of Self-Incompatibility Genotypes of Japanese Plum. Nucleotide Sequences EF177346.1, EF177347.1, EF177348.1, EF177349.1. Available online: https://www.ncbi.nlm.nih.gov/nuccore (accessed on 28 December 2022).
- Zhang, S.L.; Zhang, S.J.; Huang, S.X.; Heng, W.; Wu, H.Q.; Wu, J. Identification of Self-Incompatibility Genotypes of Japanese plum by S-Allele-Specific PCR Analysis. Nucleotide sequences EU113259.1, EU113260.1, EU113261.1, EU113262.1, EU113263.1, EU113264.1, EU113265.1, EU113266.1, EU113267.1. Available online: https://www.ncbi.nlm.nih.gov/nuccore (accessed on 28 December 2022).
- Wuyun, T.N.; Li, H.G. Isolation of Six SFB New Genes in the Prunus armeniaca (China Apricot) Nucleotide Sequence FJ377732.1. Available online: https://www.ncbi.nlm.nih.gov/nuccore (accessed on 28 December 2022).
- Wu Yun, T.N.; Li, H.G. Isolation of Six New S-RNase Genes in the Prunus armeniaca. Nucleotide Sequence GU574195.1. Available online: https://www.ncbi.nlm.nih.gov/nuccore (accessed on 28 December 2022).
- Na, T.; Yun, W.; Li, H.G. Isolation of Six New S-RNase Genes in the Prunus salicina. Nucleotide Sequence GU968758.1. Available online: https://www.ncbi.nlm.nih.gov/nuccore (accessed on 28 December 2022).
- Ushijima, K.; Sassa, H.; Tao, R.; Yamane, H.; Dandekar, A.M.; Gradziel, T.M.; Hirano, H. Cloning and characterization of cDNAs encoding S-RNases from almond (Prunus dulcis): Primary structural features and sequence diversity of the S-RNases in Rosaceae. Mol. Gen. Genet. 1998, 260, 261–268. [Google Scholar] [CrossRef]
- Edgar, R.C. Muscle5: High-accuracy alignment ensembles enable unbiased assessments of sequence homology and phylogeny. Nat. Commun. 2022, 13, 6968. [Google Scholar] [CrossRef] [PubMed]
- Guerra, M.E.; Rodrigo, J.; López-Corrales, M.; Wünsch, A. S-RNase genotyping and incompatibility group assignment by PCR and pollination experiments in Japanese plum. Plant Breed. 2009, 128, 304–311. [Google Scholar] [CrossRef]
- Okie, W.R.; Hancock, J.F. Plums. In Temperate Fruit Crop Breeding, Hancock, J.F., Ed.; Springer: Dordrecht, Germany, 2008; pp. 337–357. [Google Scholar]
- Faust, M.; Surányi, D. Origin and Dissemination of Plums. In Horticultural Reviews; Janick, J., Ed.; John Wiley & Sons Inc.: New York, NY, USA, 1999; pp. 179–231. [Google Scholar]
- Okie, W.R.; Weinberger, J.H. Plums. In Fruit Breeding, Volume I: Tree and Tropical Fruits; Janick, J., Moore, J.N., Eds.; John Wiley and Sons, Inc.: New York, NY, USA, 1996; Volume 1, pp. 559–608. [Google Scholar]
- Wei, X.W.; Shen, F.; Zhang, Q.; Liu, N.; Zhang, Y.; Xu, M.; Liu, S.; Zhang, Y.; Ma, X.; Liu, W. Genetic diversity analysis of Chinese plum (Prunus salicina L.) based on whole-genome resequencing. Tree Genet. Genomes 2021, 17, 26. [Google Scholar] [CrossRef]
- Liu, C.; Feng, C.; Peng, W.; Hao, J.; Wang, J.; Pan, J.; He, Y. Chromosome-level draft genome of a diploid plum (Prunus salicina). GigaScience 2020, 9, giaa13. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Shen, F.; Chen, Y.; Cao, K.; Wang, L. Chromosome-scale genome assembly and population genomics provide insights into the adaptation, domestication, and flavonoid metabolism of Chinese plum. Plant J. 2021, 108, 1174–1192. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.; Li, S.; Zhang, Y.; Chen, M.; Wen, B.; Jiang, S.; Chen, X.; Fu, X.; Li, D.; Wu, H.; et al. Chromosome-level genome assemblies of five Prunus species and genome-wide association studies for key agronomic traits in peach. Hortic. Res. 2021, 8, 213. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Cock, P.J.A.; Chilton, J.M.; Grüning, B.; Johnson, J.E.; Soranzo, N. NCBI BLAST+ integrated into Galaxy. GigaScience 2015, 4, 39. [Google Scholar] [CrossRef] [Green Version]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Cech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses. Nucleic Acids Res. 2018, 48, W395–W402. [Google Scholar]
- Andrews, S. FastQC: A Quality Control Tool for High Througput Sequence Data. Available online: http://www.bioinformataics.babraham.ac.uk/proyects/fastqc (accessed on 28 December 2022).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Breese, M.R.; Liu, Y. NGSUtils: A software suite for analyzing and manipulating next-generation sequencing datasets. Bioinformatics 2013, 29, 494–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Merisov, J.P. Integrative genomics viewer. Nat. Biotech. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Cao, K.; Li, Y.; Deng, C.H.; Gardiner, S.E.; Zhu, G.; Fang, W.; Chen, C.; Wang, X.; Wang, L. Comparative population genomics identified genomic regions and candidate genes associated with fruit domestication traits in peach. Plant Biotechnol. J. 2019, 17, 1954–1970. [Google Scholar] [CrossRef] [Green Version]
- Fang, B.; Xu, Y.; Yu, J. The complete chloroplast genome sequence of Prunus salicina ‘Wushan plum’. Mitochondrial DNA B. 2021, 26, 1243–1244. [Google Scholar] [CrossRef]
- Wang, X.; Liu, S.; Zuo, H.; Zheng, W.; Zhang, S.; Huang, Y.; Pingcuo, G.; Ying, H.; Zhao, F.; Li, Y.; et al. Genomic basis of high-altitude adaptation in Tibetan Prunus fruit trees. Curr. Biol. 2021, 31, 3848–3860. [Google Scholar] [CrossRef]
- Cao, K.; Peng, Z.; Zhao, X.; Li, Y.; Liu, K.; Arús, P.; Fang, W.; Chen, C.; Wang, X.; Wu, J.; et al. Chromosome level genome assemblies of four wild peach species provide insights into genome evolution and genetic basis of stress resistance. BMC Biol. 2022, 139. [Google Scholar] [CrossRef]
- Xu, Y.; Fang, B.; Wang, Y.; Liu, J.; Liu, C.; Yu, J. Phylogenomic analysis and development of molecular markers for the determination of twelve plum cultivars (Prunus, Rosaceae). BMC Genomics 2022, 23, 745. [Google Scholar] [CrossRef]
- Fiol, A.; Garcí a-Gómez, B.E.; Jurado-Ruiz, F.; Alexiou, K.; Howard, W.; Aranzana, M.J. Characterization of Japanese Plum (Prunus salicina) PsMYB10 Alleles Reveals Structural Variation and Polymorphisms Correlating with Fruit Skin Color. Frontiers Plant Sci. 2021, 12, 655267. [Google Scholar] [CrossRef] [PubMed]
- Fiol, A.; Jurado-Ruiz, F.; López-Girona, E.; Aranzana, M.J. An efficient CRISPR-Cas9 enrichment sequencing strategy for characterizing complex and highly duplicated genomic regions. A case study in the Prunus salicina LG3 MYB10 genes cluster. Plant Methods 2022, 18, 105. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.-Z.; Wang, K.-L.; Dai, H.; Zhou, D.-R.; Jiang, C.-C.; Espley, R.V.; Deng, C.; Lin, Y.-J.; Pan, S.-L.; Ye, X.-F. The genome of low-chill Chinese plum “Sanyueli” (Prunus salicina Lindl.) provides insights into the regulation of the chilling requirement of flower buds. Mol. Ecol. Resour. 2022, 22, 1919–1938. [Google Scholar] [CrossRef] [PubMed]
- Numaguchi, K.; Akagi, T.; Kitamura, Y.; Ishikawa, R.; Ishii, T. Interspecific introgression and natural selection in the evolution of Japanese apricot (Prunus mume). The Plant J. 2020, 104, 1551–1567. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hedhly, A.; Guerra, M.E.; Grimplet, J.; Rodrigo, J. S-Locus Genotyping in Japanese Plum by High Throughput Sequencing Using a Synthetic S-Loci Reference Sequence. Int. J. Mol. Sci. 2023, 24, 3932. https://doi.org/10.3390/ijms24043932
Hedhly A, Guerra ME, Grimplet J, Rodrigo J. S-Locus Genotyping in Japanese Plum by High Throughput Sequencing Using a Synthetic S-Loci Reference Sequence. International Journal of Molecular Sciences. 2023; 24(4):3932. https://doi.org/10.3390/ijms24043932
Chicago/Turabian StyleHedhly, Afif, María Engracia Guerra, Jerome Grimplet, and Javier Rodrigo. 2023. "S-Locus Genotyping in Japanese Plum by High Throughput Sequencing Using a Synthetic S-Loci Reference Sequence" International Journal of Molecular Sciences 24, no. 4: 3932. https://doi.org/10.3390/ijms24043932
APA StyleHedhly, A., Guerra, M. E., Grimplet, J., & Rodrigo, J. (2023). S-Locus Genotyping in Japanese Plum by High Throughput Sequencing Using a Synthetic S-Loci Reference Sequence. International Journal of Molecular Sciences, 24(4), 3932. https://doi.org/10.3390/ijms24043932