

All-Atom Molecular Dynamics Simulations Indicated the Involvement of a Conserved Polar Signaling Channel in the Activation Mechanism of the Type I Cannabinoid Receptor



Abstract
:1. Introduction
2. Results and Discussion
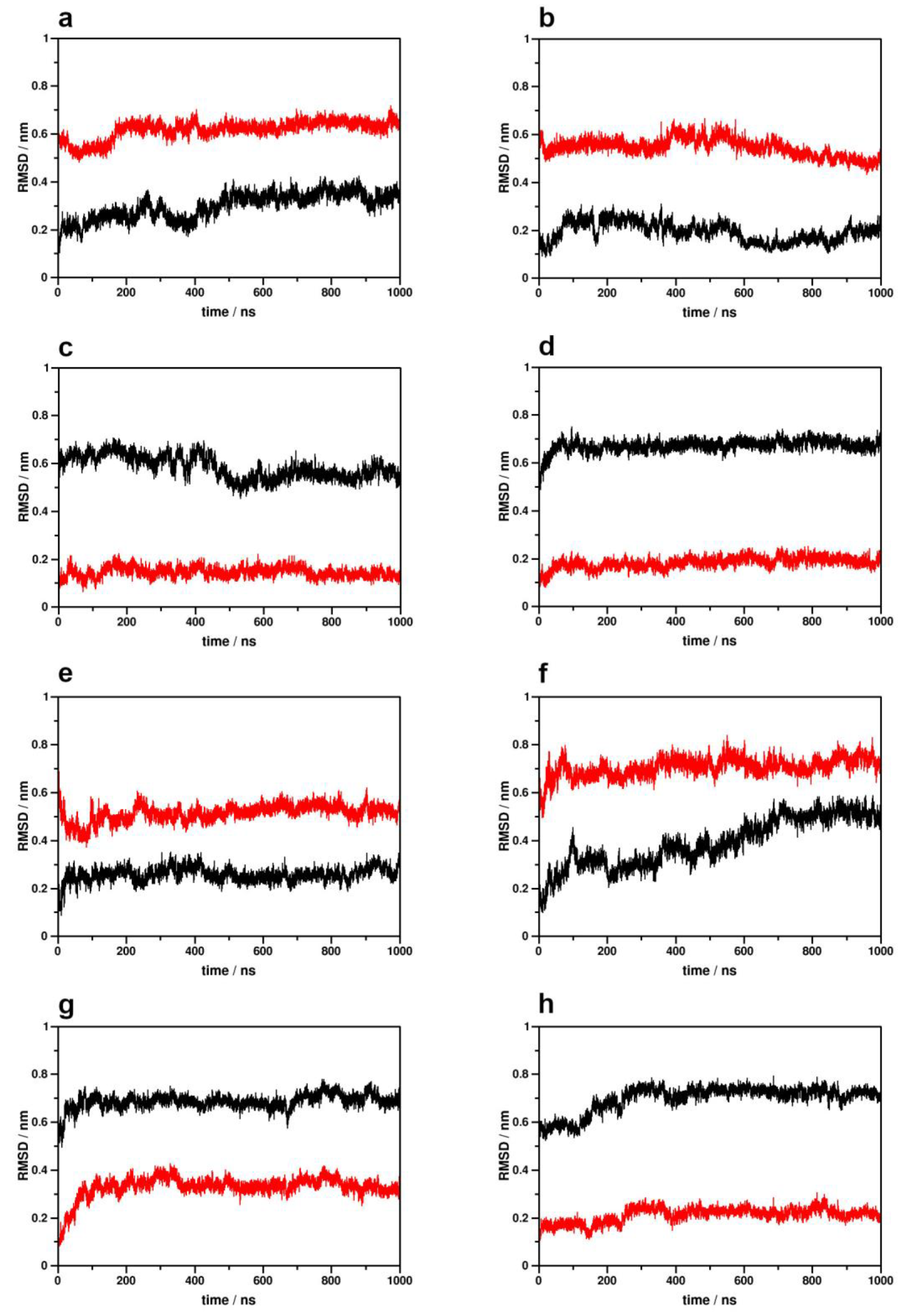
2.1. Simulation System Integrity
2.2. Transmembrane Helix and Loop Dynamics
2.3. Intramolecular Interactions
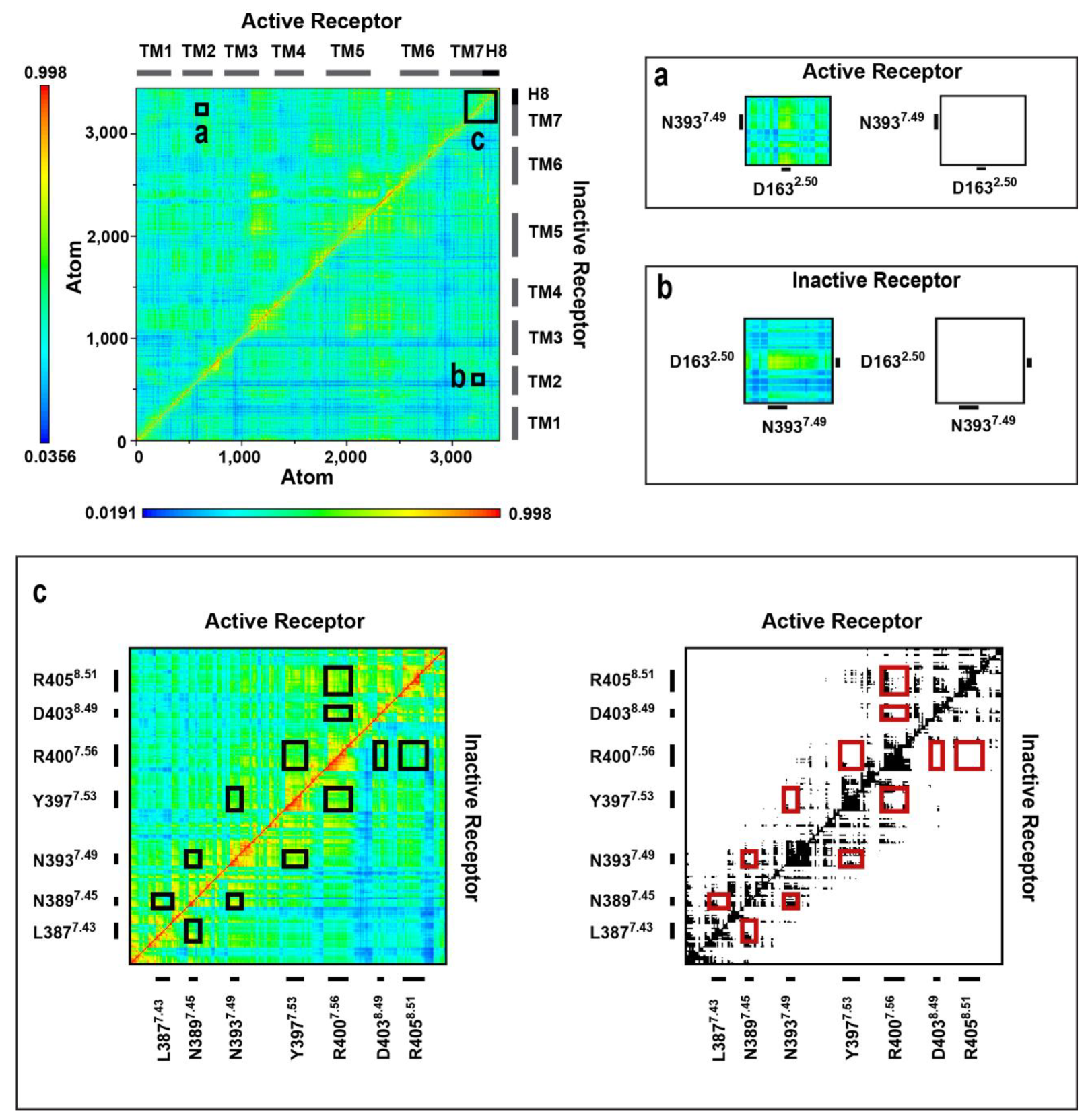
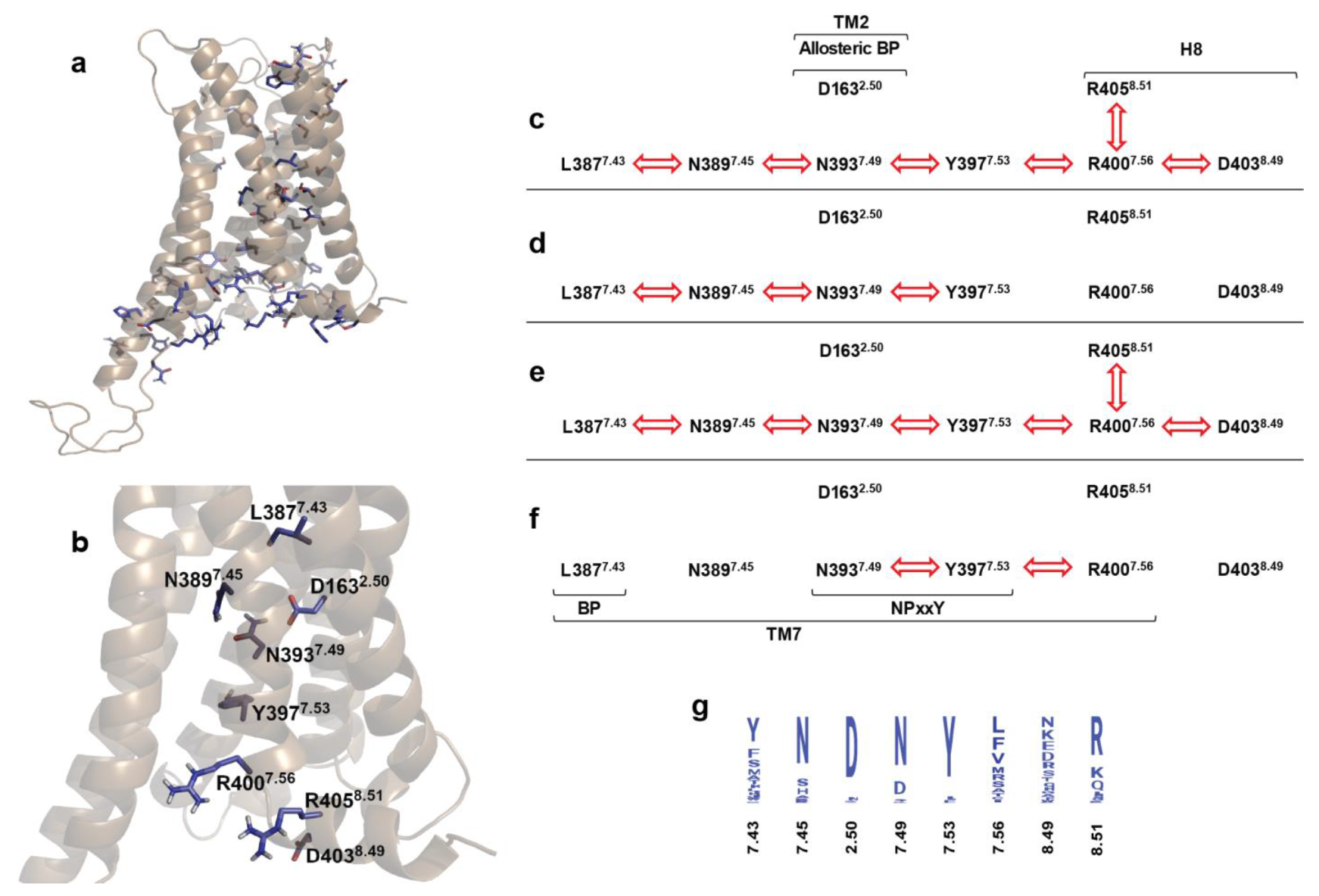
2.4. Correlated Side Chain Motions in the Transmembrane Domain
2.5. Alternative Signaling States
3. Conclusions
4. Methods
4.1. System Building
4.2. MD Simulations
4.3. MD Trajectory Analysis
4.4. Sequence Alignment and Conservation Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef] [Green Version]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D.E. Trends in GPCR Drug Discovery: New Agents, Targets and Indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef]
- Congreve, M.; de Graaf, C.; Swain, N.A.; Tate, C.G. Impact of GPCR Structures on Drug Discovery. Cell 2020, 181, 81–91. [Google Scholar] [CrossRef]
- Matsuda, L.A.; Lolait, S.J.; Brownstein, M.J.; Young, A.C.; Bonner, T.I. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 1990, 346, 561–564. [Google Scholar] [CrossRef]
- Aizpurua-Olaizola, O.; Elezgarai, I.; Rico-Barrio, I.; Zarandona, I.; Etxebarria, N.; Usobiaga, A. Targeting the endocannabinoid system: Future therapeutic strategies. Drug. Discov. Today 2017, 22, 105–110. [Google Scholar] [CrossRef]
- Al-Zoubi, R.; Morales, P.; Reggio, P.H. Structural Insights into CB1 Receptor Biased Signaling. Int. J. Mol. Sci. 2019, 20, 1837. [Google Scholar] [CrossRef] [Green Version]
- Pándy-Szekeres, G.; Munk, C.; Tsonkov, T.M.; Mordalski, S.; Harpsøe, K.; Hauser, A.S.; Bojarski, A.J.; Gloriam, D.E. GPCRdb in 2018: Adding GPCR structure models and ligands. Nucleic. Acids. Res. 2018, 46, D440–D446. [Google Scholar] [CrossRef] [Green Version]
- Cvicek, V.; Goddard, W.A., III; Abrol, R. Structure-Based Sequence Alignment of the Transmembrane Domains of All Human GPCRs: Phylogenetic, Structural and Functional Implications. PLoS Comput. Biol. 2016, 12, e1004805. [Google Scholar] [CrossRef] [Green Version]
- Latorraca, N.R.; Venkatakrishnan, A.J.; Dror, R.O. GPCR Dynamics: Structures in Motion. Chem. Rev. 2017, 117, 139–155. [Google Scholar] [CrossRef]
- Pert, C.B.; Pasternak, G.; Snyder, S.H. Opiate agonists and antagonists discriminated by receptor binding in brain. Science 1973, 182, 1359–1361. [Google Scholar] [CrossRef]
- Palczewski, K.; Kumasaka, T.; Hori, T.; Behnke, C.A.; Motoshima, H.; Fox, B.A.; Le Trong, I.; Teller, D.C.; Okada, T.; Stenkamp, R.E.; et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science 2000, 289, 739–745. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Chun, E.; Thompson, A.A.; Chubukov, P.; Xu, F.; Katritch, V.; Han, G.W.; Roth, C.B.; Heitman, L.H.; Ijzerman, A.P.; et al. Structural basis for allosteric regulation of GPCRs by sodium ions. Science 2012, 337, 232–236. [Google Scholar] [CrossRef] [Green Version]
- Stoddart, L.A.; Kellam, B.; Briddon, S.J.; Hill, S.J. Effect of a toggle switch mutation in TM6 of the human adenosine A3 receptor on Gi protein-dependent signalling and Gi-independent receptor internalization. Br. J. Pharmacol. 2014, 171, 3827–3844. [Google Scholar] [CrossRef]
- Katritch, V.; Fenalti, G.; Abola, E.E.; Roth, B.L.; Cherezov, V.; Stevens, R.C. Allosteric sodium in class A GPCR signaling. Trends Biochem. Sci. 2014, 39, 233–244. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Yang, D.; Wu, M.; Guo, Y.; Guo, W.; Zhong, L.; Cai, X.; Dai, A.; Jang, W.; Shakhnovich, E.I.; et al. Common activation mechanism of class A GPCRs. Elife. 2019, 8, e50279. [Google Scholar] [CrossRef]
- Filipek, S. Molecular switches in GPCRs. Curr. Opin. Struct. Biol. 2019, 55, 114–120. [Google Scholar] [CrossRef]
- Mitra, A.; Sarkar, A.; Szabó, M.R.; Borics, A. Correlated Motions of Conserved Polar Motifs Lay out a Plausible Mechanism of G Protein-Coupled Receptor Activation. Biomolecules 2021, 11, 670. [Google Scholar] [CrossRef]
- Mitra, A.; Sarkar, A.; Borics, A. Universal properties and specificities of the β2-adrenergic receptor–Gs protein complex activation mechanism revealed by all-atom molecular dynamics simulations. Int. J. Mol. Sci. 2021, 22, 10423. [Google Scholar] [CrossRef]
- Fenalti, G.; Giguere, P.M.; Katritch, V.; Huang, X.P.; Thompson, A.A.; Cherezov, V.; Roth, B.L.; Stevens, R.C. Molecular Control of δ-Opioid Receptor Signaling. Nature 2014, 506, 191–196. [Google Scholar] [CrossRef] [Green Version]
- Jongejan, A.; Bruysters, M.; Ballesteros, J.A.; Haaksma, E.; Bakker, R.A.; Pardo, L.; Leurs, R. Linking Agonist Binding to Histamine H1 Receptor Activation. Nat. Chem. Biol. 2005, 1, 98–103. [Google Scholar] [CrossRef]
- Liu, R.; Nahon, D.; le Roy, B.; Lenselink, E.B.; Ijzerman, A.P. Scanning Mutagenesis in a Yeast System Delineates the Role of the NPxxY(x)5,6F Motif and Helix 8 of the Adenosine A2B Receptor in G Protein Coupling. Biochem. Pharmacol. 2015, 95, 290–300. [Google Scholar] [CrossRef]
- Hothersall, J.D.; Torella, R.; Humphreys, S.; Hooley, M.; Brown, A.; McMurray, G.; Nickolls, S.A. Residues W320 and Y328 within the Binding Site of the μ-Opioid Receptor Influence Opiate Ligand Bias. Neuropharmacology 2017, 118, 46–58. [Google Scholar] [CrossRef]
- Sealfon, S.C.; Chi, L.; Ebersole, B.J.; Rodic, V.; Zhang, D.; Ballesteros, J.A.; Weinstein, H. Related Contribution of Specific Helix 2 and 7 Residues to Conformational Activation of the Serotonin 5-HT2A Receptor. J. Biol. Chem. 1995, 270, 16683–16688. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Ozdener, F.; Li, J.G.; Chen, C.; de Riel, J.K.; Weinstein, H.; Liu-Chen, L.Y. Functional Role of the Spatial Proximity of Asp1142.50 in TMH 2 and Asn3327.49 in TMH 7 of the Mu Opioid Receptor. FEBS Lett. 1999, 447, 318–324. [Google Scholar] [CrossRef] [Green Version]
- Galés, C.; Kowalski-Chauvel, A.; Dufour, M.N.; Seva, C.; Moroder, L.; Pradayrol, L.; Vaysse, N.; Fourmy, D.; Silvente-Poirot, S. Mutation of Asn-391 within the Conserved NPxxY Motif of the Cholecystokinin B Receptor Abolishes Gq Protein Activation without Affecting Its Association with the Receptor. J. Biol. Chem. 2000, 275, 17321–17327. [Google Scholar] [CrossRef] [Green Version]
- Barak, L.S.; Tiberi, M.; Freedman, N.J.; Kwatra, M.M.; Lefkowitz, R.J.; Caron, M.G. A Highly Conserved Tyrosine Residue in G Protein-Coupled Receptors is Required for Agonist-Mediated Beta 2-Adrenergic Receptor Sequestration. J. Biol. Chem. 1994, 269, 2790–2795. [Google Scholar] [CrossRef]
- Prioleau, C.; Visiers, I.; Ebersole, B.J.; Weinstein, H.; Sealfon, S.C. Conserved Helix 7 Tyrosine Acts as a Multistate Conformational Switch in the 5-HT2C Receptor. Identification of a Novel “LOCKED-ON” Phenotype and Double Revertant Mutations. J. Biol. Chem. 2002, 277, 36577–36584. [Google Scholar]
- Kalatskaya, I.; Schüssler, S.; Blaukat, A.; Müller-Esterl, W.; Jochum, M.; Proud, D.; Faussner, A. Mutation of Tyrosine in the Conserved NPxxY Sequence Leads to Constitutive Phosphorylation and Internalization, but Not Signaling of the Human B2 Bradykinin Receptor. J. Biol. Chem. 2004, 279, 31268–31276. [Google Scholar] [CrossRef] [Green Version]
- Suomivuori, C.M.; Latorraca, N.R.; Wingler, L.M.; Eismann, S.; King, M.C.; Kleinhenz, A.L.W.; Skiba, M.A.; Staus, D.P.; Kruse, A.C.; Lefkowitz, R.J.; et al. Molecular mechanism of biased signaling in a prototypical G protein-coupled receptor. Science 2020, 367, 881–887. [Google Scholar] [CrossRef]
- Qu, Q.; Huang, W.; Aydin, D.; Paggi, J.M.; Seven, A.B.; Wang, H.; Chakraborty, S.; Che, T.; DiBerto, J.F.; Robertson, M.J.; et al. Insights into distinct signaling profiles of the µOR activated by diverse agonists. Nat. Chem. Biol. 2022; Epub ahead of print. [Google Scholar] [CrossRef]
- Krishna Kumar, K.; Shalev-Benami, M.; Robertson, M.J.; Hu, H.; Banister, S.D.; Hollingsworth, S.A.; Latorraca, N.R.; Kato, H.E.; Hilger, D.; Maeda, S.; et al. Structure of a Signaling Cannabinoid Receptor 1-G Protein Complex. Cell 2019, 176, 448–458. [Google Scholar] [CrossRef] [Green Version]
- Hua, T.; Vemuri, K.; Pu, M.; Qu, L.; Han, G.W.; Wu, Y.; Zhao, S.; Shui, W.; Li, S.; Korde, A.; et al. Crystal Structure of the Human Cannabinoid Receptor CB1. Cell 2016, 167, 750–762. [Google Scholar] [CrossRef] [Green Version]
- Tao, Q.; Abood, M.E. Mutation of a highly conserved aspartate residue in the second transmembrane domain of the cannabinoid receptors, CB1 and CB2, disrupts G-protein coupling. J. Pharmacol. Exp. Ther. 1998, 285, 651–658. [Google Scholar]
- Dror, R.O.; Arlow, D.H.; Maragakis, P.; Mildorf, T.J.; Pan, A.C.; Xu, H.; Borhani, D.W.; Shaw, D.E. Activation mechanism of the beta2-adrenergic receptor. Proc. Natl. Acad. Sci USA 2011, 108, 18684–18689. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Warne, T.; Nehmé, R.; Pandey, S.; Dwivedi-Agnihotri, H.; Chaturvedi, M.; Edwards, P.C.; García-Nafría, J.; Leslie, A.G.W.; Shukla, A.K.; et al. Molecular basis of β-arrestin coupling to formoterol-bound β1-adrenoceptor. Nature 2020, 583, 862–866. [Google Scholar] [CrossRef]
- Huang, W.; Masureel, M.; Qu, Q.; Janetzko, J.; Inoue, A.; Kato, H.E.; Robertson, M.J.; Nguyen, K.C.; Glenn, J.S.; Skiniotis, G.; et al. Structure of the neurotensin receptor 1 in complex with β-arrestin 1. Nature 2020, 579, 303–308. [Google Scholar] [CrossRef]
- Ma, X.; Hu, Y.; Batebi, H.; Heng, J.; Xu, J.; Liu, X.; Niu, X.; Li, H.; Hildebrand, P.W.; Jin, C.; et al. Analysis of β2AR-Gs and β2AR-Gi complex formation by NMR spectroscopy. Proc. Natl. Acad. Sci. USA 2020, 117, 23096–23105. [Google Scholar] [CrossRef]
- Rasmussen, S.G.; DeVree, B.T.; Zou, Y.; Kruse, A.C.; Chung, K.Y.; Kobilka, T.S.; Thian, F.S.; Chae, P.S.; Pardon, E.; Calinski, D.; et al. Crystal structure of the β2 adrenergic receptor–Gs protein complex. Nature 2011, 477, 549–555. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Manglik, A.; Venkatakrishnan, A.J.; Laeremans, T.; Feinberg, E.N.; Sanborn, A.L.; Kato, H.E.; Livingston, K.E.; Thorsen, T.S.; Kling, R.C.; et al. Structural Insights into µ-Opioid Receptor Activation. Nature 2015, 524, 315–321. [Google Scholar] [CrossRef] [Green Version]
- Wall, M.A.; Coleman, D.E.; Lee, E.; Iñiguez-Lluhi, J.A.; Posner, B.A.; Gilman, A.G.; Sprang, S.R. The structure of the G protein heterotrimer Giα1β1γ2. Cell 1995, 83, 1047–1058. [Google Scholar] [CrossRef] [Green Version]
- Zhan, X.; Gimenez, L.E.; Gurevich, V.V.; Spiller, B.W. Crystal Structure of Arrestin-3 Reveals the Basis of the Difference in Receptor Binding Between Two Non-visual Subtypes. J. Mol. Biol. 2011, 406, 467–478. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.; Zhou, X.E.; Gao, X.; He, Y.; Liu, W.; Ishchenko, A.; Barty, A.; White, T.A.; Yefanov, O.; Han, G.W.; et al. Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature 2015, 523, 561–567. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and AutoDockTools4: Automated docking with selective receptor flexiblity. J. Comp. Chem. 2009, 16, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Hua, T.; Vemuri, K.; Nikas, S.P.; Laprairie, R.B.; Wu, Y.; Qu, L.; Pu, M.; Korde, A.; Jiang, S.; Ho, J.H.; et al. Crystal structures of agonist-bound human cannabinoid receptor CB1. Nature 2017, 547, 468–471. [Google Scholar] [CrossRef] [Green Version]
- Fiser, A.; Do, R.K.; Sali, A. Modeling of loops in protein structures. Prot. Sci. 2000, 9, 1753–1773. [Google Scholar]
- Johansson, M.U.; Zoete, V.; Michielin, O.; Guex, N. Defining and searching for structural motifs using DeepView/Swiss-PdbViewer. BMC Bioinform. 2012, 13, 173. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A Web-based Graphical User Interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Howlett, A.C.; Champion-Dorow, T.M.; McMahon, L.L.; Westlake, T.M. The cannabinoid receptor: Biochemical and cellular properties in neuroblastoma cells. Pharmacol. Biochem. Behav. 1991, 40, 565–569. [Google Scholar] [CrossRef]
- Song, C.; Howlett, A.C. Rat brain cannabinoid receptors are N-linked glycosylated proteins. Life Sci. 1995, 56, 1983–1989. [Google Scholar] [CrossRef]
- Ruehle, S.; Wager-Miller, J.; Straiker, A.; Farnsworth, J.; Murphy, M.N.; Loch, S.; Monory, K.; Mackie, K.; Lutz, B. Discovery and characterization of two novel CB1 receptor splice variants with modified N-termini in mouse. J. Neurochem. 2017, 142, 521–533. [Google Scholar] [CrossRef] [Green Version]
- Rapino, C.; Castellucci, A.; Lizzi, A.R.; Sabatucci, A.; Angelucci, C.B.; Tortolani, D.; Rossi, G.; D’Andrea, G.; Maccarrone, M. Modulation of Endocannabinoid-Binding Receptors in Human Neuroblastoma Cells by Tunicamycin. Molecules 2019, 24, 1432. [Google Scholar] [CrossRef] [Green Version]
- Oddi, S.; Dainese, E.; Sandiford, S.; Fezza, F.; Lanuti, M.; Chiurchiù, V.; Totaro, A.; Catanzaro, G.; Barcaroli, D.; De Laurenzi, V.; et al. Effects of palmitoylation of Cys415 in helix 8 of the CB1 cannabinoid receptor on membrane localization and signalling. Br. J. Pharmacol. 2012, 165, 2635–2651. [Google Scholar] [CrossRef] [Green Version]
- Jin, W.; Brown, S.; Roche, J.P.; Hsieh, C.; Celver, J.P.; Kovoor, A.; Chavkin, C.; Mackie, K. Distinct domains of the CB1 cannabinoid receptor mediate desensitization and internalization. J. Neurosci. 1999, 19, 3773–3780. [Google Scholar] [CrossRef] [Green Version]
- Howlett, A.C.; Blume, L.C.; Dalton, G.D. CB1 cannabinoid receptors and their associated proteins. Curr. Med. Chem. 2010, 17, 1382–1393. [Google Scholar] [CrossRef] [Green Version]
- Pike, L.J.; Han, X.; Chung, K.N.; Gross, R.W. Lipid rafts are enriched in arachidonic acid and plasmenylethanolamine and their composition is independent of caveolin-1 expression: A quantitative electrospray ionization/mass spectrometric analysis. Biochemistry 2002, 41, 2075–2088. [Google Scholar] [CrossRef]
- Ingólfsson, H.I.; Melo, M.N.; van Eerden, F.J.; Arnarez, C.; Lopez, C.A.; Wassenaar, T.A.; Periole, X.; de Vries, A.H.; Tieleman, D.P.; Marrink, S.J. Lipid organization of the plasma membrane. J. Am. Chem. Soc. 2014, 136, 14554–14559. [Google Scholar] [CrossRef]
- Huang, J.; MacKerell, A.D., Jr. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [Green Version]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C.; Postma, J.P.M.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Lange, O.F.; Grubmüller, H. Generalized correlation for biomolecular dynamics. Proteins 2006, 62, 1053–1061. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Sys. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2-a multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand Disposition | Na+ Penetration | TM6 Disposition | NPxxY Disposition | Correlated Motions in Segment 1 | Correlated Motions in Segment 2 | Signaling State | |
|---|---|---|---|---|---|---|---|
| active CB1-Gi complex, replica 1 | moderate | no | moderate | stable | complete | complete | canonical |
| active CB1-Gi complex, replica 2 | large | no | stable | transition to intermediate | complete | complete | canonical |
| inactive CB1-Gi complex, replica 1 | moderate | no | stable | stable | complete | incomplete | N/A |
| inactive CB1-Gi complex, replica 2 | moderate | yes | stable | stable | complete | incomplete | N/A |
| active CB1-β-Arr-2 complex, replica 1 | moderate | no | moderate | transition to intermediate | complete | complete | transition to alternative |
| active CB1-β-Arr-2 complex, replica 2 | large | no | transition to inactive | transition to intermediate | complete | incomplete | transition to alternative |
| inactive CB1-β-Arr-2 complex, replica 1 | large | yes | transition to intermediate | transition to intermediate | complete | incomplete | N/A |
| inactive CB1-β-Arr-2 complex, replica 2 | moderate | no | stable | transition to intermediate | incomplete | incomplete | N/A |
| Interactions | Residues Involved | Gi Protein Complex | β-Arrestin-2 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Active State | Inactive State | Active State | Inactive State | ||||||
| Replica 1 | Replica 2 | Replica 1 | Replica 2 | Replica 1 | Replica 2 | Replica 1 | Replica 2 | ||
| salt bridges | |||||||||
| DRY—H8 | R2143.50; D4038.49 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| intra-DRY | D2133.49; R2143.50 | 0.0 | 0.0 | 25.6 | 25.3 | 24.4 | 3.6 | 27.33 | 27.24 |
| DRY—TM6 | R2143.50; D3386.30 | 0.0 | 0.0 | 16.5 | 30.5 | 0.0 | 0.0 | 17.2 | 26.1 |
| H-bonds | |||||||||
| DRY—H8 | R2143.50; S4018.47 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.1 | 0.0 | 0.0 |
| R2143.50; D4038.49 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.1 | 0.0 | 0.0 | |
| R2143.50; R4058.51 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | |
| intra-DRY | D2133.49; R2143.50 | 0.1 | 0.0 | 99.9 | 99.8 | 94.9 | 20.5 | 99.8 | 99.9 |
| DRY—ICL2 | D2133.49; R220ICL2-T229ICL2 | 96.1 | 99.8 | 99.9 | 99.6 | 99.5 | 99.7 | 99.8 | 99.7 |
| DRY—TM5 | R2143.50; Y2945.58 | 96.3 | 64.4 | 0.0 | 0.0 | 0.1 | 0.2 | 0.0 | 0.0 |
| DRY—TM6 | R2143.50; D3386.30 | 0.0 | 0.0 | 37.8 | 98.8 | 0.0 | 0.0 | 43.5 | 99.6 |
| CWxP—TM7 | C3556.47-W3566.48; N3897.45 | 6.9 | 24.1 | 11.6 | 13.8 | 14.1 | 42.3 | 0.1 | 0.1 |
| Pathway-Specific Distance/Dihedral Angle | Canonical Active State | Alternative Active State | Active CB1-Gi Complex, Replica 1 | Active CB1-Gi Complex, Replica 2 | Active CB1-β-Arr-2 Complex, Replica 1 | Active CB1-β-Arr-2 Complex, Replica 2 |
|---|---|---|---|---|---|---|
| F1702.57—F2003.36 | ~1.00 nm | ~1.50 nm | canonical * | canonical * | canonical * | canonical * |
| D1632.50—S1993.35 | ~1.00 nm | ~0.50 nm | canonical | canonical | canonical | canonical |
| F1702.57—L3877.43 | ~0.50 nm | ~1.00 nm | canonical | canonical | canonical | canonical |
| G1662.53—F1702.57 | ~0.75 nm | ~0.50 nm | canonical | canonical | canonical | canonical |
| N1341.50—T3917.47 | ~0.50 nm | ~0.25 nm | canonical * | canonical * | canonical * | canonical *, large fluctuations |
| N1341.50—S3907.46 | ~0.25 nm | ~0.60 nm | alternative | alternative | switching between the two states | alternative, large fluctuations |
| L1592.46—P3947.50 | ~0.79 nm | ~0.64 nm | canonical | canonical | canonical, large fluctuations | switching between the two states |
| Y3977.53 χ1 | gauche- | trans | canonical | alternative * | alternative | alternative |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarkar, A.; Mitra, A.; Borics, A. All-Atom Molecular Dynamics Simulations Indicated the Involvement of a Conserved Polar Signaling Channel in the Activation Mechanism of the Type I Cannabinoid Receptor. Int. J. Mol. Sci. 2023, 24, 4232. https://doi.org/10.3390/ijms24044232
Sarkar A, Mitra A, Borics A. All-Atom Molecular Dynamics Simulations Indicated the Involvement of a Conserved Polar Signaling Channel in the Activation Mechanism of the Type I Cannabinoid Receptor. International Journal of Molecular Sciences. 2023; 24(4):4232. https://doi.org/10.3390/ijms24044232
Chicago/Turabian StyleSarkar, Arijit, Argha Mitra, and Attila Borics. 2023. "All-Atom Molecular Dynamics Simulations Indicated the Involvement of a Conserved Polar Signaling Channel in the Activation Mechanism of the Type I Cannabinoid Receptor" International Journal of Molecular Sciences 24, no. 4: 4232. https://doi.org/10.3390/ijms24044232
APA StyleSarkar, A., Mitra, A., & Borics, A. (2023). All-Atom Molecular Dynamics Simulations Indicated the Involvement of a Conserved Polar Signaling Channel in the Activation Mechanism of the Type I Cannabinoid Receptor. International Journal of Molecular Sciences, 24(4), 4232. https://doi.org/10.3390/ijms24044232